
Infectious Bursal Disease Virus-Host Interactions: Multifunctional Viral Proteins that Perform Multiple and Differing Jobs


{kind=link}
Abstract
:1. Introduction
2. Virus Characteristics
2.1. Virus Structure of IBDV
2.2. Genetic Variation of IBDV
3. Host and Target Cells
4. Cellular Receptors and the Key Elements Associated with the Entry of IBDV
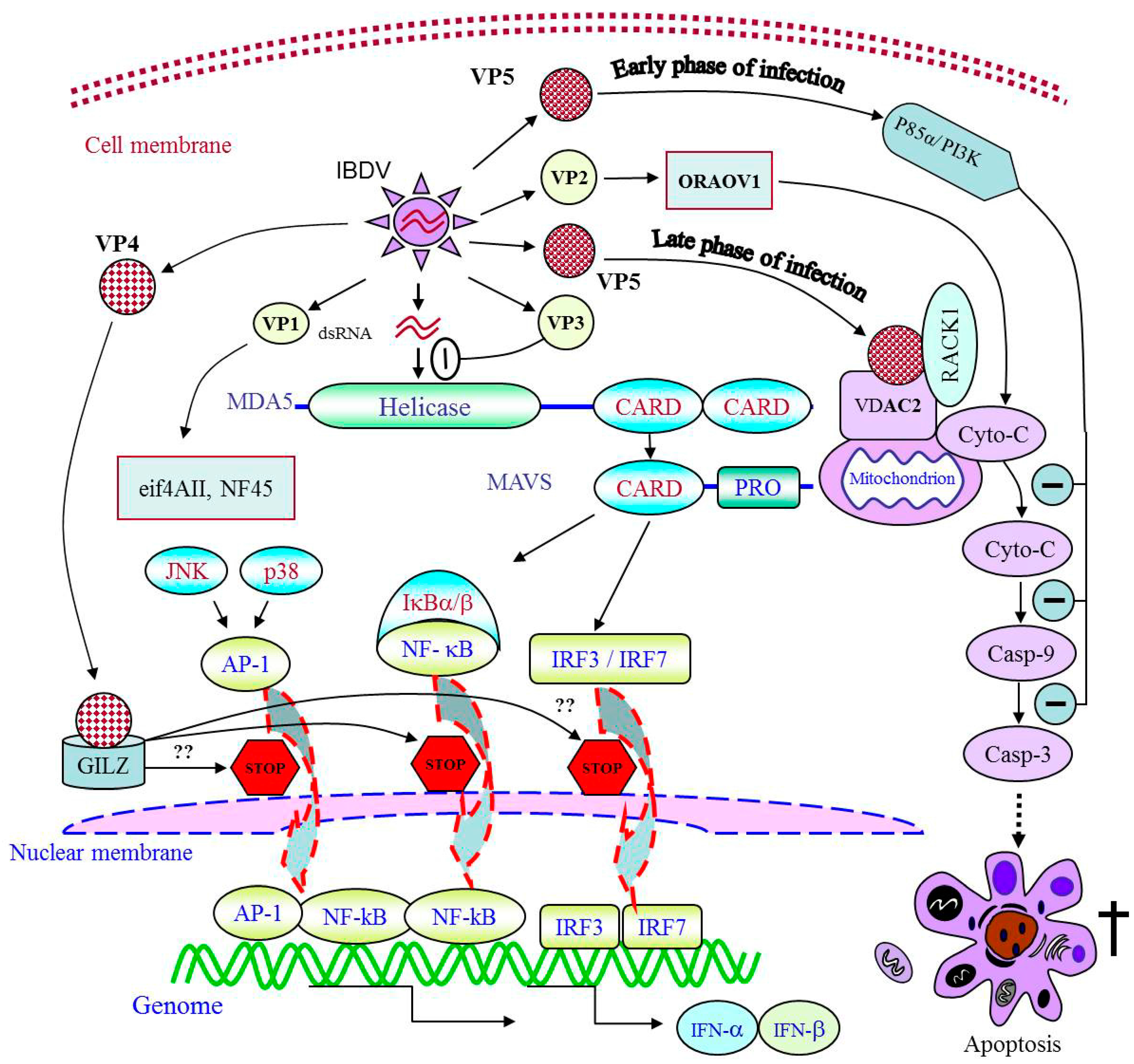
5. Suppression of Host Immune Responses
5.1. Apoptosis Leads to the Depletion of Lymphoid Cells
5.2. Key Elements That Impair the Innate Immune Response
6. Cellular Factors That Affect the Replication of IBDV
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sharma, J.M.; Kim, I.; Rautenschlein, S.; Yeh, H. Infectious bursal disease virus of chickens: Pathogenesis and immunosuppression. Dev. Comp. Immunol. 2000, 24, 223–235. [Google Scholar] [CrossRef]
- Nera, K.; Kylaniemi, M.K.; Lassila, O. Bursa of Fabricius; John Wiley & Sons, Ltd.: Chichester, UK, 2015. [Google Scholar]
- Mahgoub, H.A. An overview of infectious bursal disease. Arch. Virol. 2012, 157, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Ingrao, F.; Rauw, F.; Lambrecht, B.; van den Berg, T. Infectious Bursal Disease: A complex host-pathogen interaction. Dev. Comp. Immunol. 2013, 41, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Chettle, N.; Stuart, J.C.; Wyeth, P.J. Outbreak of virulent infectious bursal disease in East Anglia. Vet. Rec. 1989, 125, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Stoute, S.T.; Jackwood, D.J.; Sommer-Wagner, S.E.; Crossley, B.M.; Woolcock, P.R.; Charlton, B.R. Pathogenicity associated with coinfection with very virulent infectious bursal disease and Infectious bursal disease virus strains endemic in the United States. J. Vet. Diagn. Investig. 2013, 25, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, A.; Ramakrishnan, S.; Sachan, S.; Kumar, B.S.; Sharma, B.K.; Kumar, V.; Palanivelu, M.; Varghese, B.P.; Kumar, A.; Saravanan, B.C.; et al. Prophylactic potential of resiquimod against very virulent infectious bursal disease virus (vvIBDV) challenge in the chicken. Vet. Microbiol. 2016, 187, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vidalain, P.O.; Tangy, F. Virus-host protein interactions in RNA viruses. Microb. Infect. 2010, 12, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Withers, D.R.; Young, J.R.; Davison, T.F. Infectious bursal disease virus-induced immunosuppression in the chick is associated with the presence of undifferentiated follicles in the recovering bursa. Viral Immunol. 2005, 18, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.M. Alteration of chicken heterophil and macrophage functions by the infectious bursal disease virus. Microb. Pathog. 1998, 25, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rauw, F.; Lambrecht, B.; van den Berg, T. Pivotal role of ChIFNγ in the pathogenesis and immunosuppression of infectious bursal disease. Avian Pathol. 2007, 36, 367–374. [Google Scholar] [CrossRef] [PubMed]
- McNeilly, F.; Walker, I.; Allan, G.M.; Adair, B.M. Bursal lymphocyte proliferation in the presence of phorbol myristate acetate: Effect of IBDV strains on the proliferation response. Avian Pathol. 1999, 28, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Barrett, S.A.; Fahey, K.J. The characterization and molecular cloning of the double-stranded RNA genome of an Australian strain of infectious bursal disease virus. Virology 1985, 143, 35–44. [Google Scholar] [CrossRef]
- Macreadie, I.G.; Azad, A.A. Expression and RNA dependent RNA polymerase activity of birnavirus VP1 protein in bacteria and yeast. Biochem. Mol. Biol. Int. 1993, 30, 1169–1178. [Google Scholar] [PubMed]
- Von Einem, U.I.; Gorbalenya, A.E.; Schirrmeier, H.; Behrens, S.E.; Letzel, T.; Mundt, E. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J. Gen. Virol. 2004, 85, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Spies, U.; Muller, H.; Becht, H. Nucleotide sequence of infectious bursal disease virus genome segment A delineates two major open reading frames. Nucleic Acids Res. 1989, 17, 7982. [Google Scholar] [CrossRef] [PubMed]
- Kibenge, F.S.; McKenna, P.K.; Dybing, J.K. Genome cloning and analysis of the large RNA segment (segment A) of a naturally avirulent serotype 2 infectious bursal disease virus. Virology 1991, 184, 437–440. [Google Scholar] [CrossRef]
- Lejal, N.; Da, C.B.; Huet, J.C.; Delmas, B. Role of Ser-652 and Lys-692 in the protease activity of infectious bursal disease virus VP4 and identification of its substrate cleavage sites. J. Gen. Virol. 2000, 81, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, N.; Caston, J.R.; Rodriguez, J.F. Host Proteolytic Activity Is Necessary for Infectious Bursal Disease Virus Capsid Protein Assembly. J. Biol. Chem. 2012, 287, 24473–24482. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, N.; Garriga, D.; Navarro, A.; Verdaguer, N.; Rodriguez, J.F.; Caston, J.R. Autoproteolytic activity derived from the infectious bursal disease virus capsid protein. J. Biol. Chem. 2009, 284, 8064–8072. [Google Scholar] [CrossRef] [PubMed]
- Saugar, I.; Luque, D.; Oña, A.; Rodriguez, J.F.; Carrascosa, J.L.; Trus, B.L.; Castón, J.R. Structural Polymorphism of the Major Capsid Protein of a Double-Stranded RNA Virus: An Amphipathic α Helix as a Molecular Switch. Structure 2005, 13, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Casado, S.; Mata, C.P.; Hernando-Perez, M.; de Pablo, P.J.; Carrascosa, J.L.; Castón, J.R. A protein with simultaneous capsid scaffolding and dsRNA-binding activities enhances the birnavirus capsid mechanical stability. Sci. Rep. 2015, 5, 13486. [Google Scholar] [CrossRef] [PubMed]
- Dobos, P.; Hill, B.J.; Hallett, R.; Kells, D.T.; Becht, H.; Teninges, D. Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J. Virol. 1979, 32, 593–605. [Google Scholar] [PubMed]
- Chevalier, C.; Galloux, M.; Pous, J.; Henry, C.; Denis, J.; da Costa, B.; Navaza, J.; Lepault, J.; Delmas, B. Structural Peptides of a Nonenveloped Virus Are Involved in Assembly and Membrane Translocation. J. Virol. 2005, 79, 12253–12263. [Google Scholar] [CrossRef] [PubMed]
- Vakharia, V.N.; He, J.; Ahamed, B.; Snyder, D.B. Molecular basis of antigenic variation in infectious bursal disease virus. Virus Res. 1994, 31, 265–273. [Google Scholar] [CrossRef]
- Brandt, M.; Yao, K.; Liu, M.; Heckert, R.A.; Vakharia, V.N. Molecular determinants of virulence, cell tropism, and pathogenic phenotype of infectious bursal disease virus. J. Virol. 2001, 75, 11974–11982. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, L.; Chen, Y.; Gao, L.; Wu, G.; Qin, L.; Wang, Y.; Ren, X.; Gao, Y.; Gao, H.; et al. Mutations of residues 249 and 256 in VP2 are involved in the replication and virulence of infectious Bursal disease virus. PLoS ONE 2013, 8, e70982. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Zierenberg, K.; Muller, H. The genome segment B encoding the RNA-dependent RNA polymerase protein VP1 of very virulent infectious bursal disease virus (IBDV) is phylogenetically distinct from that of all other IBDV strains. Arch. Virol. 2001, 146, 2481–2492. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, J.; Zheng, J.; Xu, H.; Li, L.; Yu, L. Genetic reassortment of infectious bursal disease virus in nature. Biochem. Biophys. Res. Commun. 2006, 350, 277–287. [Google Scholar] [CrossRef] [PubMed]
- He, C.Q.; Ma, L.Y.; Wang, D.; Li, G.R.; Ding, N.Z. Homologous recombination is apparent in infectious bursal disease virus. Virology 2009, 384, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J. Molecular epidemiologic evidence of homologous recombination in infectious bursal disease viruses. Avian Dis. 2012, 56, 574–577. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xiong, Z.; Yang, L.; Guan, D.; Yang, X.; Wei, P. Molecular epidemiology studies on partial sequences of both genome segments reveal that reassortant infectious bursal disease viruses were dominantly prevalent in southern China during 2000–2012. Arch. Virol. 2014, 159, 3279–3292. [Google Scholar] [CrossRef] [PubMed]
- Pitesky, M.; Cataline, K.; Crossley, B.; Poulos, M.; Ramos, G.; Willoughby, D.; Woolcock, P.; Cutler, G.; Bland, M.; Tran, J.; et al. Historical, spatial, temporal, and time-space epidemiology of very virulent infectious bursal disease in California: A retrospective study 2008–2011. Avian Dis. 2013, 57, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, R.A.; Carrasco-Medanic, R.; Zhou, H.; Lyu, S.; Wang, Y.; Woolcock, P.R.; Hoerr, F.J. Effects of challenge with very virulent infectious bursal disease virus reassortants in commercial chickens. Avian Dis. 2014, 58, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Kurukulsuriya, S.; Ahmed, K.A.; Ojkic, D.; Gunawardana, T.; Gupta, A.; Goonewardene, K.; Karunaratne, R.; Popowich, S.; Willson, P.; Tikoo, S.K.; et al. Circulating strains of variant infectious bursal disease virus may pose a challenge for antibiotic-free chicken farming in Canada. Res. Vet. Sci. 2016, 108, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, M.; Nunoya, T.; Otaki, Y.; Tajima, M.; Saito, T.; Nakamura, T. Pathogenesis of highly virulent infectious bursal disease virus infection in intact and bursectomized chickens. J. Vet. Med. Sci. 1994, 56, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- McFerran, J.B.; McNulty, M.S.; McKillop, E.R.; Connor, T.J.; McCracken, R.M.; Collins, D.S.; Allan, G.M. Isolation and serological studies with infectious bursal disease viruses from fowl, turkeys and ducks: Demonstration of a second serotype. Avian Pathol. 1980, 9, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kasanga, C.J.; Yamaguchi, T.; Wambura, P.N.; Munang’Andu, H.M.; Ohya, K.; Fukushi, H. Detection of infectious bursal disease virus (IBDV) genome in free-living pigeon and guinea fowl in Africa suggests involvement of wild birds in the epidemiology of IBDV. Virus Genes 2008, 36, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Jayasundara, J.M.; Walkden-Brown, S.W.; Katz, M.E.; Islam, A.F.; Renz, K.G.; McNally, J.; Hunt, P.W. Pathogenicity, tissue distribution, shedding and environmental detection of two strains of IBDV following infection of chickens at 0 and 14 days of age. Avian Pathol. 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aricibasi, M.; Jung, A.; Heller, E.D.; Rautenschlein, S. Differences in genetic background influence the induction of innate and acquired immune responses in chickens depending on the virulence of the infecting infectious bursal disease virus (IBDV) strain. Vet. Immunol. Immunopathol. 2010, 135, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Tippenhauer, M.; Heller, D.E.; Weigend, S.; Rautenschlein, S. The host genotype influences infectious bursal disease virus pathogenesis in chickens by modulation of T cells responses and cytokine gene expression. Dev. Comp. Immunol. 2013, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bumstead, N.; Reece, R.L.; Cook, J.K. Genetic differences in susceptibility of chicken lines to infection with infectious bursal disease virus. Poult. Sci. 1993, 72, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.L.; Sorensen, P.; Hedemand, J.E.; Laursen, S.B.; Jorgensen, P.H. Inflammatory response of different chicken lines and B haplotypes to infection with infectious bursal disease virus. Avian Pathol. 1998, 27, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Sa, E.S.M.; Rissi, D.R.; Swayne, D.E. Very Virulent Infectious Bursal Disease Virus Produces More-Severe Disease and Lesions in Specific-Pathogen-Free (SPF) Leghorns Than in SPF Broiler Chickens. Avian Dis. 2016, 60, 63–66. [Google Scholar]
- Inoue, M.; Yamamoto, H.; Matuo, K.; Hihara, H. Susceptibility of chicken monocytic cell lines to infectious bursal disease virus. J. Vet. Med. Sci. 1992, 54, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Yin, Y.; Qin, T.; Yang, Q. Chicken bone marrow-derived dendritic cells maturation in response to infectious bursal disease virus. Vet. Immunol. Immunopathol. 2015, 164, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.L.; Nemerow, G.R. Viral weapons of membrane destruction: Variable modes of membrane penetration by non-enveloped viruses. Curr. Opin. Virol. 2011, 1, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yamaguchi, T.; Setiyono, A.; Ho, T.; Matsuda, H.; Furusawa, S.; Fukushi, H.; Hirai, K. Some characteristics of a cellular receptor for virulent infectious bursal disease virus by using flow cytometry. Arch. Virol. 1998, 143, 2327–2341. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhang, H.; Teng, M.; Fan, J.M.; You, L.M.; Xiao, Z.J.; Yi, M.L.; Zhi, Y.B.; Li, X.W.; Zhang, G.P. Surface IgM on DT40 cells may be a component of the putative receptor complex responsible for the binding of infectious bursal disease virus. Avian Pathol. 2010, 39, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.W.; Lo, C.W.; Lai, S.Y.; Fan, R.J.; Lo, C.J.; Chou, Y.M.; Thiruvengadam, R.; Wang, A.H.; Wang, M.Y. Chicken heat shock protein 90 is a component of the putative cellular receptor complex of infectious bursal disease virus. J. Virol. 2007, 81, 8730–8741. [Google Scholar] [CrossRef] [PubMed]
- Delgui, L.; Ona, A.; Gutierrez, S.; Luque, D.; Navarro, A.; Caston, J.R.; Rodriguez, J.F. The capsid protein of infectious bursal disease virus contains a functional alpha 4 beta 1 integrin ligand motif. Virology 2009, 386, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.M.; Han, J.; Ginsberg, M.H. α4 integrins and the immune response. Immunol. Rev. 2002, 186, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Han, X.; Yu, Z.; Zhang, E.; Wang, L.; Liu, H. Infectious Bursal Disease Virus activates c-Src to promote α4β1 integrin-dependent viral entry via modulating downstream Akt-RhoA GTPase-actin rearrangement cascade. J. Virol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, M.C.; Rodriguez, A.J.; Colombo, M.I.; Delgui, L.R. Infectious bursal disease virus uptake involves macropinocytosis and trafficking to early endosomes in a Rab5-dependent manner. Cell. Microbiol. 2015, 17, 988–1007. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Libersou, S.; Morellet, N.; Bouaziz, S.; Da, C.B.; Ouldali, M.; Lepault, J.; Delmas, B. Infectious bursal disease virus, a non-enveloped virus, possesses a capsid-associated peptide that deforms and perforates biological membranes. J. Biol. Chem. 2007, 282, 20774–20784. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.W.; Hon, C.C.; Zeng, F.; Leung, F.C. Cell culture-adapted IBDV uses endocytosis for entry in DF-1 chicken embryonic fibroblasts. Virus Res. 2012, 165, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell. Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Futter, C.E.; White, I.J. Annexins and endocytosis. Traffic 2007, 8, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Tebar, F.; Gelabert-Baldrich, M.; Hoque, M.; Cairns, R.; Rentero, C.; Pol, A.; Grewal, T.; Enrich, C. Annexins and endosomal signaling. Meth. Enzymol. 2014, 535, 55–74. [Google Scholar] [PubMed]
- Ren, X.; Zhang, L.; Gao, Y.; Gao, H.; Wang, Y.; Liu, C.; Cui, H.; Zhang, Y.; Jiang, L.; Qi, X.; et al. Binding chicken Anx2 is beneficial for infection with infectious bursal disease virus. Virus Res. 2015, 210, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, A.C.; Lam, K.M. Apoptosis induced by infectious bursal disease virus. J. Gen. Virol. 1994, 75, 1803–1806. [Google Scholar] [CrossRef] [PubMed]
- Tham, K.M.; Moon, C.D. Apoptosis in cell cultures induced by infectious bursal disease virus following in vitro infection. Avian Dis. 1996, 40, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Shahsavandi, S.; Ebrahimi, M.M.; Sadeghi, K.; Mahravani, H. Apoptotic response of chicken embryonic fibroblast cells to infectious bursal disease virus infections reflects viral pathogenicity. In Vitro Cell. Dev. Biol. Anim. 2014, 50, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Blaho, J.A. Apoptosis during herpes simplex virus infection. Adv. Virus Res. 2007, 69, 67–97. [Google Scholar] [PubMed]
- Koyama, A.H.; Adachi, A.; Irie, H. Physiological significance of apoptosis during animal virus infection. Int. Rev. Immunol. 2003, 22, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, E.; Maraver, A.; Espinosa, I.; Fernandez-Arias, A.; Rodriguez, J.F. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 2000, 277, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Arias, A.; Martinez, S.; Rodriguez, J.F. The major antigenic protein of infectious bursal disease virus, VP2, is an apoptotic inducer. J. Virol. 1997, 71, 8014–8018. [Google Scholar] [PubMed]
- Busnadiego, I.; Maestre, A.M.; Rodriguez, D.; Rodriguez, J.F. The infectious bursal disease virus RNA-binding VP3 polypeptide inhibits PKR-mediated apoptosis. PLoS ONE 2012, 7, e46768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, T.; Liu, H.; Shuai, X. Inhibitory effect of Sargassum polysaccharide on oxidative stress induced by infectious bursa disease virus in chicken bursal lymphocytes. Int. J. Biol. Macromol. 2011, 49, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Pyo, C.; Lee, S.; Choi, S. Oxidative stress induces PKR-dependent apoptosis via IFN-gamma activation signaling in Jurkat T cells. Biochem. Biophys. Res. Commun. 2008, 377, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Vakharia, V.N. Nonstructural protein of infectious bursal disease virus inhibits apoptosis at the early stage of virus infection. J. Virol. 2006, 80, 3369–3377. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Xu, Z.; Wang, Y.; Li, X.; He, Y.; Cao, H.; Zheng, S.J. VP2 of the infectious bursal disease virus is a protease that induces apoptosis by cleaving the oral cancer overexpressed 1 protein. To be submitted for publication. 2017. [Google Scholar]
- Li, M.; Cui, X.; Shen, Y.; Dong, H.; Liang, W.; Chen, Y.; Hu, J.; Li, S.; Kong, J.; Li, H.; et al. ORAOV1 overexpression in esophageal squamous cell carcinoma and esophageal dysplasia: A possible biomarker of progression and poor prognosis in esophageal carcinoma. Hum. Pathol. 2015, 46, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zeng, X.; Wang, Z.; Ji, N.; Zhou, Y.; Liu, X.; Chen, Q. Oral cancer overexpressed 1 (ORAOV1) regulates cell cycle and apoptosis in cervical cancer HeLa cells. Mol. Cancer 2010, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Arao, T.; Kato, H.; Matsumoto, K.; Terashima, M.; Hayashi, H.; de Velasco, M.A.; Fujita, Y.; Kimura, H.; Yasuda, T.; et al. Frequent amplification of ORAOV1 gene in esophageal squamous cell cancer promotes an aggressive phenotype via proline metabolism and ROS production. Oncotarget 2014, 5, 2962–2973. [Google Scholar] [CrossRef] [PubMed]
- Zhai, C.; Li, Y.; Mascarenhas, C.; Lin, Q.; Li, K.; Vyrides, I.; Grant, C.M.; Panaretou, B. The function of ORAOV1/LTO1, a gene that is overexpressed frequently in cancer: Essential roles in the function and biogenesis of the ribosome. Oncogene 2014, 33, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Vakharia, V.N. Induction of Apoptosis in Vitro by the 17-kDa Nonstructural Protein of Infectious Bursal Disease Virus: Possible Role in Viral Pathogenesis. Virology 2001, 285, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Goodwin, M.A.; Vakharia, V.N. Generation of a mutant infectious bursal disease virus that does not cause bursal lesions. J. Virol. 1998, 72, 2647–2654. [Google Scholar] [PubMed]
- Li, Z.; Wang, Y.; Xue, Y.; Li, X.; Cao, H.; Zheng, S.J. Critical Role for Voltage-Dependent Anion Channel 2 in Infectious Bursal Disease Virus-Induced Apoptosis in Host Cells via Interaction with VP5. J. Virol. 2012, 86, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhang, Z.; Xu, Z.; Wang, B.; Li, X.; Cao, H.; Wang, Y.; Zheng, S.J. The Association of Receptor of Activated Protein Kinase C 1 (RACK1) with Infectious Bursal Disease Virus Viral Protein VP5 and Voltage-dependent Anion Channel 2 (VDAC2) Inhibits Apoptosis and Enhances Viral Replication. J. Biol. Chem. 2015, 290, 8500–8510. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Hou, L.; Zhu, S.; Wang, J.; Zhou, J.; Liu, J. Infectious bursal disease virus activates the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway by interaction of VP5 protein with the p85alpha subunit of PI3K. Virology 2011, 417, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Qi, X.; Gao, Y.; Gao, H.; Lu, X.; Wang, Y.; Bu, Z.; Wang, X. VP5-deficient mutant virus induced protection against challenge with very virulent infectious bursal disease virus of chickens. Vaccine 2010, 28, 3735–3740. [Google Scholar] [CrossRef] [PubMed]
- Palmquist, J.M.; Khatri, M.; Cha, R.M.; Goddeeris, B.M.; Walcheck, B.; Sharma, J.M. In vivo activation of chicken macrophages by infectious bursal disease virus. Viral Immunol. 2006, 19, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Ragland, W.L.; Novak, R.; El-Attrache, J.; Savic, V.; Ester, K. Chicken anemia virus and infectious bursal disease virus interfere with transcription of chicken IFN-α and IFN-γ mRNA. J. Interferon Cytokine Res. 2002, 22, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; Li, X.; Li, X.; Cao, H.; Zheng, S.J. Critical roles of glucocorticoid-induced leucine zipper in infectious bursal disease virus (IBDV)-induced suppression of type I Interferon expression and enhancement of IBDV growth in host cells via interaction with VP4. J. Virol. 2013, 87, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, B.; Massetti, M.; Bruscoli, S.; Macchiarulo, A.; di Virgilio, R.; Velardi, E.; Donato, V.; Migliorati, G.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ)/NF-κB interaction: Role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res. 2007, 35, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Liu, L.J.; Zhong, B.; Liu, T.T.; Li, Y.; Yang, Y.; Ran, Y.; Li, S.; Tien, P.; Shu, H.B. WDR5 is essential for assembly of the VISA-associated signaling complex and virus-triggered IRF3 and NF-κB activation. Proc. Natl. Acad. Sci. USA 2010, 107, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Karin, M. Nuclear factor-κB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Mittelstadt, P.R.; Ashwell, J.D. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J. Biol. Chem. 2001, 276, 29603–29610. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Jia, L.; Sun, Y.; Hu, B.; Wang, L.; Lu, X.; Zhou, J. Inhibition of antiviral innate immunity by birnavirus VP3 protein via blockage of viral double-stranded RNA binding to the host cytoplasmic RNA detector MDA5. J. Virol. 2014, 88, 11154–11165. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Hur, S. How RIG-I like receptors activate MAVS. Curr. Opin. Virol. 2015, 12, 91–98. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Simon, A.K. Autophagy in the immune system. Immunology 2014, 141, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, Y.; Jia, L.; Wu, H.; Fan, C.; Sun, Y.; Ye, C.; Liao, M.; Zhou, J. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 2015, 11, 503–515. [Google Scholar] [PubMed]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef] [PubMed]
- Tacken, M.G.; Thomas, A.A.; Peeters, B.P.; Rottier, P.J.; Boot, H.J. VP1, the RNA-dependent RNA polymerase and genome-linked protein of infectious bursal disease virus, interacts with the carboxy-terminal domain of translational eukaryotic initiation factor 4AII. Arch. Virol. 2004, 149, 2245–2260. [Google Scholar] [CrossRef] [PubMed]
- Stricker, R.L.; Behrens, S.E.; Mundt, E. Nuclear factor NF45 interacts with viral proteins of infectious bursal disease virus and inhibits viral replication. J. Virol. 2010, 84, 10592–10605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ren, X.; Chen, Y.; Gao, Y.; Wang, N.; Lu, Z.; Gao, L.; Qin, L.; Wang, Y.; Gao, H.; et al. Chondroitin sulfate N-acetylgalactosaminyltransferase-2 contributes to the replication of infectious bursal disease virus via interaction with the capsid protein VP2. Viruses 2015, 7, 1474–1491. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, L.; Chen, Y.; Lu, Z.; Gao, L.; Wang, Y.; Gao, Y.; Gao, H.; Cui, H.; Li, K.; et al. Cyclophilin A Interacts with Viral VP4 and Inhibits the Replication of Infectious Bursal Disease Virus. BioMed Res. Int. 2015, 2015, 719454. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, Y.; Zheng, S.J. Infectious Bursal Disease Virus-Host Interactions: Multifunctional Viral Proteins that Perform Multiple and Differing Jobs. Int. J. Mol. Sci. 2017, 18, 161. https://doi.org/10.3390/ijms18010161
Qin Y, Zheng SJ. Infectious Bursal Disease Virus-Host Interactions: Multifunctional Viral Proteins that Perform Multiple and Differing Jobs. International Journal of Molecular Sciences. 2017; 18(1):161. https://doi.org/10.3390/ijms18010161
Chicago/Turabian StyleQin, Yao, and Shijun J. Zheng. 2017. "Infectious Bursal Disease Virus-Host Interactions: Multifunctional Viral Proteins that Perform Multiple and Differing Jobs" International Journal of Molecular Sciences 18, no. 1: 161. https://doi.org/10.3390/ijms18010161