
Cancer Cell Fusion: Mechanisms Slowly Unravel


{kind=link}
Abstract
:1. Introduction
2. Cell Fusion and Metastasis
3. Cancer Cell Fusion and Tumor Heterogeneity
4. Cancer Cell Fusion and Chemoresistance
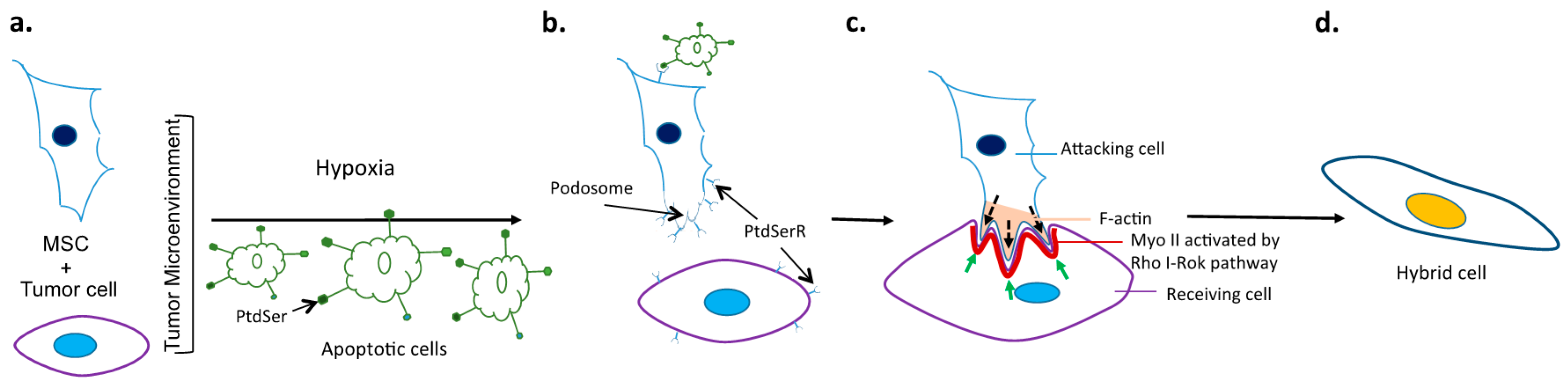
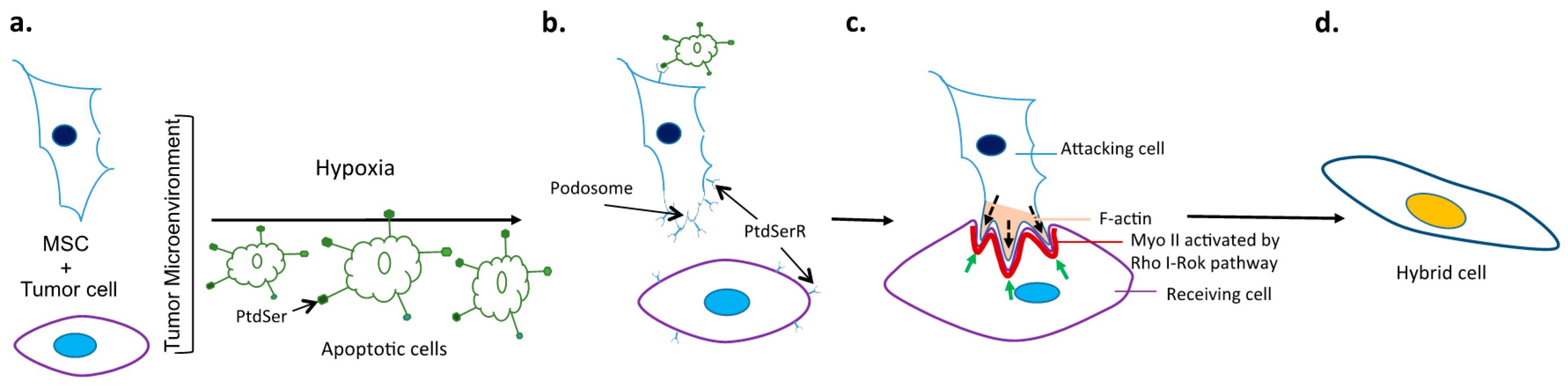
5. Potential Molecular Mechanisms and Signaling Pathways Driving Cancer Cell Fusion
Conflicts of Interest
References
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Byler, S.; Goldgar, S.; Heerboth, S.; Leary, M.; Housman, G.; Moulton, K.; Sarkar, S. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014, 34, 1071–1077. [Google Scholar] [PubMed]
- Rodenhiser, D.I.; Andrews, J.; Kennette, W.; Sadikovic, B.; Mendlowitz, A.; Tuck, A.B.; Chambers, A.F. Epigenetic mapping and functional analysis in a breast cancer metastasis model using whole-genome promoter tiling microarrays. Breast Cancer Res. 2008, 10, R62. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition—A hallmark of breast cancer metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Zallen, J.A. Dynamics of cell-cell and cell-matrix interactions in morphogenesis, regeneration and cancer. Curr. Opin. Cell Biol. 2010, 22, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Chatterjee, S.; Damle, S.G.; Sharma, A.K. Mechanisms of resistance against cancer therapeutic drugs. Curr. Pharm. Biotechnol. 2014, 15, 1105–1112. [Google Scholar] [CrossRef]
- Velasco-Velazquez, M.A.; Homsi, N.; de La Fuente, M.; Pestell, R.G. Breast cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 573–577. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Korkaya, H.; Dontu, G.; Birnbaum, D.; Wicha, M.S.; Charafe-Jauffret, E. The cancer stem cell: The breast cancer driver. Med. Sci. 2007, 23, 1133–1139. [Google Scholar]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Aichel, O. Über Zellverschmelzung mit Qualitativ Abnormer Chromosomenverteilung als Ursache der Geschwulstbildung. In Vorträge und Aufsätze über Entwicklungsmechanik der Organismen; Engelmann: Leipzig, Germany, 1911; p. 115S. [Google Scholar]
- Mekler, L.B. Creation of antineoplastic preparations on the basis of the theory of organ-tissue mosaicism of malignant cells. Farmakol. Toksikol. 1971, 34, 713–718. [Google Scholar] [PubMed]
- Goldenberg, D.M.; Pavia, R.A.; Tsao, M.C. In vivo hybridisation of human tumour and normal hamster cells. Nature 1974, 250, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Mercapide, J.; Lorico, A. Spontaneous formation of tumorigenic hybrids between breast cancer and multipotent stromal cells is a source of tumor heterogeneity. Am. J. Pathol. 2012, 180, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.; Mathur, S.R.; Mukhopadhyay, A. Fusion-derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 2013, 73, 5360–5370. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M. Fusion of bone marrow-derived cells with cancer cells: Metastasis as a secondary disease in cancer. Chin. J. Cancer 2014, 33, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Lazova, R.; Davies, S.; Backvall, H.; Ponten, F.; Brash, D.; Pawelek, J. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transpl. 2004, 34, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Sodi, S.A.; Chakraborty, A.; Platt, J.; Kolesnikova, N.; Rosemblat, S.; Keh-Yen, A.; Bolognia, J.; Rachkovsky, M.; Orlow, S.; Pawelek, J. Melanoma x macrophage fusion hybrids acquire increased melanogenesis and metastatic potential: Altered N-glycosylation as an underlying mechanism. Pigment Cell Res. 1998, 11, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K. The cancer cell—Leukocyte fusion theory of metastasis. Adv. Cancer Res. 2008, 101, 397–444. [Google Scholar] [PubMed]
- Berndt, B.; Haverkampf, S.; Reith, G.; Keil, S.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Fusion of CCL21 non-migratory active breast epithelial and breast cancer cells give rise to CCL21 migratory active tumor hybrid cell lines. PLoS ONE 2013, 8, e63711. [Google Scholar] [CrossRef] [PubMed]
- Berndt, B.; Zanker, K.S.; Dittmar, T. Cell fusion is a potent inducer of aneuploidy and drug resistance in tumor cell/normal cell hybrids. Crit. Rev. Oncog. 2013, 18, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Schwitalla, S.; Seidel, J.; Haverkampf, S.; Reith, G.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zanker, K.S. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin. Exp. Metastasis 2011, 28, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Ozel, C.; Seidel, J.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Hybrid cells derived from breast epithelial cell/breast cancer cell fusion events show a differential RAF-AKT crosstalk. Cell Commun. Signal. 2012, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc. Natl. Acad. Sci. USA 2009, 106, 9385–9390. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.; Lichtenberg, J.; Thomsen, P.; Larsson, L.I. Spontaneous fusion between cancer cells and endothelial cells. Cell. Mol. Life Sci. 2004, 61, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: A mechanism of breast cancer metastasis. FASEB J. 2015, 29, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Nagler, C.; Zanker, K.S.; Dittmar, T. Cell fusion, drug resistance and recurrence CSCs. Adv. Exp. Med. Biol. 2011, 714, 173–182. [Google Scholar] [PubMed]
- Xu, M.H.; Gao, X.; Luo, D.; Zhou, X.D.; Xiong, W.; Liu, G.X. EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLoS ONE 2014, 9, e87893. [Google Scholar] [CrossRef] [PubMed]
- McArdle, T.J.O.B.; Noubissi, F.K. An in vitro inverted vertical invasion assay to avoid manipulation of rare or sensitive cell types. J. Cancer 2016. [Google Scholar] [CrossRef]
- Chakraborty, A.K.; Sodi, S.; Rachkovsky, M.; Kolesnikova, N.; Platt, J.T.; Bolognia, J.L.; Pawelek, J.M. A spontaneous murine melanoma lung metastasis comprised of host X tumor hybrids. Cancer Res. 2000, 60, 2512–2519. [Google Scholar] [PubMed]
- Chakraborty, A.K.; Pawelek, J.; Ikeda, Y.; Miyoshi, E.; Kolesnikova, N.; Funasaka, Y.; Ichihashi, M.; Taniguchi, N. Fusion hybrids with macrophage and melanoma cells up-regulate N-acetylglucosaminyltransferase V, β1-6 branching, and metastasis. Cell Growth Differ. 2001, 12, 623–630. [Google Scholar] [PubMed]
- Pawelek, J.M. Cancer-cell fusion with migratory bone-marrow-derived cells as an explanation for metastasis: New therapeutic paradigms. Future Oncol. 2008, 4, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, Z.; Tsang, T.C.; Tang, T.; Jia, X.; He, X.; Pennington, M.E.; Badowski, M.S.; Liu, A.K.; Chen, D.; et al. Fusion of HepG2 cells with mesenchymal stem cells increases cancerassociated and malignant properties: An in vivo metastasis model. Oncol. Rep. 2014, 32, 539–547. [Google Scholar] [PubMed]
- Varley, K.E.; Mutch, D.G.; Edmonston, T.B.; Goodfellow, P.J.; Mitra, R.D. Intra-tumor heterogeneity of MLH1 promoter methylation revealed by deep single molecule bisulfite sequencing. Nucleic Acids Res. 2009, 37, 4603–4612. [Google Scholar] [CrossRef] [PubMed]
- Mekler, L.B. Hybridization of transformed cells with lymphocytes as 1 of the probable causes of the progression leading to the development of metastatic malignant cells. Vestnik Akad. Med. Nauk SSSR 1971, 26, 80–89. [Google Scholar]
- Andersen, T.L.; Boissy, P.; Sondergaard, T.E.; Kupisiewicz, K.; Plesner, T.; Rasmussen, T.; Haaber, J.; Kolvraa, S.; Delaisse, J.M. Osteoclast nuclei of myeloma patients show chromosome translocations specific for the myeloma cell clone: A new type of cancer-host partnership? J. Pathol. 2007, 211, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Lazova, R.; Qumsiyeh, M.; Cooper, D.; Pawelek, J. Donor Y chromosome in renal carcinoma cells of a female BMT recipient: Visualization of putative BMT-tumor hybrids by FISH. Bone Marrow Transpl. 2005, 35, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Clawson, G.A.; Matters, G.L.; Xin, P.; Imamura-Kawasawa, Y.; Du, Z.; Thiboutot, D.M.; Helm, K.F.; Neves, R.I.; Abraham, T. Macrophage-tumor cell fusions from peripheral blood of melanoma patients. PLoS ONE 2015, 10, e0134320. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Laberge, G.S.; Duvall, E.; Spoelstra, N.; Klump, V.; Sznol, M.; Cooper, D.; Spritz, R.A.; Chang, J.T.; Pawelek, J.M. A melanoma brain metastasis with a donor-patient hybrid genome following bone marrow transplantation: First evidence for fusion in human cancer. PLoS ONE 2013, 8, e66731. [Google Scholar] [CrossRef]
- Collisson, E.A.; Cho, R.J.; Gray, J.W. What are we learning from the cancer genome? Nat. Rev. Clin. Oncol. 2012, 9, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Russnes, H.G.; Navin, N.; Hicks, J.; Borresen-Dale, A.L. Insight into the heterogeneity of breast cancer through next-generation sequencing. J. Clin. Investig. 2011, 121, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.M.; Padilla-Nash, H.M.; Berman, D.; Murphy, K.M.; Ried, T.; Lazebnik, Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 2007, 17, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Mohr, M.; Zaenker, K.S.; Dittmar, T. Fusion in cancer: An explanatory model for aneuploidy, metastasis formation, and drug resistance. Methods Mol. Biol. 2015, 1313, 21–40. [Google Scholar]
- Freeman, B.T.; Jung, J.P.; Ogle, B.M. Single-cell RNA-seq reveals activation of unique gene groups as a consequence of stem cell-parenchymal cell fusion. Sci. Rep. 2016, 6, 23270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Merchak, K.; Lee, W.; Grande, J.P.; Cascalho, M.; Platt, J.L. Cell fusion connects oncogenesis with tumor evolution. Am. J. Pathol. 2015, 185, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Ha, S.A.; Yang, Y.S.; Kim, J.W. p-Glycoprotein ABCB5 and YB-1 expression plays a role in increased heterogeneity of breast cancer cells: Correlations with cell fusion and doxorubicin resistance. BMC Cancer 2010, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, S.; Li, C.; Ng, K.T.; Kong, C.W.; Cheng, J.; Cheng, S.H.; Li, R.A.; Lo, C.M.; Man, K.; et al. Fusion with stem cell makes the hepatocellular carcinoma cells similar to liver tumor-initiating cells. BMC Cancer 2015, 16, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, T.M.; Johnson-Camacho, K.; Peto, M.; Wang, N.J.; Macey, T.A.; Korkola, J.E.; Koppie, T.M.; Corless, C.L.; Gray, J.W.; Spellman, P.T. Exome Sequencing of cell-free DNA from metastatic cancer patients identifies clinically actionable mutations distinct from primary disease. PLoS ONE 2015, 10, e0136407. [Google Scholar] [CrossRef] [PubMed]
- Garay, J.P.; Gray, J.W. Omics and therapy—A basis for precision medicine. Mol. Oncol. 2012, 6, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Maisel, D.; Birzele, F.; Voss, E.; Nopora, A.; Bader, S.; Friess, T.; Goller, B.; Laifenfeld, D.; Weigand, S.; Runza, V. Targeting tumor cells with anti-CD44 antibody triggers macrophage-mediated immune modulatory effects in a cancer xenograft model. PLoS ONE 2016, 11, e0159716. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter-Hufford, A.E.; Lee, C.S.; Kinchen, J.M.; Sokolowski, J.D.; Arandjelovic, S.; Call, J.A.; Klibanov, A.L.; Yan, Z.; Mandell, J.W.; Ravichandran, K.S. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 2013, 497, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Pajcini, K.V.; Pomerantz, J.H.; Alkan, O.; Doyonnas, R.; Blau, H.M. Myoblasts and macrophages share molecular components that contribute to cell–cell fusion. J. Cell Biol. 2008, 180, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ravichandran, K.S. Dock180-ELMO cooperation in Rac activation. Methods Enzymol. 2006, 406, 388–402. [Google Scholar] [PubMed]
- Park, D.; Tosello-Trampont, A.C.; Elliott, M.R.; Lu, M.; Haney, L.B.; Ma, Z.; Klibanov, A.L.; Mandell, J.W.; Ravichandran, K.S. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 2007, 450, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, L.; Fu, H.; Yan, J.; Wang, Y.; Guo, H.; Hao, X.; Xu, X.; Jin, T.; Zhang, N. Association between Galphai2 and ELMO1/Dock180 connects chemokine signalling with Rac activation and metastasis. Nat. Commun. 2013, 4, 1706. [Google Scholar] [CrossRef] [PubMed]
- Abu-Thuraia, A.; Gauthier, R.; Chidiac, R.; Fukui, Y.; Screaton, R.A.; Gratton, J.P.; Cote, J.F. Axl phosphorylates Elmo scaffold proteins to promote Rac activation and cell invasion. Mol. Cell. Biol. 2015, 35, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Spurdle, A.B. Prioritizing candidate genetic modifiers of BRCA1 and BRCA2 using a combinatorial analysis of global expression and polymorphism association studies of breast cancer. Methods Mol. Biol. 2010, 653, 23–34. [Google Scholar] [PubMed]
- Feng, H.; Hu, B.; Vuori, K.; Sarkaria, J.N.; Furnari, F.B.; Cavenee, W.K.; Cheng, S.Y. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene 2014, 33, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Ryu, Y.K.; Ji, Y.H.; Kang, J.H.; Moon, E.Y. Hypoxia/reoxygenation-experienced cancer cell migration and metastasis are regulated by Rap1- and Rac1-GTPase activation via the expression of thymosin β-4. Oncotarget 2015, 6, 9820–9833. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Bao, Y.; Li, Z.; Li, J.; Gong, M.; Lam, S.; Wang, J.; Marzese, D.M.; Donovan, N.; Tan, E.Y.; et al. RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J. Clin. Investig. 2014, 124, 5291–5304. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Saci, A.; Cantley, L.C.; Carpenter, C.L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 2011, 42, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, J.S.; Hansen, M.D.; Nelson, W.J. Spatio-temporal regulation of Rac1 localization and lamellipodia dynamics during epithelial cell-cell adhesion. Dev. Cell 2002, 3, 259–270. [Google Scholar] [CrossRef]
- Bosco, E.E.; Nakai, Y.; Hennigan, R.F.; Ratner, N.; Zheng, Y. NF2-deficient cells depend on the Rac1-canonical Wnt signaling pathway to promote the loss of contact inhibition of proliferation. Oncogene 2010, 29, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Ren, Y.; Ng, W.P.; Li, S.; Son, S.; Kee, Y.S.; Zhang, S.; Zhang, G.; Fletcher, D.A.; Robinson, D.N.; et al. Mechanical tension drives cell membrane fusion. Dev. Cell 2015, 32, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell–cell fusion mechanisms. Trends Genet. 2013, 29, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Vignery, A. Macrophage fusion: Are somatic and cancer cells possible partners? Trends Cell Biol. 2005, 15, 188–193. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noubissi, F.K.; Ogle, B.M. Cancer Cell Fusion: Mechanisms Slowly Unravel. Int. J. Mol. Sci. 2016, 17, 1587. https://doi.org/10.3390/ijms17091587
Noubissi FK, Ogle BM. Cancer Cell Fusion: Mechanisms Slowly Unravel. International Journal of Molecular Sciences. 2016; 17(9):1587. https://doi.org/10.3390/ijms17091587
Chicago/Turabian StyleNoubissi, Felicite K., and Brenda M. Ogle. 2016. "Cancer Cell Fusion: Mechanisms Slowly Unravel" International Journal of Molecular Sciences 17, no. 9: 1587. https://doi.org/10.3390/ijms17091587