
Isothiocyanates Are Promising Compounds against Oxidative Stress, Neuroinflammation and Cell Death that May Benefit Neurodegeneration in Parkinson’s Disease


Abstract
:
1. Introduction
2. Parkinson’s Disease
Mechanisms Involved in Parkinson’s Disease
3. Neuroinflammation in the Pathogenesis of PD
3.1. Microglia as a Mediator of Neuroinflammation in PD
3.2. Astrocytes as Mediator of Neuroinflammation in PD
3.3. T Cell Infiltration as a Mediator of Neuroinflammation in PD
4. Isothiocyanates and PD
4.1. Sulforaphane
4.2. Erucin
4.3. Phenethyl Isothiocyanate
4.4. 6-(Methylsulfinyl)hexyl Isothiocyanate
4.5. Novel Compounds
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barbosa, M.; Valentão, P.; Andrade, P.B. Bioactive compounds from macroalgae in the new millennium: Implications for neurodegenerative diseases. Mar. Drugs 2014, 12, 4934–4972. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial Activation and Chronic Neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, R.; Matus, S.; Bargsted, L.; Hetz, C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 2014, 35, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Litvan, I.; Halliday, G.; Hallett, M.; Goetz, C.G.; Rocca, W.; Duyckaerts, C.; Ben-shlomo, Y.; Dickson, D.W.; Lang, A.E.; Chesselet, M.; et al. The Etiopathogenesis of Parkinson Disease and Suggestions for Future Research. Part I. J. Neuropathol. Exp. Neurol. 2007, 66, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Cuzzocrea, S. New therapeutic strategy for Parkinson’s and Alzheimer’s disease. Curr. Med. Chem. 2010, 17, 2764–2774. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.; Rothwell, N. Citokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J.; Becker, K.J.; Hallenbeck, J.M. Inflammation after stroke: Is it harmful? Arch. Neurol. 2001, 58, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Novío, S.; Cartea, M.E.; Soengas, P.; Freire-Garabal, M.; Núñez-Iglesias, M.J. Effects of Brassicaceae Isothiocyanates on Prostate Cancer. Molecules 2016, 21, E626. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo, S.M.; Binda, N.S.; Nogueira-Machado, J.A.; Vieira-Filho, S.A.; Caligiorne, R.B. The antioxidant properties of organosulfur compounds (sulforaphane). Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kim, I.-S.; More, S.V.; Kim, B.-W.; Choi, D.-K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 2014, 31, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhäuser, C. Nuclear factor κB is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef] [PubMed]
- Minarini, A.; Milelli, A.; Fimognari, C.; Simoni, E.; Turrini, E.; Tumiatti, V. Exploring the effects of isothiocyanates on chemotherapeutic drugs. Expert Opin. Drug Metab. Toxicol. 2014, 10, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Noyan Ashraf, M.H.; Facci, M.; Wang, R.; Paterson, P.G.; Ferrie, A.; Juurlink, B.H.J. Dietary approach to attenuate oxidative stress, hypertension, and inflammation in the cardiovascular system. Proc. Natl. Acad. Sci. USA 2004, 101, 7094–7099. [Google Scholar] [CrossRef] [PubMed]
- Senanayake, G.V.K.; Banigesh, A.; Wu, L.; Lee, P.; Juurlink, B.H.J. The dietary phase 2 protein inducer sulforaphane can normalize the kidney epigenome and improve blood pressure in hypertensive rats. Am. J. Hypertens. 2012, 25, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Gangopadhyay, H.; Das, D.K. Broccoli: A unique vegetable that protects mammalian hearts through the redox cycling of the thioredoxin superfamily. J. Agric. Food Chem. 2008, 56, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Tarozzi, A.; Cantelli-Forti, G.; Hrelia, P. Neuroprotection by 6-(methylsulfinyl)hexyl isothiocyanate in a 6-hydroxydopamine mouse model of Parkinson’s disease. Brain Res. 2014, 1589, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. Neurotoxicology 2013, 36, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kerns, M.L.; DePianto, D.; Dinkova-Kostova, A.T.; Talalay, P.; Coulombe, P.A. Reprogramming of keratin biosynthesis by sulforaphane restores skin integrity in epidermolysis bullosa simplex. Proc. Natl. Acad. Sci. USA 2007, 104, 14460–14465. [Google Scholar] [CrossRef] [PubMed]
- Savitt, J.; Dawson, V.; Dawson, T. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.; Schmidt, M.; Lee, V.; Trojanowski, J.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoets, G.; Verleden, S.; Santens, P.; Miatton, M.; de Reuck, J. Predictors of cognitive impairment in advanced Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.W.; Nicholas, A.P. Nonmotor symptoms in parkinson’s disease. Expanding the view of Parkinson’s disease beyond a pure motor, pure dopaminergic problem. Neurol. Clin. 2013, 31, S1–S16. [Google Scholar] [CrossRef] [PubMed]
- Francardo, V.; Recchia, A.; Popovic, N.; Andersson, D.; Nissbrandt, H.; Cenci, M.A. Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L-DOPA in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neurobiol. Dis. 2011, 42, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.-D.V.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Go, Y.M. Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 2010, 12, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Olanow, C.W. The pathogenesis of cell death in Parkinson’s disease. Neurology 2006, 66, S24–S36. [Google Scholar] [CrossRef] [PubMed]
- Wahner, A.D.; Bronstein, J.M.; Bordelon, Y.M.; Ritz, B. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 2007, 69, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, Y.; Qin, Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Zavos, C.; Polyzos, S.A.; Deretzi, G.; Vardaka, E.; Giartza-Taxidou, E.; Katsinelos, P.; Rapti, E.; Chatzopoulos, D.; Tzilves, D.; et al. Helicobacter pylori infection and Parkinson’s disease: Apoptosis as an underlying common contributor. Eur. J. Neurol. 2012, 19, e56. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Jain, S. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Jellinger, K.A. Cell death mechanisms in Parkinson’s disease. J. Neural Transm. 2000, 107, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Stojkovska, I.; Wagner, B.M.; Morrison, B.E. Parkinson’s disease and enhanced inflammatory response. Exp. Biol. Med. 2015, 240, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.; Bell, M.; Brown, H.; Matyszak, M. Inflammation in the nervous system. Curr. Opin. Neurobiol. 1995, 5, 636–641. [Google Scholar] [CrossRef]
- Barone, F.C.; Feuerstein, G.Z. Inflammatory Mediators and Stroke: New Opportunities for Novel Therapeutics. J. Cereb. Blood Flow Metab. 1999, 19, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of α-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5, e8784. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.; Itagaki, S.; Boyes, B.; McGeer, E. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernán, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-U.; Kim, Y.-D.; Cho, H.-J.; Chung, S.-W. Is Neuroinflammation Involved in the Development of Dementia in Patients with Parkinson’s Disease? Intern. Med. 2013, 52, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.E.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.-Y.; Choi, D.-K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Kohutnicka, M.; Lewandowska, E.; Kurkowska-Jastrzebska, I.; Członkowski, A.; Członkowska, A. Microglial and astrocytic involvement in a murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Immunopharmacology 1998, 39, 167–180. [Google Scholar] [CrossRef]
- McLaughlin, P.; Zhou, Y.; Ma, T.; Liu, J.; Zhang, W.; Hong, J.-S.; Kovacs, M.; Zhang, J. Proteomic analysis of microglial contribution to mouse strain-dependent dopaminergic neurotoxicity. Glia 2006, 53, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1 β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Koppula, S.; Kumar, H.; Kim, I.S.; Choi, D.-K. Reactive oxygen species and inhibitors of inflammatory enzymes, NADPH oxidase, and iNOS in experimental models of Parkinson’s disease. Mediat. Inflamm. 2012, 2012, 823902. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, I.; Galea, I.; Perry, V. What is the blood-brain barrier (not)? Trends Immunol. 2007, 28, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.; Tieu, K. Astrocytes and therapeutics for Parkinson’s disease. Neurotherapeutics 2010, 7, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Tieu, K.; Cohen, O.; Choi, D.; Vila, M.; Jackson-Lewis, V.; Teismann, P.; Przedborski, S. Glial cell response: A pathogenic factor in Parkinson’s disease. J. Neurovirol. 2002, 8, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Carrillo-de Sauvage, M.-A.; Ceyzériat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Wong, S.C.; Tan, E.K. Evidence of inflammatory system involvement in Parkinson’s disease. BioMed Res. Int. 2014, 2014, 308654. [Google Scholar] [CrossRef] [PubMed]
- Mosley, R.L.; Hutter-Saunders, J.A.; Stone, D.K.; Gendelman, H.E. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009381. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated α-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Tinarelli, C.; Schulz, R.J.; Polidori, M.C. Nutraceuticals in cognitive impairment and Alzheimer’s disease. Front. Pharmacol. 2014, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Yaya, E.E. Phytoalexins from Brassicaceae: News from the front. Phytochemistry 2010, 71, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Kapusta-Duch, J.; Kopeć, A.; Piatkowska, E.; Borczak, B.; Leszczyńska, T. The beneficial effects of Brassica vegetables on human health. Rocz. Państwowego Zakładu Hig. 2012, 63, 389–395. [Google Scholar]
- Verkerk, R.; Schreiner, M.; Krumbein, A.; Ciska, E.; Holst, B.; Rowland, I.; de Schrijver, R.; Hansen, M.; Gerhäuser, C.; Mithen, R.; et al. Glucosinolates in Brassica vegetables: The influence of the food supply chain on intake, bioavailability and human health. Mol. Nutr. Food Res. 2009, 53 (Suppl. 2), S219. [Google Scholar] [CrossRef] [PubMed]
- Bones, A.M.; Rossiter, J.T. The enzymic and chemically induced decomposition of glucosinolates. Phytochemistry 2006, 67, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Eto, H.; Kuboi, T. Tissue printing for myrosinase activity in roots of turnip and Japanese radish and horseradish: A technique for localizing myrosinases. Plant Sci. 2001, 160, 425–431. [Google Scholar] [CrossRef]
- Ishida, M.; Hara, M.; Fukino, N.; Kakizaki, T.; Morimitsu, Y. Glucosinolate metabolism, functionality and breeding for the improvement of Brassicaceae vegetables. Breed. Sci. 2014, 64, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef] [PubMed]
- Drobnica, L.; Kristian, P. The Chemistry of the -NCS Group; Patai, S., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1977. [Google Scholar]
- Zhang, Y.; Kolm, R.H.; Mannervik, B.; Talalay, P. Reversible conjugation of isothiocyanates with glutathione catalyzed by human glutathione transferases. Biochem. Biophys. Res. Commun. 1995, 206, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.L.; Levine, S. Nrf2, a master regulator of detoxification and also antioxidant, anti-inflammatory and other cytoprotective mechanisms, is raised by health promoting factors. Sheng Li Xue Bao 2015, 67, 1–18. [Google Scholar] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in Nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.C.; Sykiotis, G.P.; Bohmann, D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Model. Mech. 2011, 4, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cui, W.; Li, G.; Yuan, S.; Xu, D.; Hoi, M.P.M.; Lin, Z.; Dou, J.; Han, Y.; Lee, S.M.Y. Baicalein protects against 6-OHDA-induced neurotoxicity through activation of Keap1/Nrf2/HO-1 and involving PKCα and PI3K/AKT signaling pathways. J. Agric. Food Chem. 2012, 60, 8171–8182. [Google Scholar] [CrossRef] [PubMed]
- Jakel, R.J.; Townsend, J.A.; Kraft, A.D.; Johnson, J.A. Nrf2-Mediated protection against 6-hydroxydopamine. Brain Res. 2007, 1144, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; García, C.; Fernández-Ruiz, J.; Grochot-Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.; Ponsford, A.; Sanderson, C. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.G.-H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Pawate, S.; Shen, Q.; Fan, F.; Bhat, N.R. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J. Neurosci. Res. 2004, 77, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hayden, M.S. New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S128–S138. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.-O.; Kipp, M.; Lucius, R.; Pufe, T.; Wruck, C.J. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflamm. Res. 2009, 59, 443–450. [Google Scholar] [CrossRef] [PubMed]
- McWalter, G.K.; Higgins, L.G.; McLellan, L.I.; Henderson, C.J.; Song, L.; Thornalley, P.J.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J. Nutr. 2004, 134, 3499S–3506S. [Google Scholar] [PubMed]
- Han, J.M.; Lee, Y.J.; Lee, S.Y.; Kim, E.M.; Moon, Y.; Kim, H.W.; Hwang, O. Protective effect of sulforaphane against dopaminergic cell death. J. Pharmacol. Exp. Ther. 2007, 321, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Iori, R.; De Nicola, G.; Bramanti, P.; Mazzon, E. Anti-Inflammatory and anti-apoptotic effects of (RS)-glucoraphanin bioactivated with myrosinase in murine sub-acute and acute MPTP-induced Parkinson’s disease. Bioorg. Med. Chem. 2013, 21, 5532–5547. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; You, B.; Jo, E.-K.; Han, S.-K.; Simon, M.I.; Lee, S.J. NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2010, 107, 14851–14856. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wu, R.; Wu, T.; Khor, T.-O.; Wang, H.; Kong, A.-N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Khor, T.O.; Saw, C.L.L.; Lin, W.; Wu, T.; Huang, Y.; Kong, A.-N.T. Role of Nrf2 in suppressing LPS-induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Mol. Pharm. 2010, 7, 2185–2193. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Hrelia, S.; Angeloni, C.; Cantelli-Forti, G.; Hrelia, P. Sulforaphane as an inducer of glutathione prevents oxidative stress-induced cell death in a dopaminergic-like neuroblastoma cell line. J. Neurochem. 2009, 111, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Tao, R.; Yu, S.-Z.; Jin, H. Sulforaphane protects against 6-hydroxydopamine-induced cytotoxicity by increasing expression of heme oxygenase-1 in a PI3K/Akt-dependent manner. Mol. Med. Rep. 2012, 5, 847–851. [Google Scholar] [PubMed]
- Deng, C.; Tao, R.; Yu, S.-Z.; Jin, H. Inhibition of 6-hydroxydopamine-induced endoplasmic reticulum stress by sulforaphane through the activation of Nrf2 nuclear translocation. Mol. Med. Rep. 2012, 6, 215–219. [Google Scholar] [PubMed]
- Vauzour, D.; Buonfiglio, M.; Corona, G.; Chirafisi, J.; Vafeiadou, K.; Angeloni, C.; Hrelia, S.; Hrelia, P.; Spencer, J.P.E. Sulforaphane protects cortical neurons against 5-S-cysteinyl-dopamine-induced toxicity through the activation of ERK1/2, Nrf-2 and the upregulation of detoxification enzymes. Mol. Nutr. Food Res. 2010, 54, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Galuppo, M.; Montaut, S.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. An overview on neuroprotective effects of isothiocyanates for the treatment of neurodegenerative diseases. Fitoterapia 2015, 106, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Rajasekar, N.; Hanif, K.; Nath, C.; Shukla, R. Sulforaphane Ameliorates Okadaic Acid-Induced Memory Impairment in Rats by Activating the Nrf2/HO-1 Antioxidant Pathway. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Riedl, K.; Bella, D.; Schwartz, S.J.; Stevens, J.F.; Ho, E. Comparison of isothiocyanate metabolite levels and histone deacetylase activity in human subjects consuming broccoli sprouts or broccoli supplement. J. Agric. Food Chem. 2011, 59, 10955–10963. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, N.; Coldham, N.; Sauer, M.J.; Ioannides, C. Up-Regulation of the CYP1 family in rat and human liver by the aliphatic isothiocyanates erucin and sulforaphane. Toxicology 2008, 252, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanlon, N.; Coldham, N.; Sauer, M.J.; Ioannides, C. Modulation of rat pulmonary carcinogen-metabolising enzyme systems by the isothiocyanates erucin and sulforaphane. Chem. Biol. Interact. 2009, 177, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchini, A.; Traka, M.H. Biological profile of erucin: A new promising anticancer agent from cruciferous vegetables. Toxins 2010, 2, 593–612. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, H.; Soroka, Y.; Zlotkin-Frušić, M.; Gilhar, A.; Milner, Y.; Tamir, S. Isothiocyanates inhibit psoriasis-related proinflammatory factors in human skin. Inflamm. Res. 2012, 61, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lee, K.W.; Park, J.H.Y. Erucin exerts anti-inflammatory properties in murine macrophages and mouse skin: Possible mediation through the inhibition of NFκB signaling. Int. J. Mol. Sci. 2013, 14, 20564–20577. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Sturm, C.; Piegholdt, S.; Wolf, I.; Esatbeyoglu, T.; De Nicola, G.; Iori, R.; Rimbach, G. Myrosinase-treated glucoerucin is a potent inducer of the Nrf2 target gene heme oxygenase 1—Studies in cultured HT-29 cells and mice. J. Nutr. Biochem. 2015, 26, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Morroni, F.; Bolondi, C.; Sita, G.; Hrelia, P.; Djemil, A.; Cantelli-Forti, G. Neuroprotective effects of erucin against 6-hydroxydopamine-induced oxidative damage in a dopaminergic-like neuroblastoma cell line. Int. J. Mol. Sci. 2012, 13, 10899–10910. [Google Scholar] [CrossRef] [PubMed]
- Eklind, K.I.; Morse, M.A.; Chung, F.L. Distribution and metabolism of the natural anticarcinogen phenethyl isothiocyanate in A/J mice. Carcinogenesis 1990, 11, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Morris, M.E.; Ji, Y.; Morris, M.E.M.E.; Ji, Y.; Morris, M.E. Determination of phenethyl isothiocyanate in human plasma and urine by ammonia derivatization and liquid chromatography-tandem mass spectrometry. Anal. Biochem. 2003, 323, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P. Anticarcinogenic activities of organic isothiocyanates: Chemistry and mechanisms. Cancer Res. 1994, 54, 1976s–1981s. [Google Scholar] [PubMed]
- Hecht, S.S. Chemoprevention by isothiocyanates. J. Cell. Biochem. Suppl. 1995, 22, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.; Won, Y.K.Y.; Ong, C.N.C.; Whiteman, M. β-Phenylethyl and 8-methylsulphinyloctyl isothiocyanates, constituents of watercress, suppress LPS induced production of nitric oxide and prostaglandin E2 in RAW 264.7 macrophages. Nitric Oxide 2005, 12, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Adesida, A.; Edwards, L.; Thornalley, P. Inhibition of human leukaemia 60 cell growth by mercapturic acid metabolites of phenylethyl isothiocyanate. Food Chem. Toxicol. 1996, 34, 385–392. [Google Scholar] [CrossRef]
- Qin, C.-Z.; Zhang, X.; Wu, L.-X.; Wen, C.-J.; Hu, L.; Lv, Q.-L.; Shen, D.-Y.; Zhou, H.-H. Advances in molecular signaling mechanisms of β-phenethyl isothiocyanate antitumor effects. J. Agric. Food Chem. 2015, 63, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Ribnicky, D.; Kurmukov, A.G.; Raskin, I. In vitro and in vivo anti-inflammatory activity of a seed preparation containing phenethylisothiocyanate. J. Pharmacol. Exp. Ther. 2006, 317, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, S.; Park, S.; Eom, S.; Gu, G.; Kim, S.; Youn, H. Phenethyl isothiocyanate regulates inflammation through suppression of the TRIF-dependent signaling pathway of Toll-like receptors. Life Sci. 2013, 92, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Boyanapalli, S.S.S.; Paredes-gonzalez, X.; Fuentes, F.; Zhang, C.; Guo, Y.; Pung, D.; Lay, C.; Saw, L.; Kong, A.T. Nrf2 Knockout Attenuates the Anti-Inflammatory Effects of Phenethyl Isothiocyanate and Curcumin. Chem. Res. Toxicol. 2014, 27, 2036–2043. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Hou, D.-X.; Morinaga, O.; Shoyama, Y. Molecular Mechanisms Underlying Anti-Inflammatory Actions of 6-(Methylsulfinyl)hexyl Isothiocyanate Derived from Wasabi (Wasabia japonica). Adv. Pharmacol. Sci. 2012, 2012, 614046. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Fujii, M.; Hou, D.-X. Inhibition of lipopolysaccharide-induced cyclooxygenase-2 transcription by 6-(methylsulfinyl) hexyl isothiocyanate, a chemopreventive compound from Wasabia japonica (Miq.) Matsumura, in mouse macrophages. Biochem. Pharmacol. 2005, 70, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Noshita, T.; Kidachi, Y.; Funayama, H.; Kiyota, H.; Yamaguchi, H.; Ryoyama, K. Anti-nitric oxide production activity of isothiocyanates correlates with their polar surface area rather than their lipophilicity. Eur. J. Med. Chem. 2009, 44, 4931–4936. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Uto, T.; Tanigawa, S.; Yamada-Kato, T.; Fujii, M.; Hou, D.-X. Microarray-based determination of anti-inflammatory genes targeted by 6-(methylsulfinyl)hexyl isothiocyanate in macrophages. Exp. Ther. Med. 2010, 1, 33–40. [Google Scholar] [PubMed]
- Morimitsu, Y.; Nakagawa, Y.; Hayashi, K.; Fujii, H.; Kumagai, T.; Nakamura, Y.; Osawa, T.; Horio, F.; Itoh, K.; Iida, K.; et al. A sulforaphane analogue that potently activates the Nrf2-dependent detoxification pathway. J. Biol. Chem. 2002, 277, 3456–3463. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Kume, T.; Muto, C.; Takada-Takatori, Y.; Izumi, Y.; Sugimoto, H.; Akaike, A. Glutathione biosynthesis via activation of the nuclear factor E2-related factor 2 (Nrf2)—Antioxidant-response element (ARE) pathway is essential for neuroprotective effects of sulforaphane and 6-(methylsulfinyl) hexyl isothiocyanate. J. Pharmacol. Sci. 2011, 115, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Son, H.J.; Park, K.D.; Han, S.H.; Shin, N.; Kim, J.H.; Kim, H.R.; Kim, D.J.; Hwang, O. A Novel Compound ITC-3 Activates the Nrf2 Signaling and Provides Neuroprotection in Parkinson’s Disease Models. Neurotox. Res. 2015, 28, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Wellejus, A.; Elbrønd-Bek, H.; Kelly, N.M.; Weidner, M.S.; Jørgensen, S.H. 4-Iodophenyl isothiocyanate: A neuroprotective compound. Restor. Neurol. Neurosci. 2012, 30, 21–38. [Google Scholar] [PubMed]
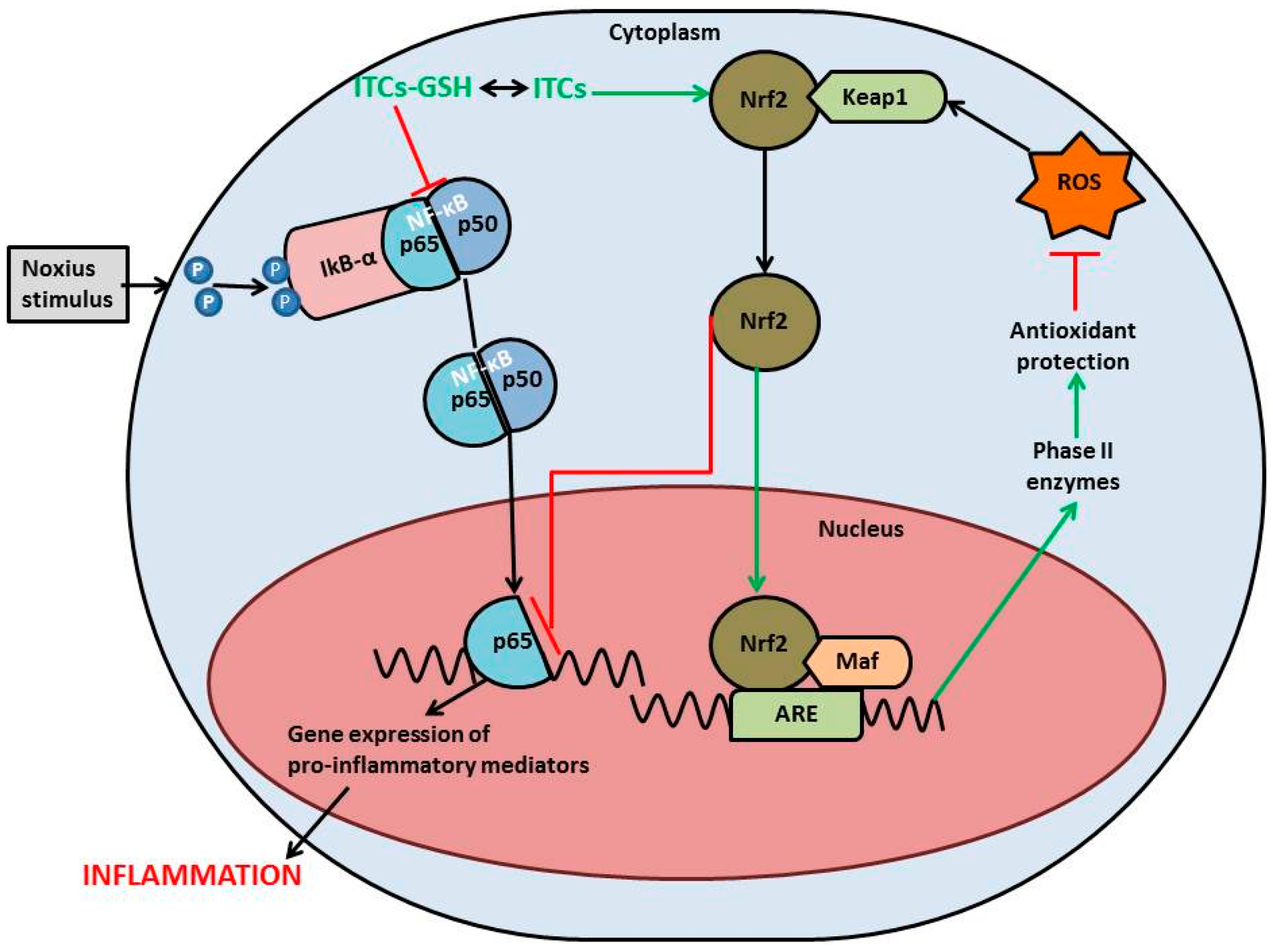

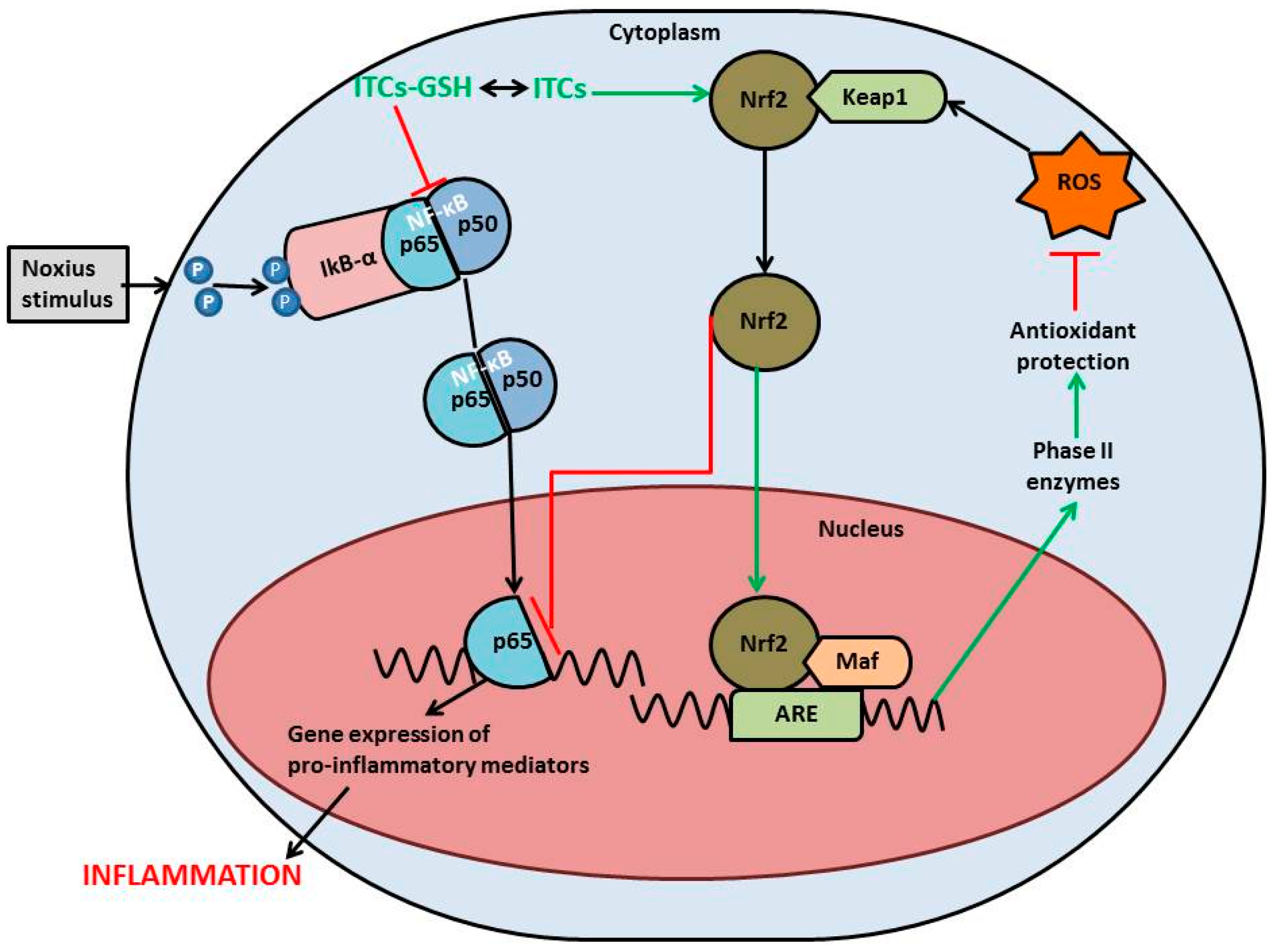
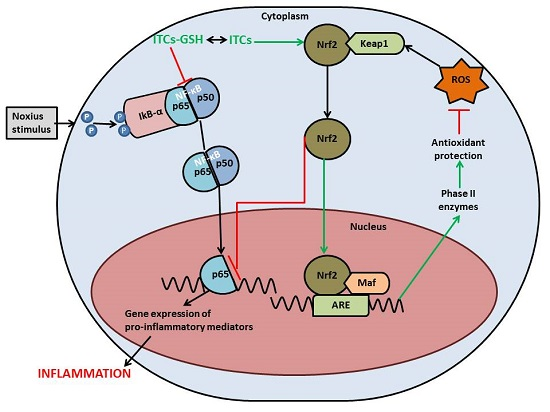{kind=link}
{kind=link}
| Model | Damage | Isothiocyanates | References | ||
|---|---|---|---|---|---|
| Toxins | Doses | Molecules and Sources | Doses | ||
| Primary peritoneal macrophage from NrF2−/− mice | LPS 1 | 1 µg/mL | SFN 2 | 5–40 µM | [97] |
| SH-SY5Y cell 3 | H2O2 4 | 300 µmol/L | SFN | 0.63–5 µmol/L | [99] |
| 6-OHDA 5 | 100 µmol/L | ||||
| PC12 6 cell line | 6-OHDA | 80 µM | SFN | 5 µM | [100] |
| Primary cortical neurons | 5-S-cysteinyl-dopamine | 100 µM | SFN | 100 nM | [102] |
| CATH.a cell 8 | BH4 7 | 200 µM | SFN | 0.5–5 µM | [93] |
| 6-OHDA | 50 µM | ||||
| MPP+ 9 | 2 mM | ||||
| AMPT 10 | 100 µM | ||||
| SK-N-BE(2)C cell 11 | BH4 | 200 µM | |||
| 6-OHDA | 50 µM | ||||
| MPP+ | 2 mM | ||||
| AMPT | 100 µM | ||||
| RAW 264.7 12 | LPS | 1 mg/L | ER 13 | 2.5–5 µmol/L | [110] |
| HT-29 cell 14 | GER 15 | 25 µmol/L | [111] | ||
| GRA 16 | |||||
| SIN 17 | |||||
| GST 18 | |||||
| SH-SY5Y cell | 6-OHDA | 200 µmol/L | ER | 1.25–5 µmol/L | [112] |
| Human plasma and urine | Watercress | 100 g | [114] | ||
| RAW 264.7 | LPS | 1 µg/mL | PEITC 19 | 100 mM | [117] |
| MSO 20 | |||||
| RAW 264.7 | LPS | 500 ng/mL | SFN | 0.4–50 µM | [12] |
| RAW 264.7 | LPS | 1 µg/mL | Barbarea verna | 1–20 µM | [120] |
| HL60 cell line 21 | PEITC metabolites | 0.1–100 µM | [118] | ||
| RAW 264.7 | LPS | 110 ng/mL | PEITC | 20 µM | [121] |
| Peritoneal macrophages of C57BL/6 mice | LPS | 1 µg/mL | PEITC | 5–10 µM | [122] |
| Peritoneal macrophages of NrF2−/− mice | |||||
| Primary cortical neurons from E18 Wistar rats | Glutamate | 20 µM | 4-IPITC 22 | 0.1–10 µM | [130] |
| H2O2 | 45 µM | ||||
| Mesencephalon neurons | MPP+ | 4 µM | |||
| RAW 264.7 | LPS | 40 ng/mL | 6-MITC 23 | 16 µM | [124] |
| RAW 264.7 | LPS | 40 ng/mL | 6-MITC | 8 µM | [126] |
| RL34 cells 24 | 6-MITC | 25 µmol | [127] | ||
| Primary neurons of rat striatum | Paraquat | 200 µM | SFN | 0.01–1 µM | [128] |
| H2O2 | 30 µM | ||||
| Paraquat | 200 µM | 6-MITC | |||
| H2O2 | 30 µM | ||||
| CATH.a cell | MPP+ | ITC-3 | 0.05–1 µM | [129] | |
| BH4 | |||||
| BV-2 cells | LPS | 0.2 µg/mL | 1–20 µM | ||
| Model | Damage | Isothiocyanates | References | ||
|---|---|---|---|---|---|
| Toxins | Doses | Molecules and Sources | Doses | ||
| NrF2−/− mice | MPTP 1 | 30 mg/kg | SFN 2 | 50 mg/kg | [95] |
| Nox2−/− mice | Spinal nerve transection | SFN | 10 mg/kg | [96] | |
| 50 mg/kg | |||||
| CX3CR13+/GFP mice | 10 mg/kg | ||||
| 50 mg/kg | |||||
| NrF2−/− mice | Broccoli seeds | 15% on weight | [92] | ||
| C57BL/6 mice | 6-OHDA 4 | 2 µL, 4 µg/mL | SFN | 5 mg/kg | [18] |
| Sprague-Dawley rats | OKA 5 | 200 ng | SFN | 5 mg/kg | [104] |
| ICR mice | TPA 6 | 5 nmol | ER 7 | 100–300 nmol | [110] |
| C57BL/6 mice | GER 8 | 20 mg/kg | [111] | ||
| GRA 9 | |||||
| SIN 10 | |||||
| GST 11 | |||||
| Wistar rats | λ-Carrageenan | 100 µL, 1% | PEITC 12 oil | 200 mg/kg | [120] |
| Dark agouti rats | Myelin oligodendrocyte glycoprotein | 0.7 mg/mL | 10–40 mg/kg | [130] | |
| C57BL/6 mice | MPTP | 20 mg/kg | 5 mg/kg | ||
| C57BL/6 mice | MPTP | 40 mg/kg | RS-GRA | 10 mg/kg | [94] |
| 20 mg/kg | |||||
| ICR mice | SFN | 25 µmol | [127] | ||
| 6-MITC 13 | |||||
| NrF2−/− mice | |||||
| C57BL/6 mice | 6-OHDA | 2 µL, 4 µg/mL | 6-MITC | 5 mg/kg | [17] |
| C57BL/6 mice | MPTP | 20 mg/kg | ITC-3 | 30 mg/kg | [129] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sita, G.; Hrelia, P.; Tarozzi, A.; Morroni, F. Isothiocyanates Are Promising Compounds against Oxidative Stress, Neuroinflammation and Cell Death that May Benefit Neurodegeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 1454. https://doi.org/10.3390/ijms17091454
Sita G, Hrelia P, Tarozzi A, Morroni F. Isothiocyanates Are Promising Compounds against Oxidative Stress, Neuroinflammation and Cell Death that May Benefit Neurodegeneration in Parkinson’s Disease. International Journal of Molecular Sciences. 2016; 17(9):1454. https://doi.org/10.3390/ijms17091454
Chicago/Turabian StyleSita, Giulia, Patrizia Hrelia, Andrea Tarozzi, and Fabiana Morroni. 2016. "Isothiocyanates Are Promising Compounds against Oxidative Stress, Neuroinflammation and Cell Death that May Benefit Neurodegeneration in Parkinson’s Disease" International Journal of Molecular Sciences 17, no. 9: 1454. https://doi.org/10.3390/ijms17091454