
PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
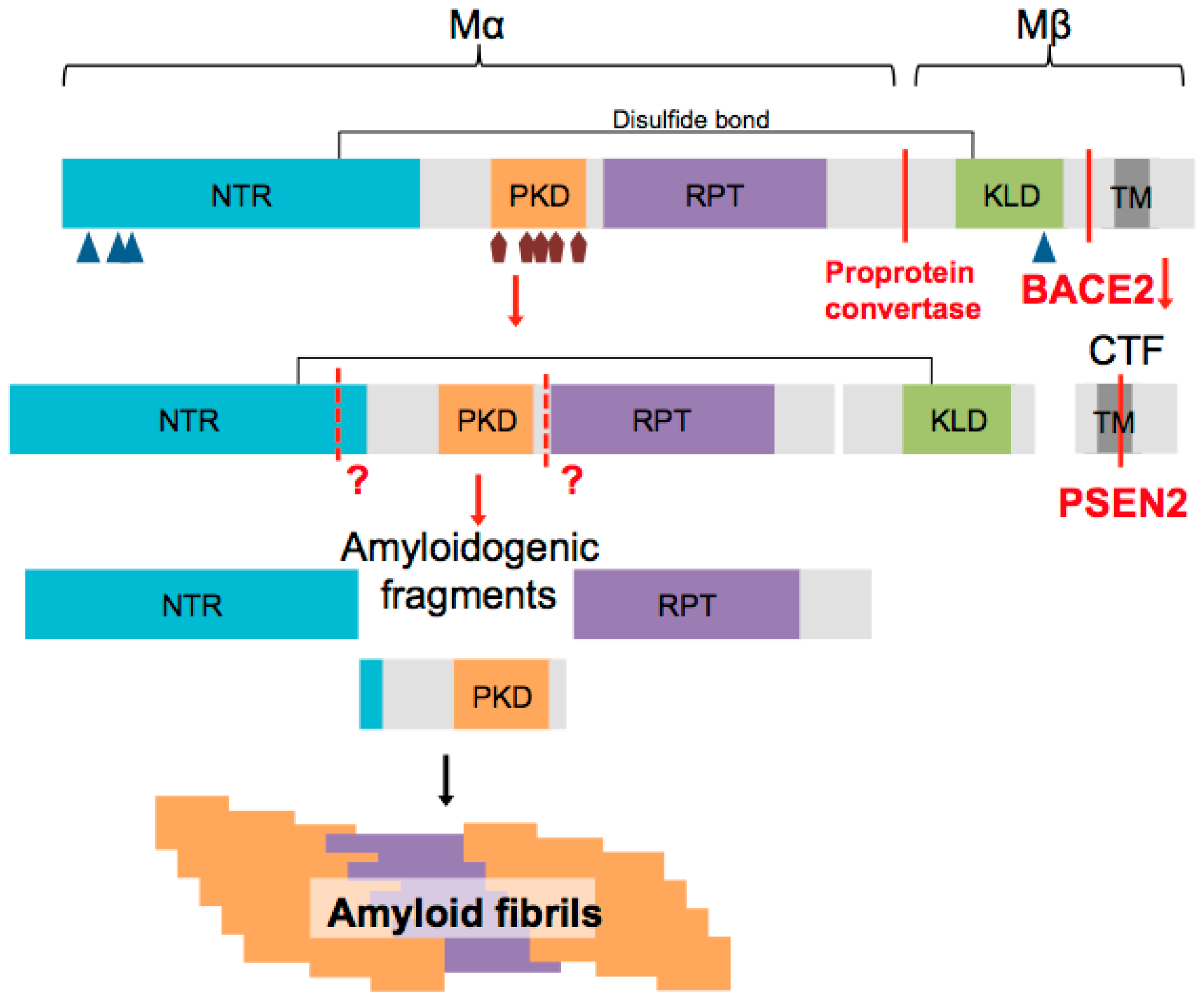
2. PMEL Protein Structure
3. PMEL Forms Physiological Amyloids
4. PMEL Processing
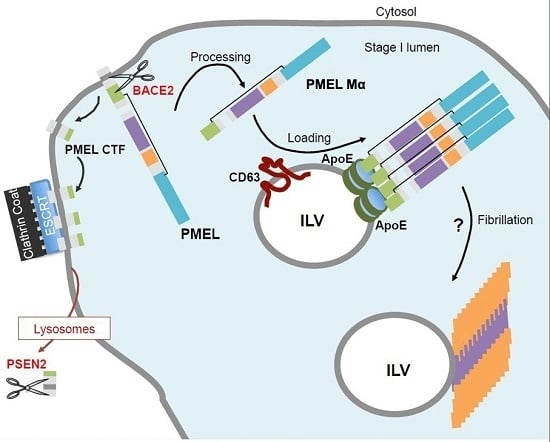
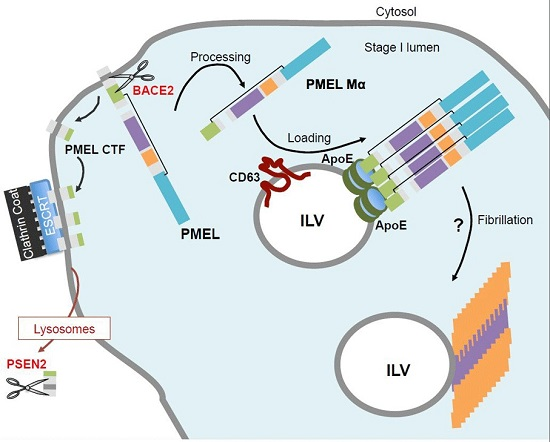
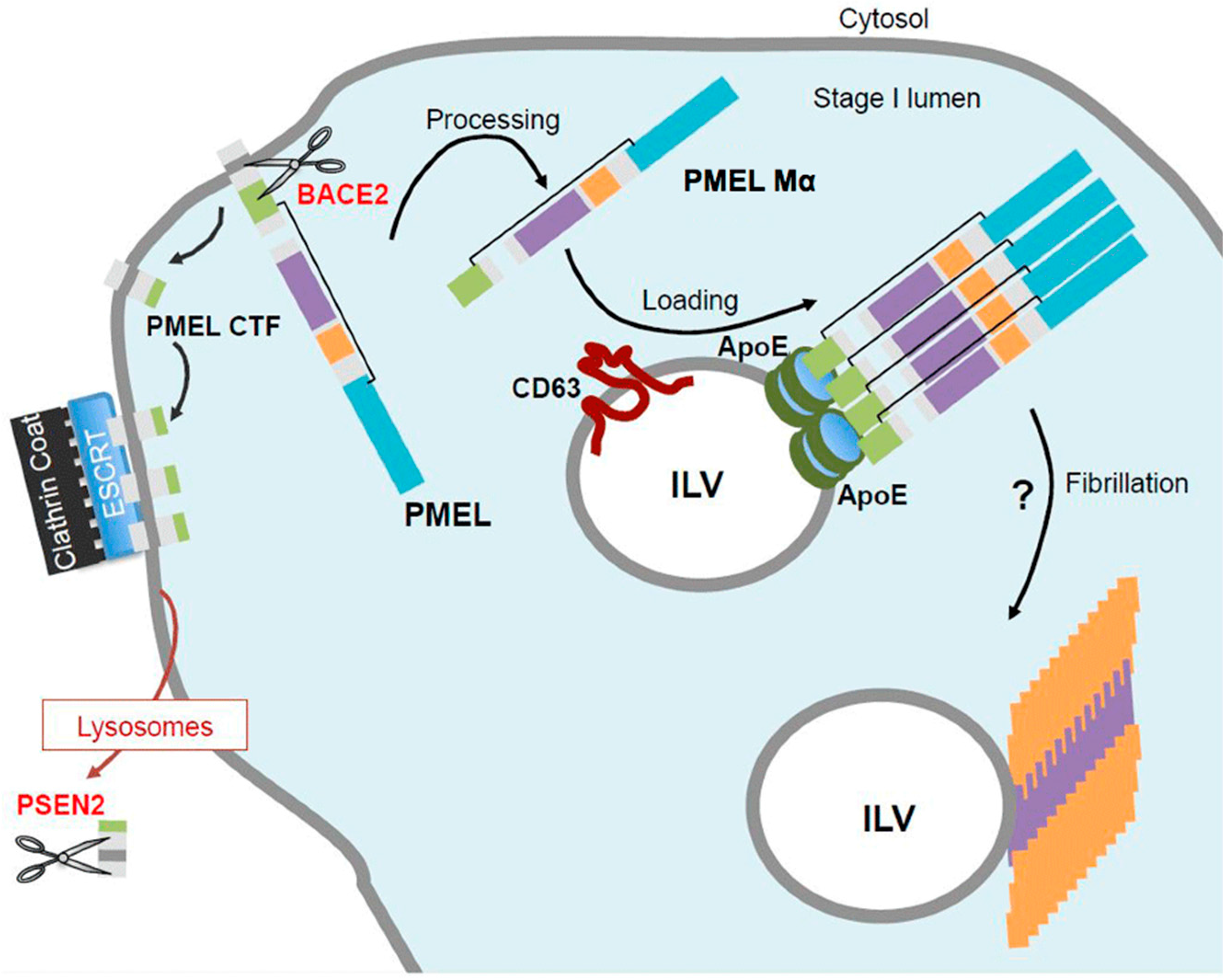
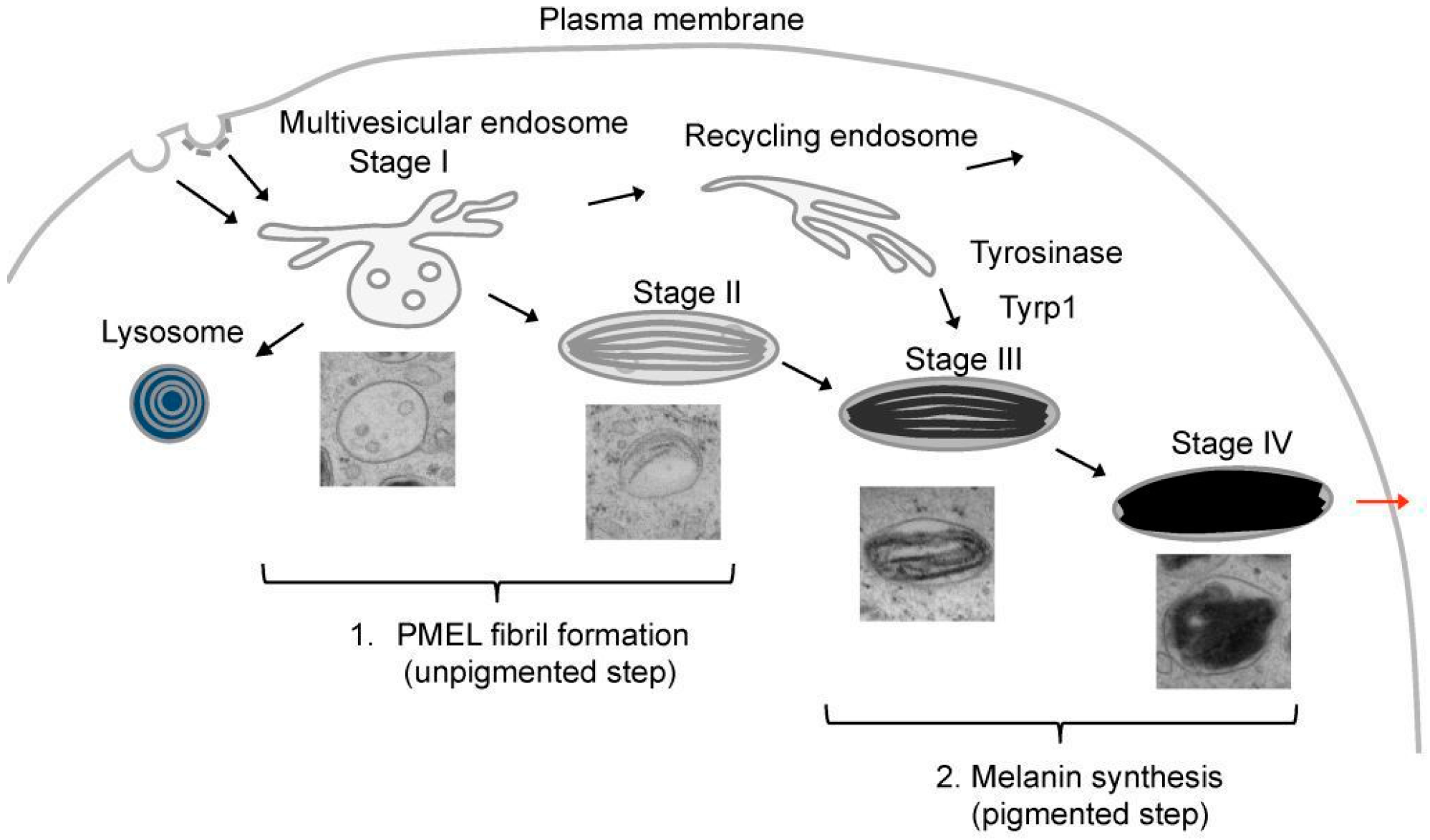
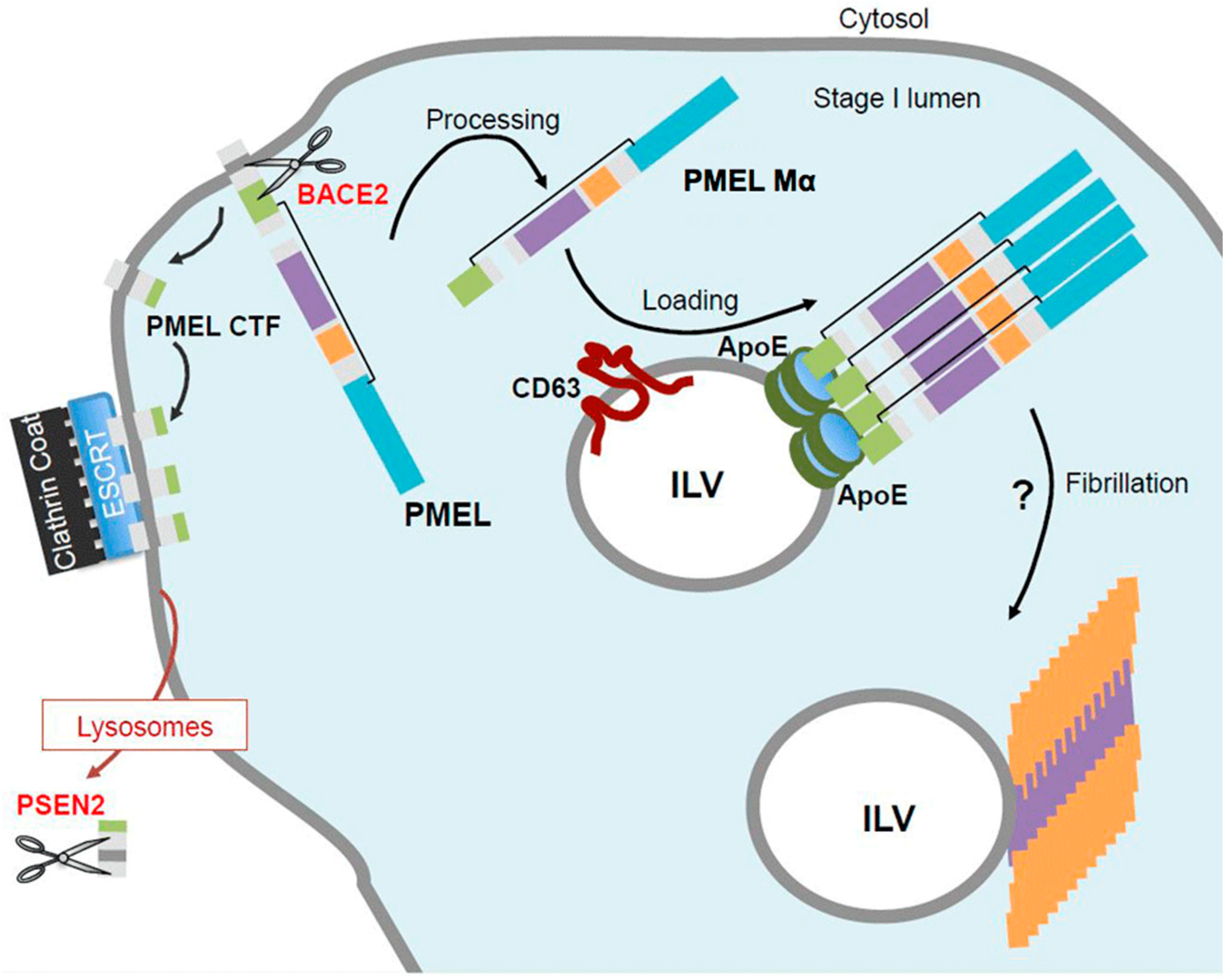
5. PMEL Trafficking and Sorting
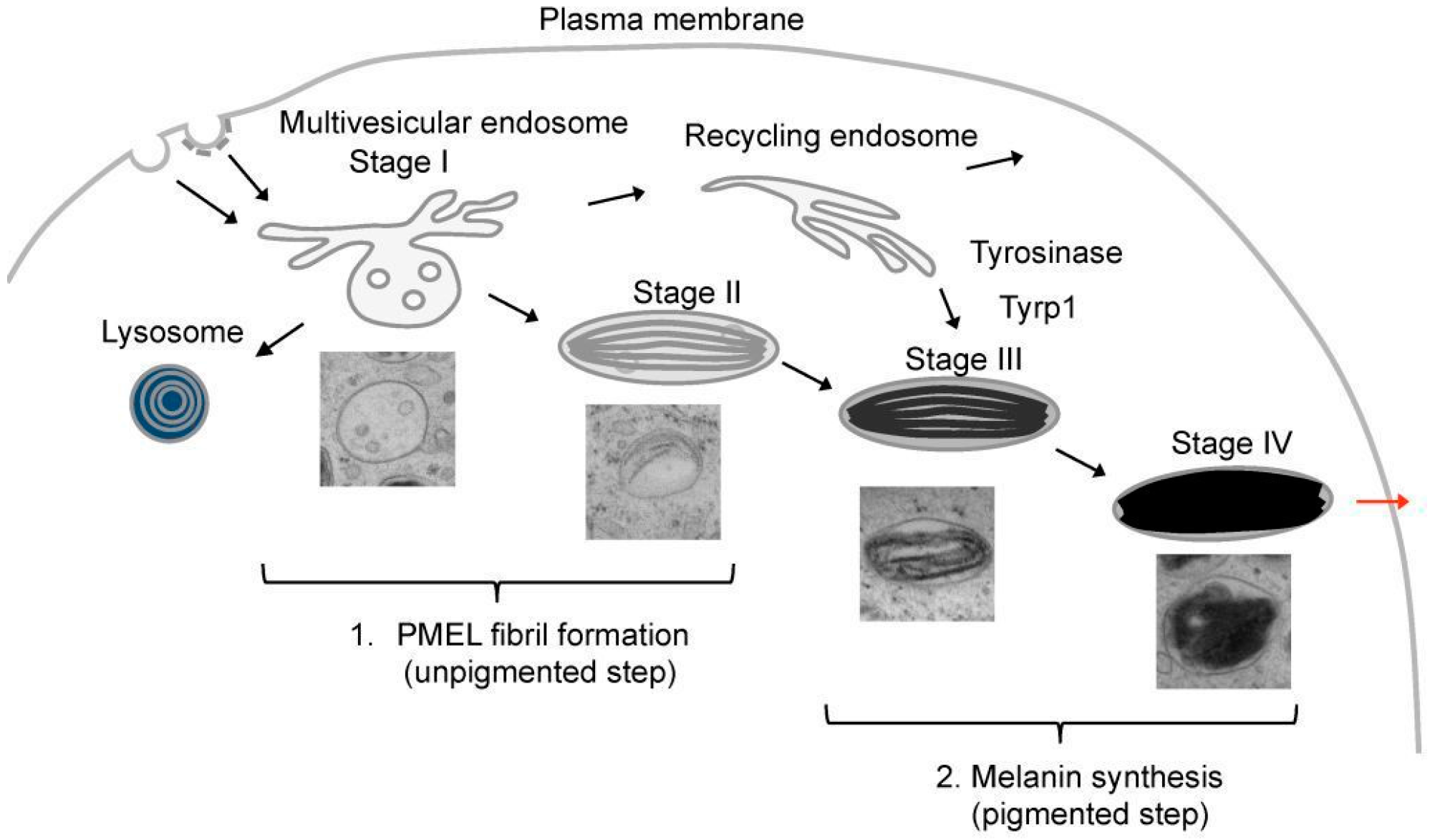
6. PMEL Fibrillation in Vivo
7. PMEL Fibrillation in Vitro
8. PMEL Fibrils: Implication for Melanogenesis
9. PMEL as a Physiological Model for Pathological Amyloids
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis-pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Vieira, W.D.; Potterf, B.; Sakai, C.; Imokawa, G.; Hearing, V.J. Modulation of melanogenic protein expression during the switch from eu- to pheomelanogenesis. J. Cell Sci. 1995, 108, 2301–2309. [Google Scholar] [PubMed]
- Furumura, M.; Sakai, C.; Potterf, S.B.; Vieira, W.D.; Barsh, G.S.; Hearing, V.J. Characterization of genes modulated during pheomelanogenesis using differential display. Proc. Natl. Acad. Sci. USA 1998, 95, 7374–7378. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Marks, M.S. Melanosomes-dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Orlow, S.J.; Zhou, B.K.; Boissy, R.E.; Pifko-Hirst, S. Identification of a mammalian melanosomal matrix glycoprotein. J. Investig. Dermatol. 1993, 101, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.K.; Kobayashi, T.; Donatien, P.D.; Bennett, D.C.; Hearing, V.J.; Orlow, S.J. Identification of a melanosomal matrix protein encoded by the murine si (silver) locus using “organelle scanning”. Proc. Natl. Acad. Sci. USA 1994, 91, 7076–7080. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. Pmel: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; Tenza, D.; Lemmon, M.A.; Kerje, S.; Raposo, G.; Andersson, L.; Marks, M.S. Mutations in or near the transmembrane domain alter PMEL amyloid formation from functional to pathogenic. PLoS Genet. 2011, 7, e1002286. [Google Scholar] [CrossRef] [PubMed]
- Hurbain, I.; Geerts, W.J.; Boudier, T.; Marco, S.; Verkleij, A.J.; Marks, M.S.; Raposo, G. Electron tomography of early melanosomes: Implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc. Natl. Acad. Sci. USA 2008, 105, 19726–19731. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Tenza, D.; Murphy, D.M.; Berson, J.F.; Marks, M.S. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 2001, 152, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Seiji, M.; Fitzpatrick, T.B.; Simpson, R.T.; Birbeck, M.S. Chemical composition and terminology of specialized organelles (melanosomes and melanin granules) in mammalian melanocytes. Nature 1963, 197, 1082–1084. [Google Scholar] [CrossRef] [PubMed]
- Theos, A.C.; Truschel, S.T.; Raposo, G.; Marks, M.S. The Silver locus product Pmel17/gp100/Silv/ME20: Controversial in name and in function. Pigment Cell Res. 2005, 18, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Nichols, S.E.; Harper, D.C.; Berson, J.F.; Marks, M.S. A novel splice variant of Pmel17 expressed by human melanocytes and melanoma cells lacking some of the internal repeats. J. Investig. Dermatol. 2003, 121, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, M.; Bateman, A.; Clarke, J.; Hamill, S.J.; Sandford, R.; Thomas, R.L.; Chothia, C. The structure of a PKD domain from polycystin-1: Implications for polycystic kidney disease. EMBO J. 1999, 18, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.C.; Theos, A.C.; Herman, K.E.; Tenza, D.; Raposo, G.; Marks, M.S. Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes. J. Biol. Chem. 2008, 283, 2307–2322. [Google Scholar] [CrossRef] [PubMed]
- Valencia, J.C.; Rouzaud, F.; Julien, S.; Chen, K.G.; Passeron, T.; Yamaguchi, Y.; Abu-Asab, M.; Tsokos, M.; Costin, G.E.; Yamaguchi, H.; et al. Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J. Biol. Chem. 2007, 282, 11266–11280. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Watt, B.; Spruce, L.A.; Seeholzer, S.H.; Marks, M.S. The Kringle-like Domain Facilitates Post-endoplasmic Reticulum Changes to Premelanosome Protein (PMEL) Oligomerization and Disulfide Bond Configuration and Promotes Amyloid Formation. J. Biol. Chem. 2016, 291, 3595–3612. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Tamaki, K.; Hearing, V.J. The secreted form of a melanocyte membrane-bound glycoprotein (Pmel17/gp100) is released by ectodomain shedding. FASEB J. 2010, 24, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Vieira, M.N.; De Felice, F.G. Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life 2007, 59, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Reed, M.N.; Kotilinek, L.A.; Grant, M.K.; Forster, C.L.; Qiang, W.; Shapiro, S.L.; Reichl, J.H.; Chiang, A.C.; Jankowsky, J.L.; et al. Quaternary structure defines a large class of amyloid-β oligomers neutralized by sequestration. Cell Rep. 2015, 11, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Berson, J.F.; Theos, A.C.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 2003, 161, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Muller, J.; Vieira, W.D.; Rouzaud, F.; Kikuchi, K.; Tamaki, K.; Hearing, V.J. The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers. J. Biol. Chem. 2006, 281, 21198–21208. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Shewmaker, F.; Hu, K.N.; McPhie, P.; Tycko, R.; Wickner, R.B. Repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH. J. Biol. Chem. 2011, 286, 8385–8393. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Shewmaker, F.; Hu, K.N.; McPhie, P.; Tycko, R.; Wickner, R.B. The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis. Proc. Natl. Acad. Sci. USA 2009, 106, 13731–13736. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; van Niel, G.; Fowler, D.M.; Hurbain, I.; Luk, K.C.; Stayrook, S.E.; Lemmon, M.A.; Raposo, G.; Shorter, J.; Kelly, J.W.; et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 2009, 284, 35543–35555. [Google Scholar] [CrossRef] [PubMed]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef] [PubMed]
- Maresh, G.A.; Marken, J.S.; Neubauer, M.; Aruffo, A.; Hellstrom, I.; Hellstrom, K.E.; Marquardt, H. Cloning and expression of the gene for the melanoma-associated ME20 antigen. DNA Cell Biol. 1994, 13, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Maresh, G.A.; Wang, W.C.; Beam, K.S.; Malacko, A.R.; Hellstrom, I.; Hellstrom, K.E.; Marquardt, H. Differential processing and secretion of the melanoma-associated ME20 antigen. Arch. Biochem. Biophys. 1994, 311, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Nufer, O.; Guldbrandsen, S.; Degen, M.; Kappeler, F.; Paccaud, J.P.; Tani, K.; Hauri, H.P. Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. J. Cell Sci. 2002, 115, 619–628. [Google Scholar] [PubMed]
- Theos, A.C.; Berson, J.F.; Theos, S.C.; Herman, K.E.; Harper, D.C.; Tenza, D.; Sviderskaya, E.V.; Lamoreux, M.L.; Bennett, D.C.; Raposo, G.; et al. Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell 2006, 17, 3598–3612. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, R.M.; Vigneron, N.; Rahner, C.; Cresswell, P. Proprotein convertases process Pmel17 during secretion. J. Biol. Chem. 2011, 286, 9321–9337. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, G.; Song, W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006, 20, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Rochin, L.; Hurbain, I.; Serneels, L.; Fort, C.; Watt, B.; Leblanc, P.; Marks, M.S.; de Strooper, B.; Raposo, G.; van Niel, G. BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10658–10663. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Hozumi, Y.; Suzuki, T. ADAM protease inhibitors reduce melanogenesis by regulating PMEL17 processing in human melanocytes. J. Dermatol. Sci. 2015, 78, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Maruyama, H.; Huelsmann, C.; Baches, S.; Weggen, S.; Koo, E.H. Formation of Pmel17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for γ-secretase. J. Biol. Chem. 2009, 284, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; de Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted location of PSEN2 γ-secretase determines substrate specificity and generates an intracellular Aβ pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Theos, A.C.; Truschel, S.T.; Tenza, D.; Hurbain, I.; Harper, D.C.; Berson, J.F.; Thomas, P.C.; Raposo, G.; Marks, M.S. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 2006, 10, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Bergam, P.; Di Cicco, A.; Hurbain, I.; Lo Cicero, A.; Dingli, F.; Palmulli, R.; Fort, C.; Potier, M.C.; Schurgers, L.J.; et al. Apolipoprotein E regulates amyloid formation within endosomes of pigment cells. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. 2010, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; le Naour, F.; Silvie, O.; Milhiet, P.E.; Boucheix, C.; Rubinstein, E. Lateral organization of membrane proteins: Tetraspanins spin their web. Biochem. J. 2009, 420, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.R.; Watt, B.; Fard, S.S.; Tenza, D.; Mannstrom, P.; Narfstrom, K.; Ekesten, B.; Ito, S.; Wakamatsu, K.; Larsson, J.; et al. Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 2011, 7, e1002285. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, R.M.; Vigneron, N.; Hee, J.S.; Graham, M.; Cresswell, P. Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils. Mol. Biol. Cell 2013. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.; Bonetti, C.; Surace, E.M.; Marigo, V.; Raposo, G. The ocular albinism type 1 (OA1) G-protein-coupled receptor functions with MART-1 at early stages of melanogenesis to control melanosome identity and composition. Hum. Mol. Genet. 2009, 18, 4530–4545. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.N.; McGlinchey, R.P.; Wickner, R.B.; Tycko, R. Segmental polymorphism in a functional amyloid. Biophys. J. 2011, 101, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, C.M.; McGlinchey, R.P.; Lee, J.C. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc. Natl. Acad. Sci. USA 2010, 107, 21447–21452. [Google Scholar] [CrossRef] [PubMed]
- Louros, N.N.; Iconomidou, V.A. Identification of an amyloid fibril forming segment of human Pmel17 repeat domain (RPT domain). Biopolymers 2016, 106, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Lee, J.C. Lysophospholipid-containing membranes modulate the fibril formation of the repeat domain of a human functional amyloid, pmel17. J. Mol. Biol. 2014, 426, 4074–4086. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. Biology of amyloid: Structure, function, and regulation. Structure 2010, 18, 1244–1260. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.K.; Platt, J.T.; Kim, K.K.; Kwon, B.S.; Bennett, D.C.; Pawelek, J.M. Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur. J. Biochem. 1996, 236, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.H.; Hou, L.; Moellmann, G.; Kuklinska, E.; Antol, K.; Fraser, M.; Halaban, R.; Kwon, B.S. Characterization and subcellular localization of human Pmel 17/silver, a 110-kDa (pre) melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J. Investig. Dermatol. 1996, 106, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Esparza, M.; Jimenez-Cervantes, C.; Bennett, D.C.; Lozano, J.A.; Solano, F.; Garcia-Borron, J.C. The mouse silver locus encodes a single transcript truncated by the silver mutation. Mamm. Genome 1999, 10, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.C.; Thigpen, L.W. The silver mouse: A recessive color variation. J. Heredity 1930, 21, 495–498. [Google Scholar]
- Bennett, D.C.; Lamoreux, M.L. The color loci of mice—A genetic century. Pigment Cell Res. 2003, 16, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Keeling, L.; Andersson, L.; Schutz, K.E.; Kerje, S.; Fredriksson, R.; Carlborg, O.; Cornwallis, C.K.; Pizzari, T.; Jensen, P. Chicken genomics: Feather-pecking and victim pigmentation. Nature 2004, 431, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Kerje, S.; Sharma, P.; Gunnarsson, U.; Kim, H.; Bagchi, S.; Fredriksson, R.; Schutz, K.; Jensen, P.; von Heijne, G.; Okimoto, R.; et al. The Dominant white, Dun and Smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics 2004, 168, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bossche, K.; Naeyaert, J.M.; Lambert, J. The quest for the mechanism of melanin transfer. Traffic 2006, 7, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Paus, R. Melanogenesis is coupled to murine anagen: Toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J. Investig. Dermatol. 1993, 101, 90–97. [Google Scholar] [CrossRef]
- Slominski, A.; Wortsman, J.; Plonka, P.M.; Schallreuter, K.U.; Paus, R.; Tobin, D.J. Hair follicle pigmentation. J. Investig. Dermatol. 2005, 124, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; Raposo, G.; Marks, M.S. Pmel17: An Amyloid Determinant of Organelle Structure. In Functional Amyloid Aggregation; Rigacci, S., Bucciantini, M., Eds.; Research Signpost: Trivandrum, India, 2010; Volume 6, pp. 89–113. [Google Scholar]
- Van Niel, G. Study of exosomes shed new light on physiology of amyloidogenesis. Cell. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Esch, F.S.; Keim, P.S.; Beattie, E.C.; Blacher, R.W.; Culwell, A.R.; Oltersdorf, T.; McClure, D.; Ward, P.J. Cleavage of amyloid β peptide during constitutive processing of its precursor. Science 1990, 248, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Hung, A.Y.; Schlossmacher, M.G.; Teplow, D.B.; Selkoe, D.J. β-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 1993, 268, 3021–3024. [Google Scholar] [PubMed]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef] [PubMed]
- Sisodia, S.S.; Koo, E.H.; Beyreuther, K.; Unterbeck, A.; Price, D.L. Evidence that β-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 1990, 248, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashier, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Lednev, I.K. Amyloid fibrils: The eighth wonder of the world in protein folding and aggregation. Biophys. J. 2014, 106, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.J.; Craft, S.; Banks, W.A. The APOE genotype: Modification of therapeutic responses in Alzheimer’s disease. Curr. Pharm. Des. 2015, 21, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Shimshek, D.R.; Jacobson, L.H.; Kolly, C.; Zamurovic, N.; Balavenkatraman, K.K.; Morawiec, L.; Kreutzer, R.; Schelle, J.; Jucker, M.; Bertschi, B. Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bissig, C.; Rochin, L.; Van Niel, G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. Int. J. Mol. Sci. 2016, 17, 1438. https://doi.org/10.3390/ijms17091438
Bissig C, Rochin L, Van Niel G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. International Journal of Molecular Sciences. 2016; 17(9):1438. https://doi.org/10.3390/ijms17091438
Chicago/Turabian StyleBissig, Christin, Leila Rochin, and Guillaume Van Niel. 2016. "PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation" International Journal of Molecular Sciences 17, no. 9: 1438. https://doi.org/10.3390/ijms17091438