
Oridonin, a Promising ent-Kaurane Diterpenoid Lead Compound

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
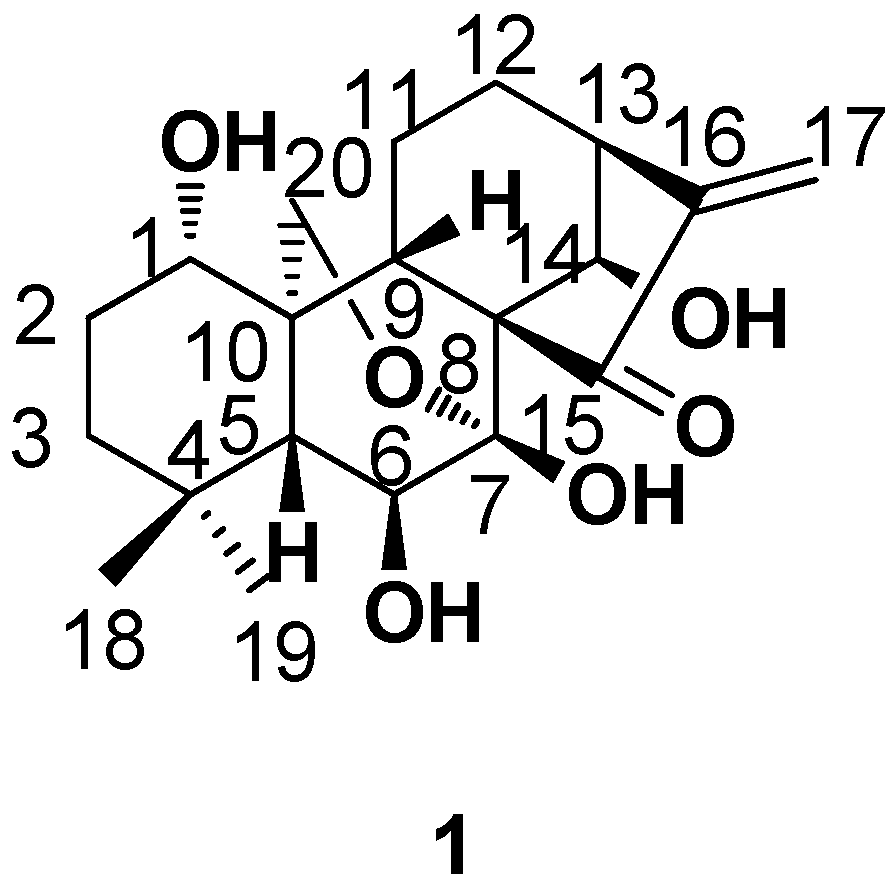
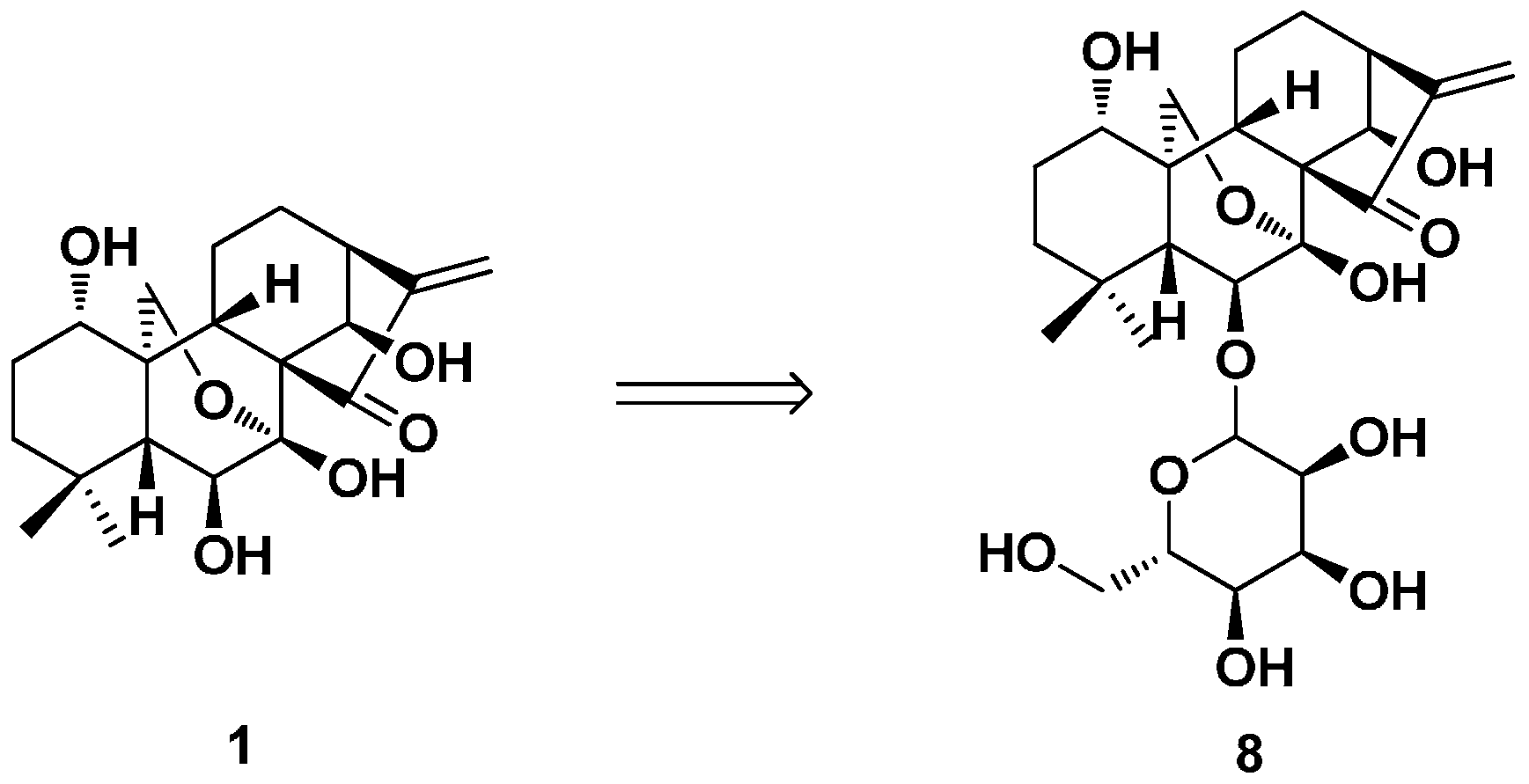
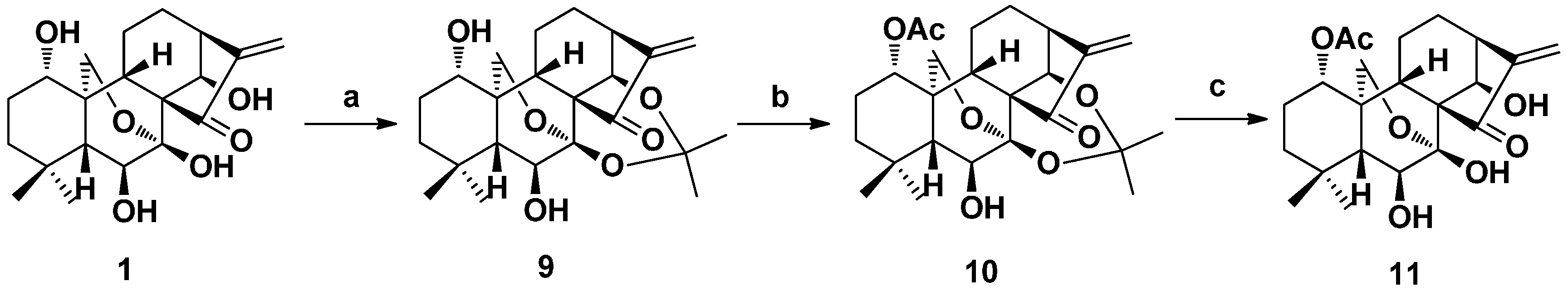
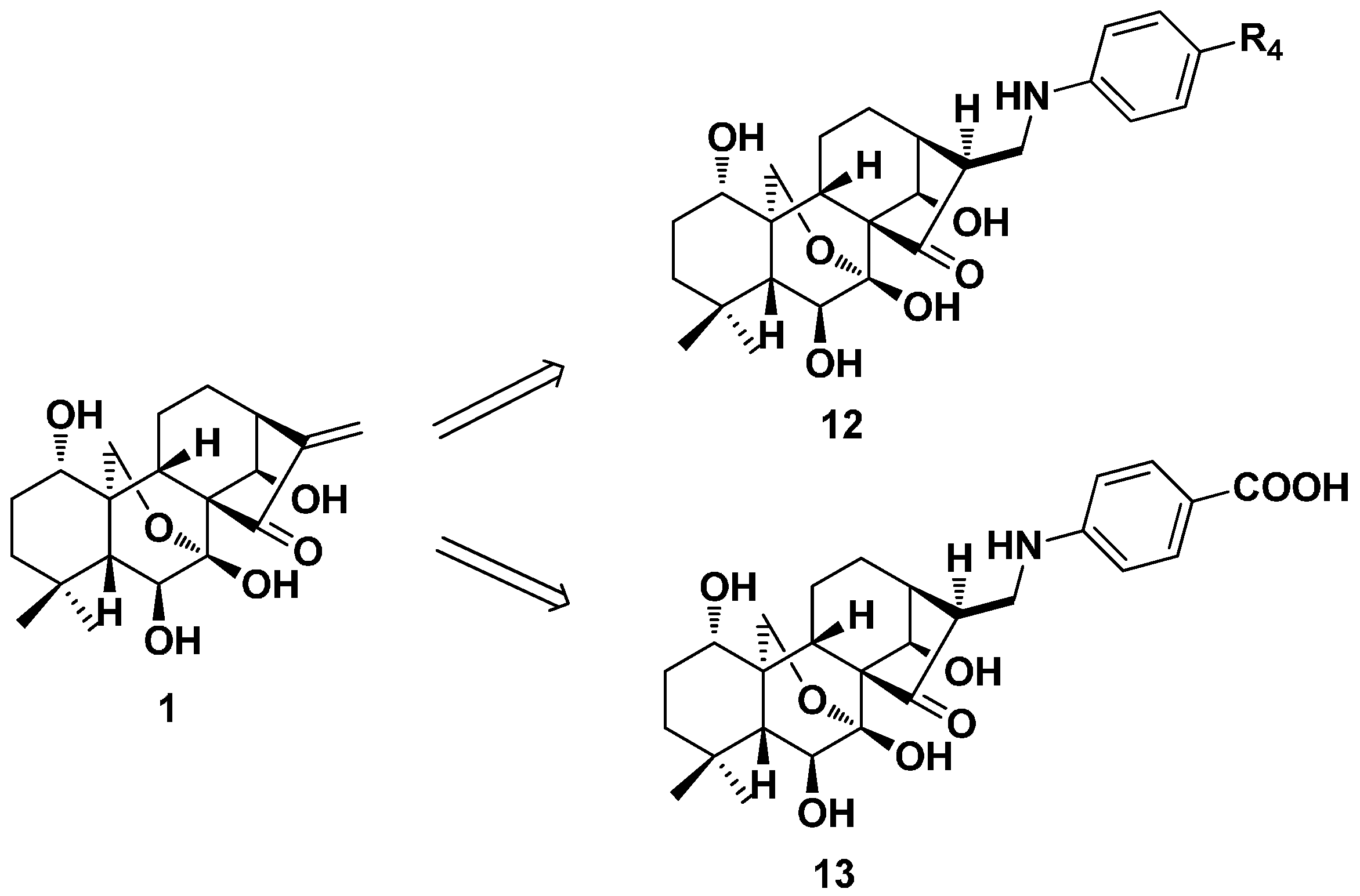
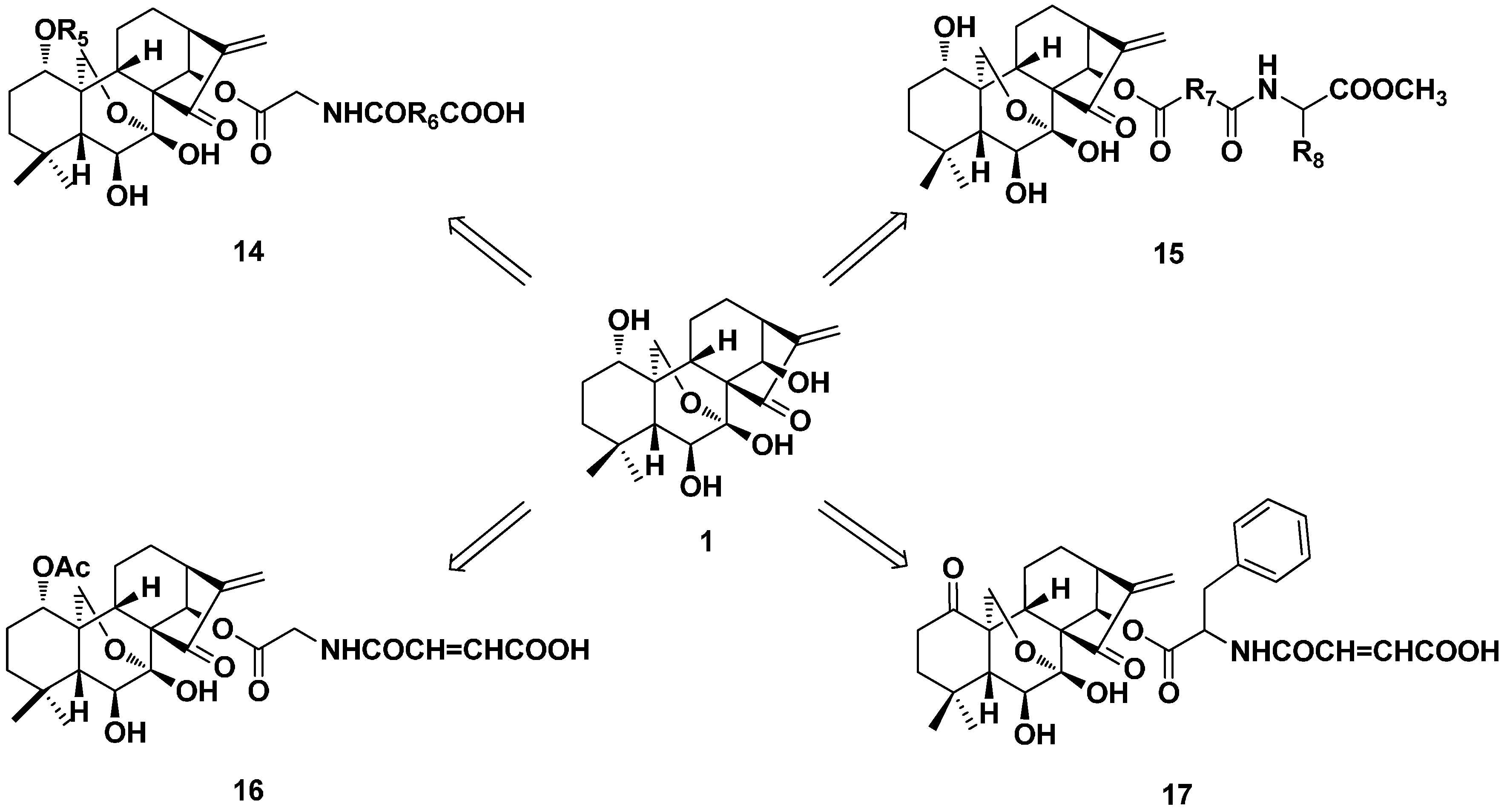
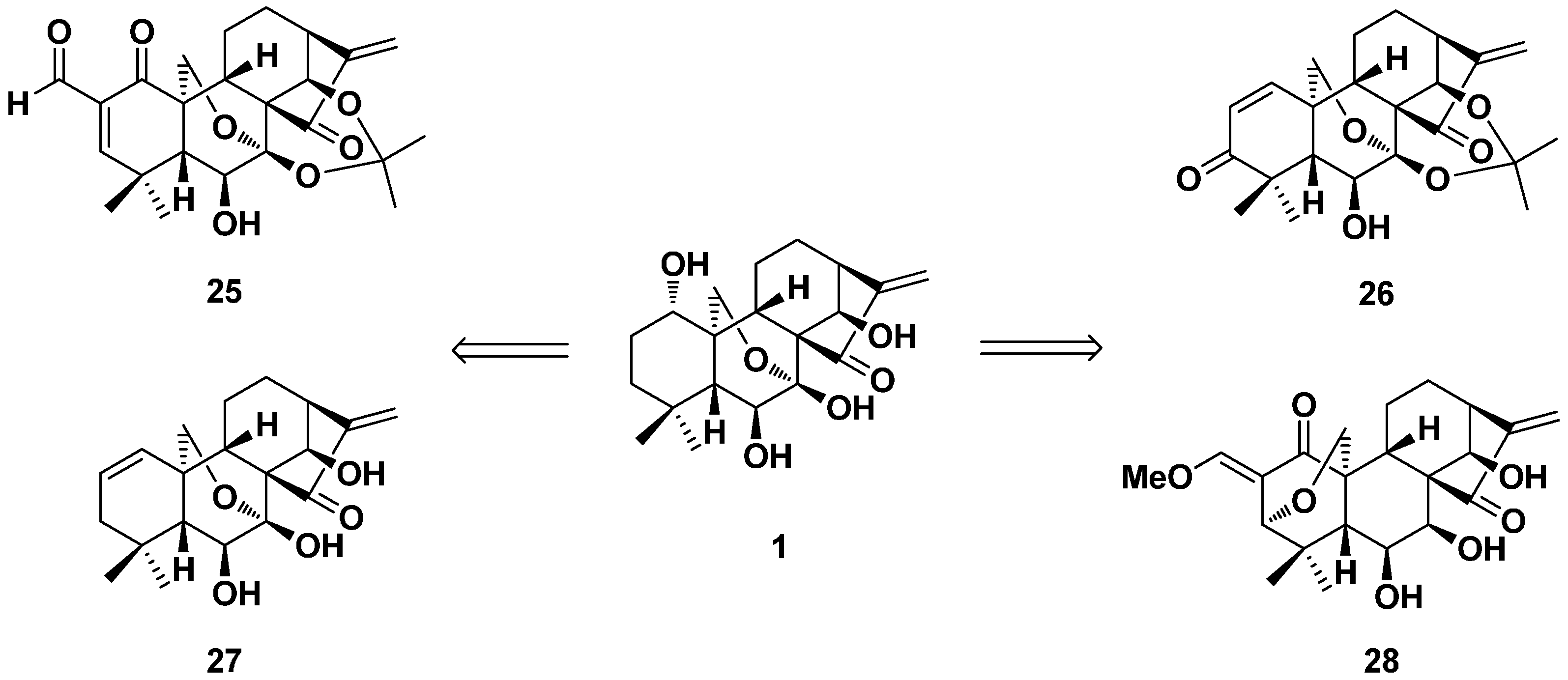
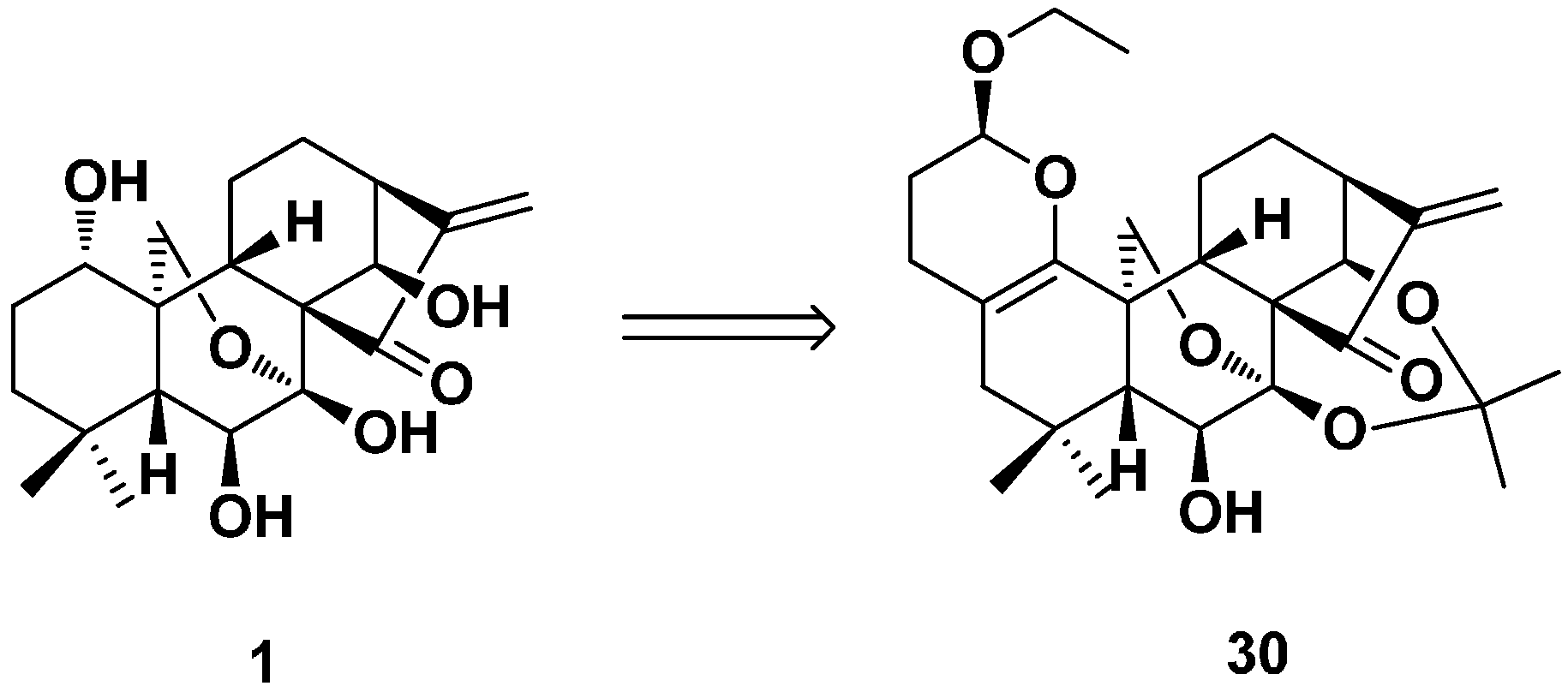
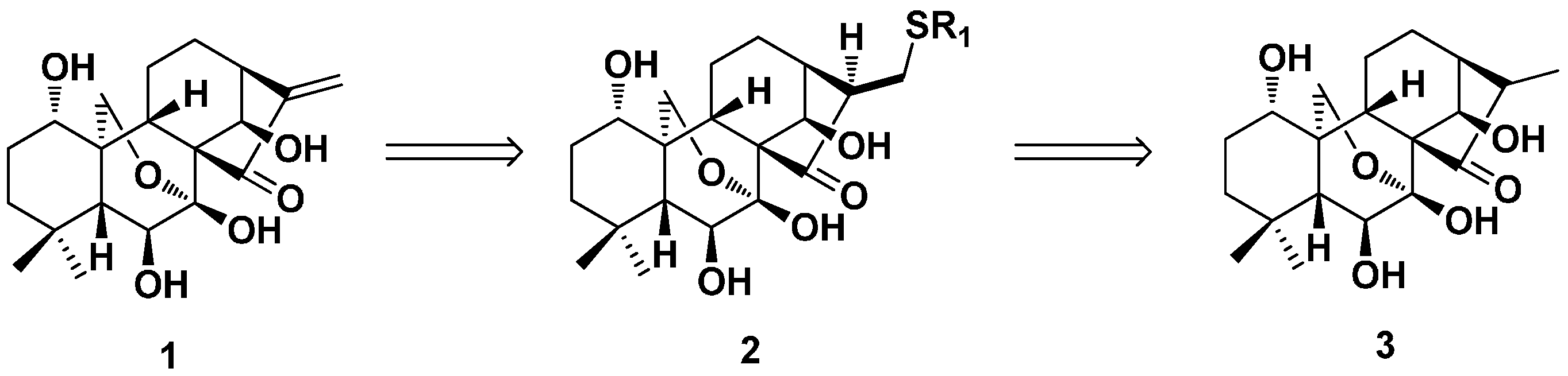
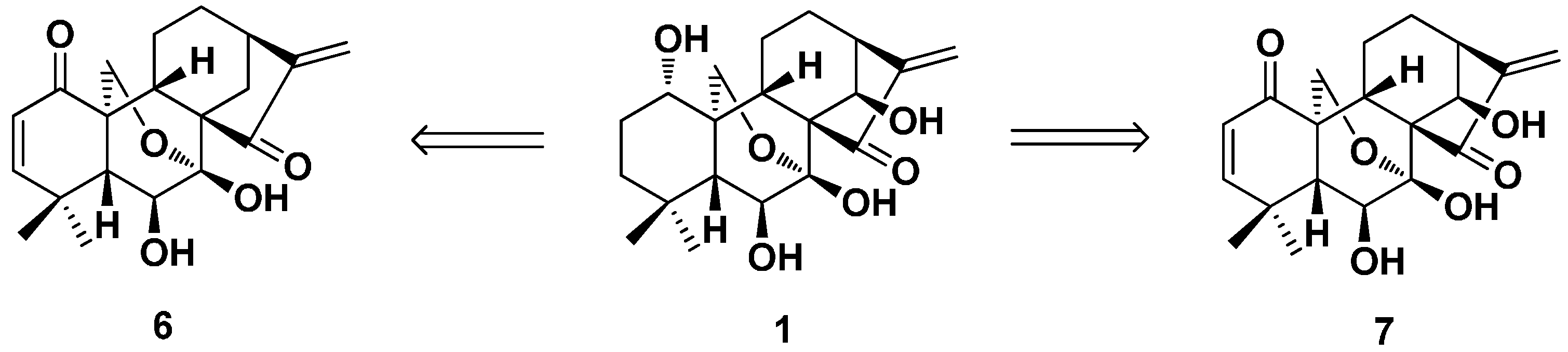
2. Structure Modification on the Core Structure of Oridonin
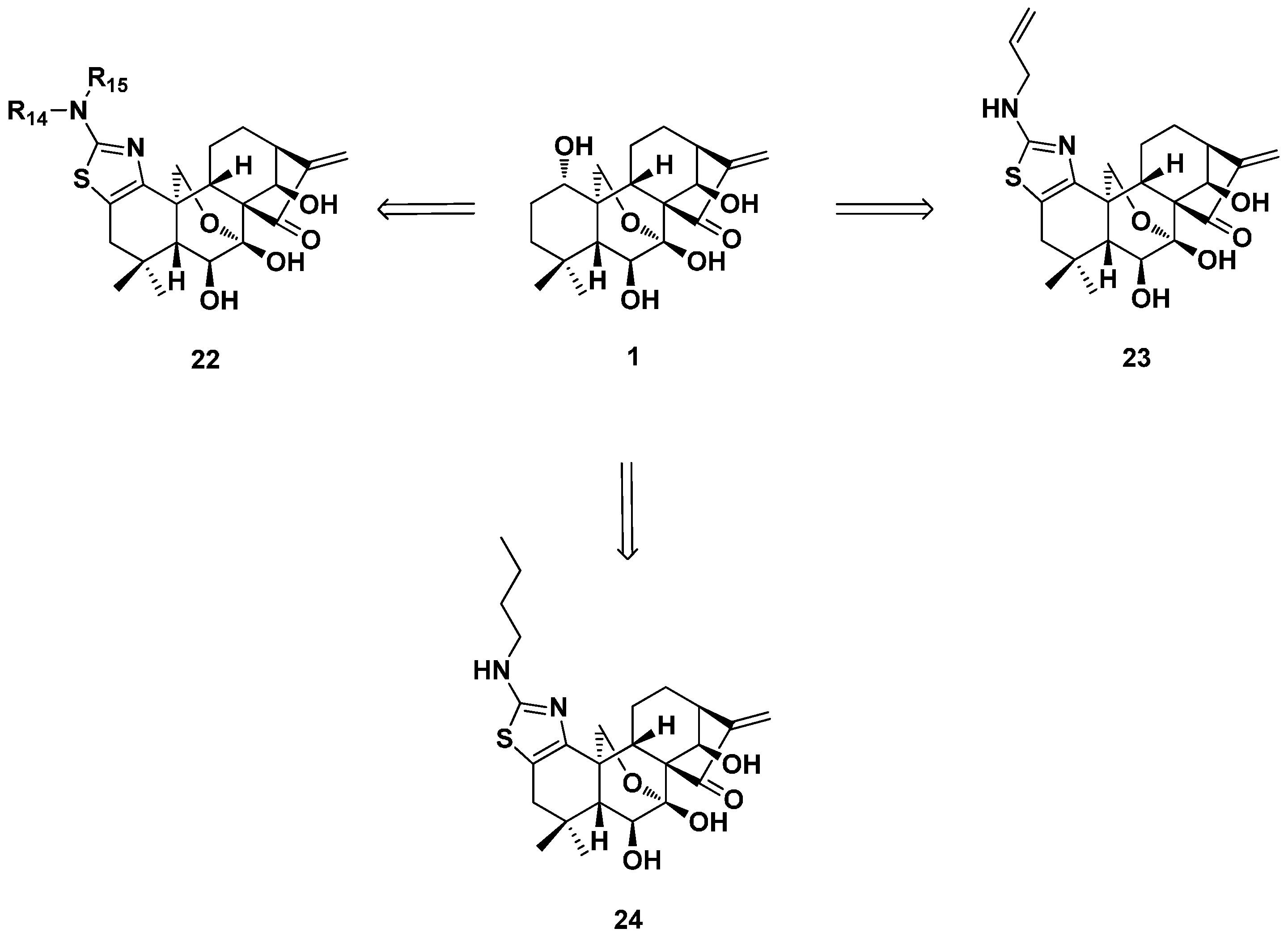
3. Oridonin as the Lead to Synthesize ent-Kaurane Diterpenoid Derivatives of Other Types
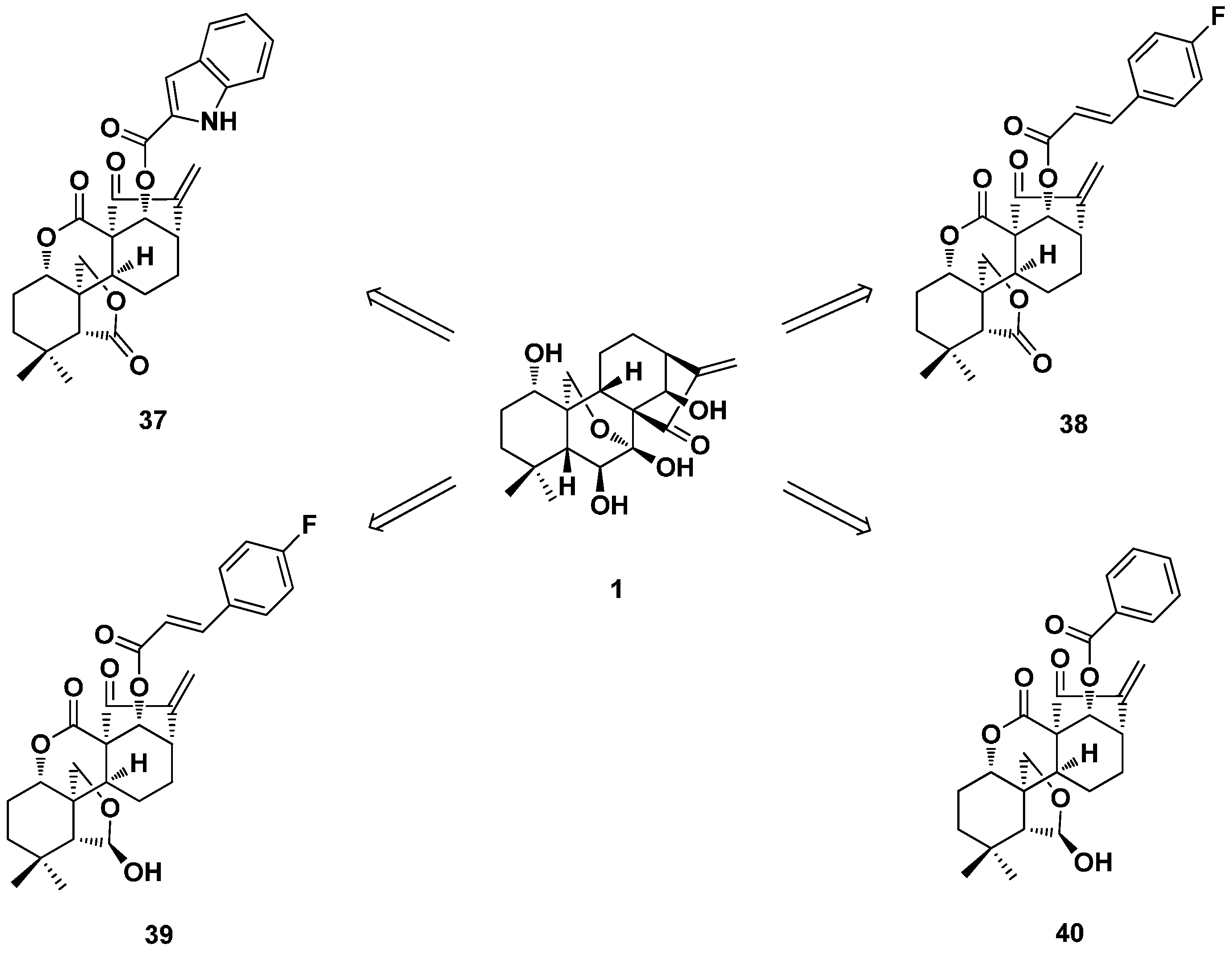
3.1. 15,16-seco-ent-Kaurane Diterpenoid Derivatives
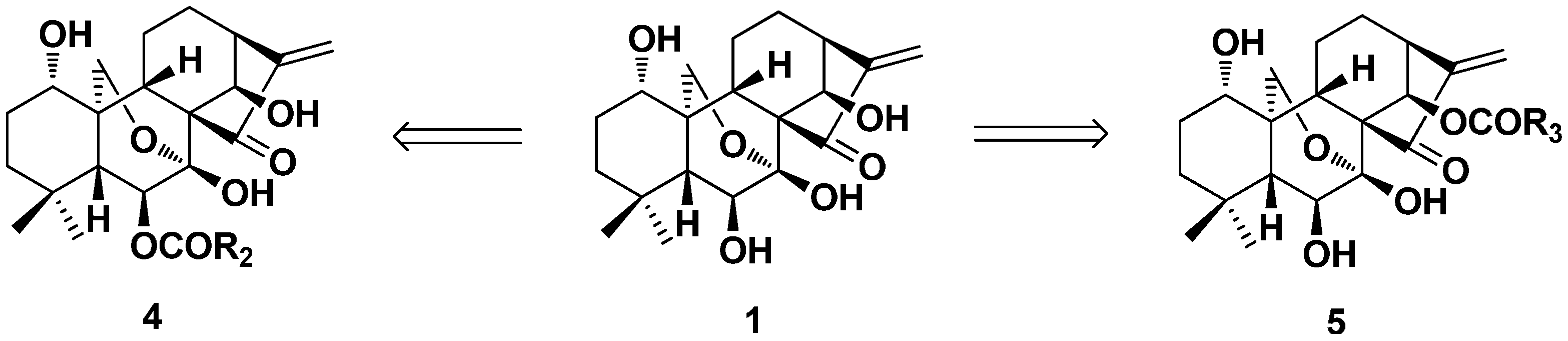
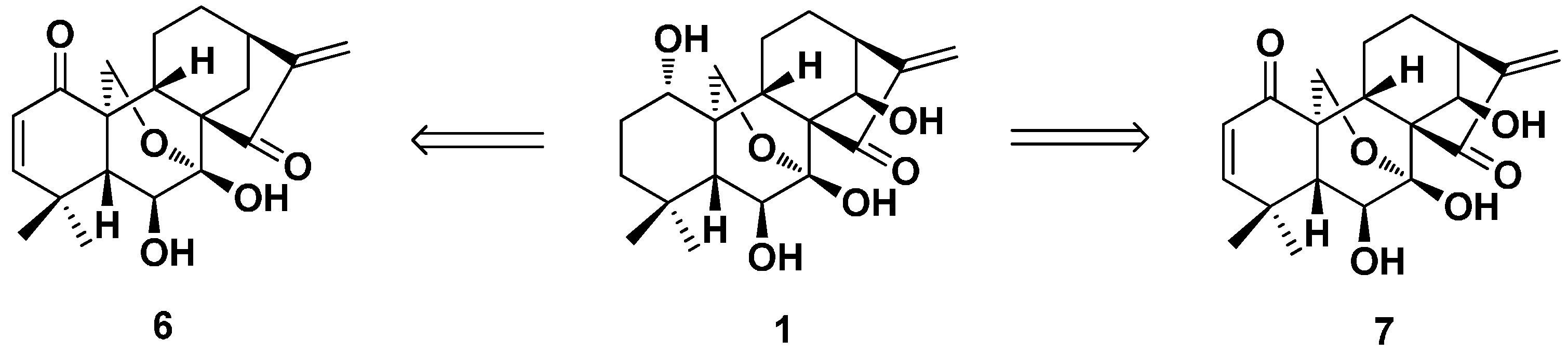
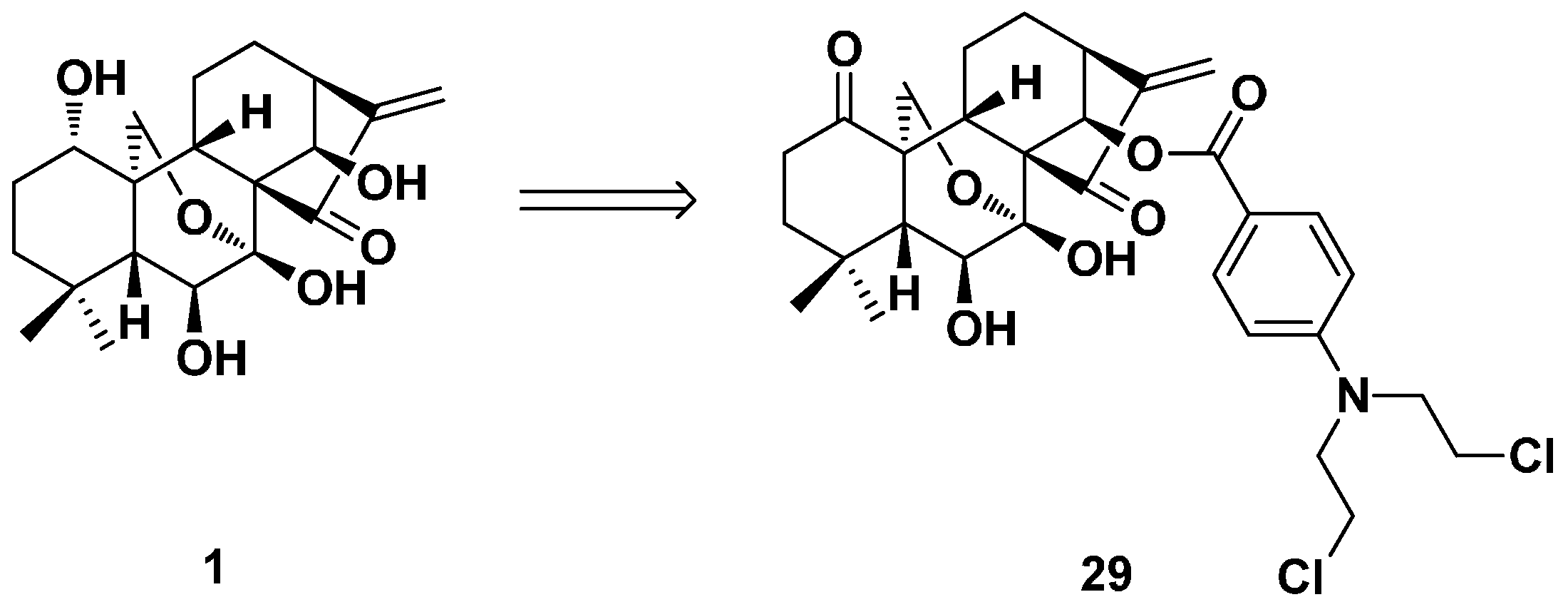
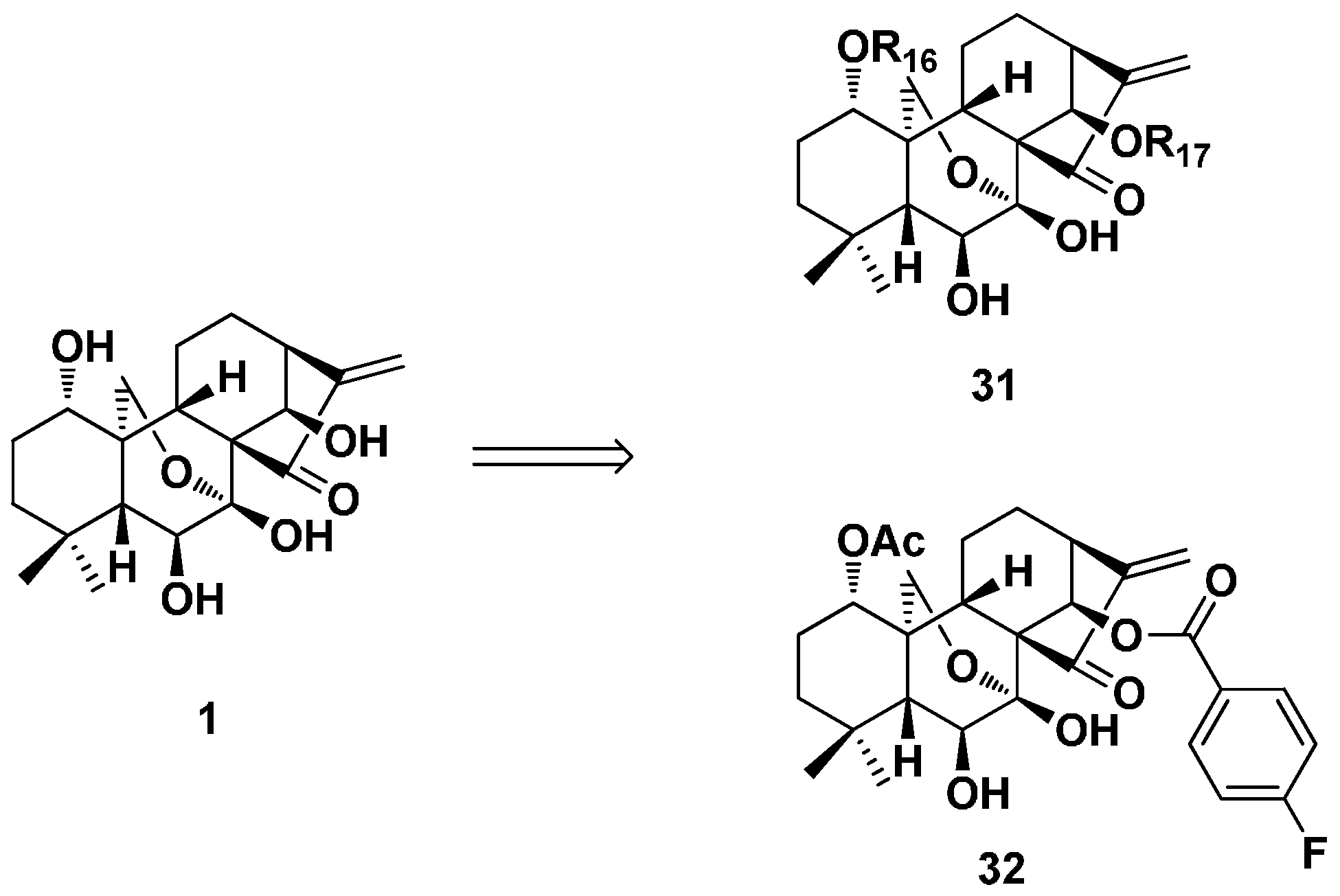
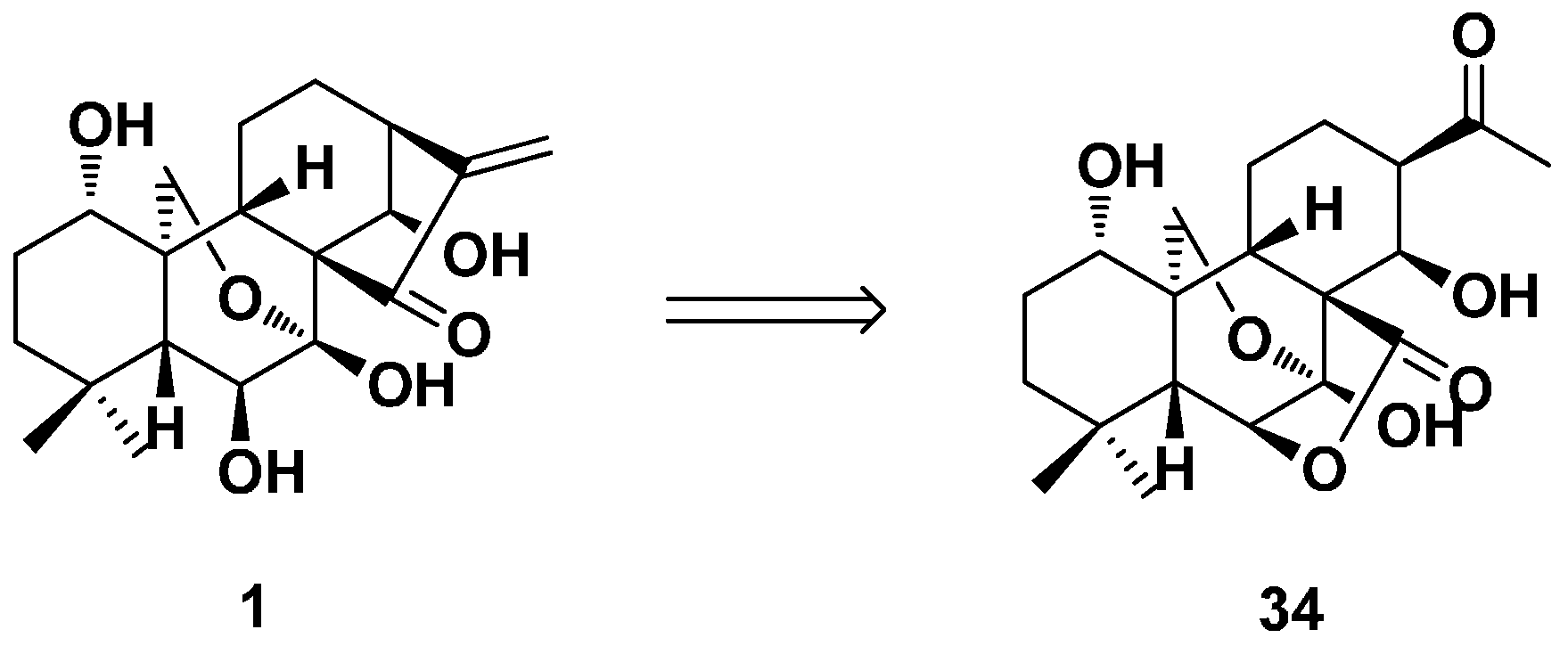
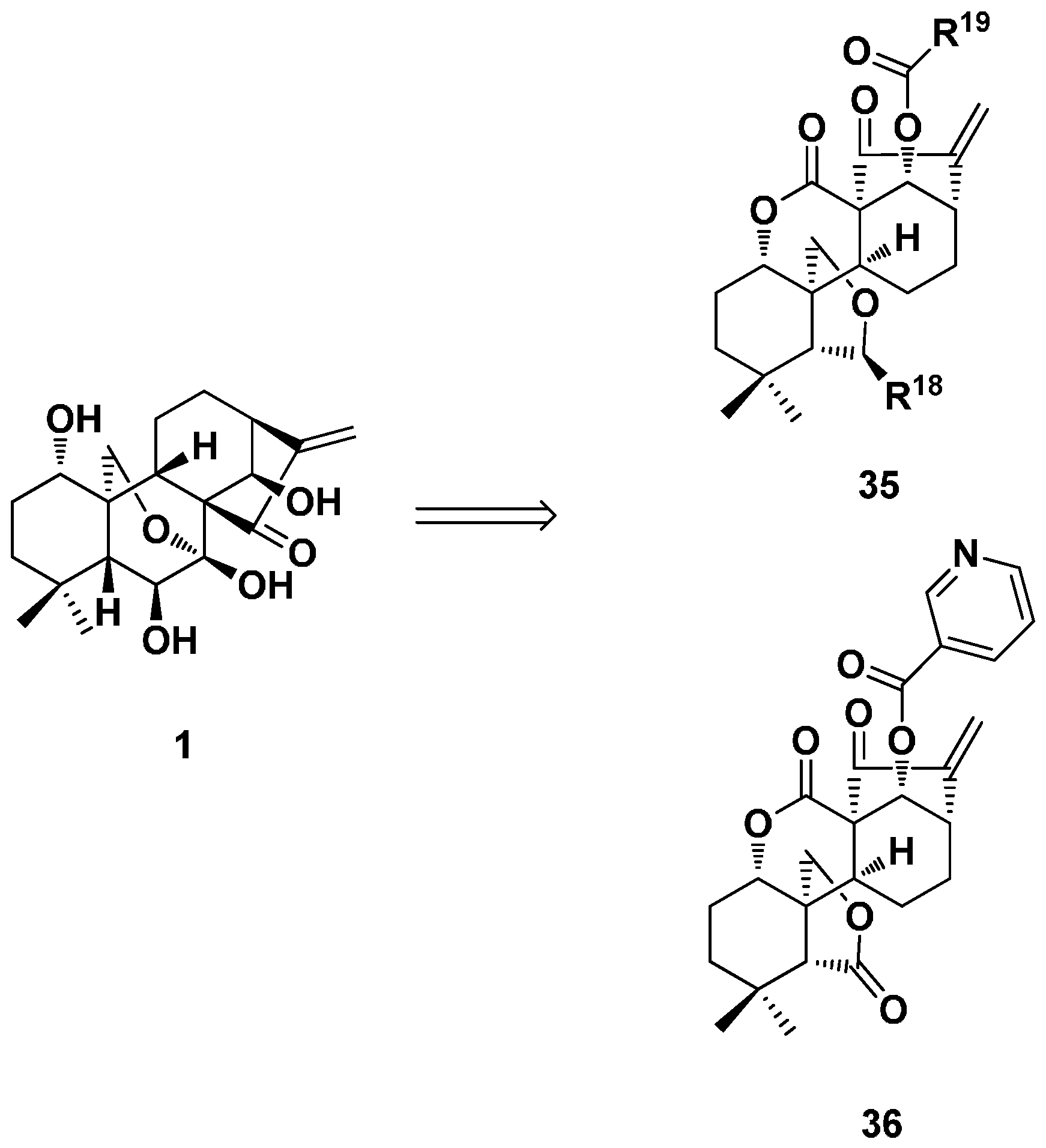
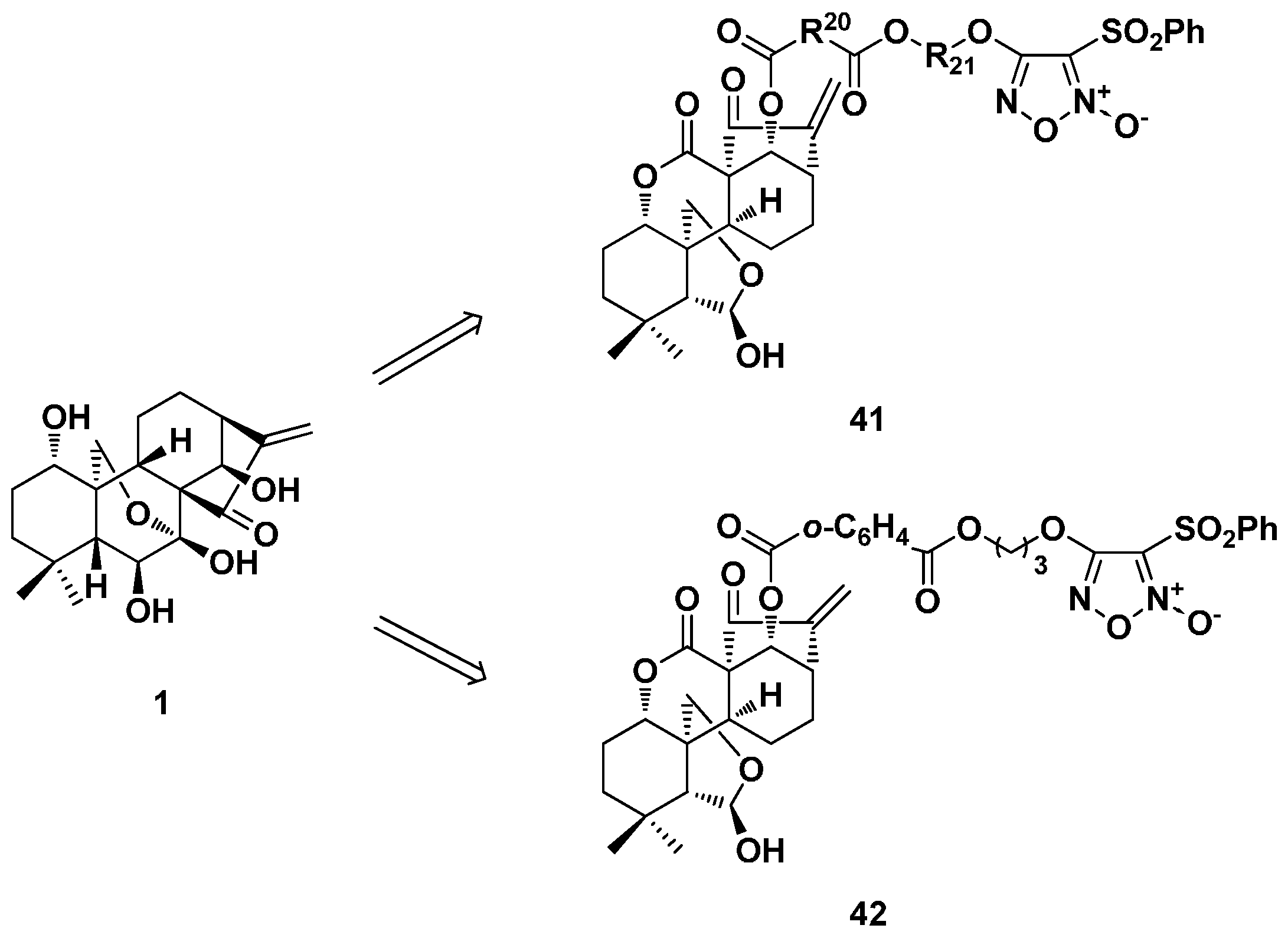
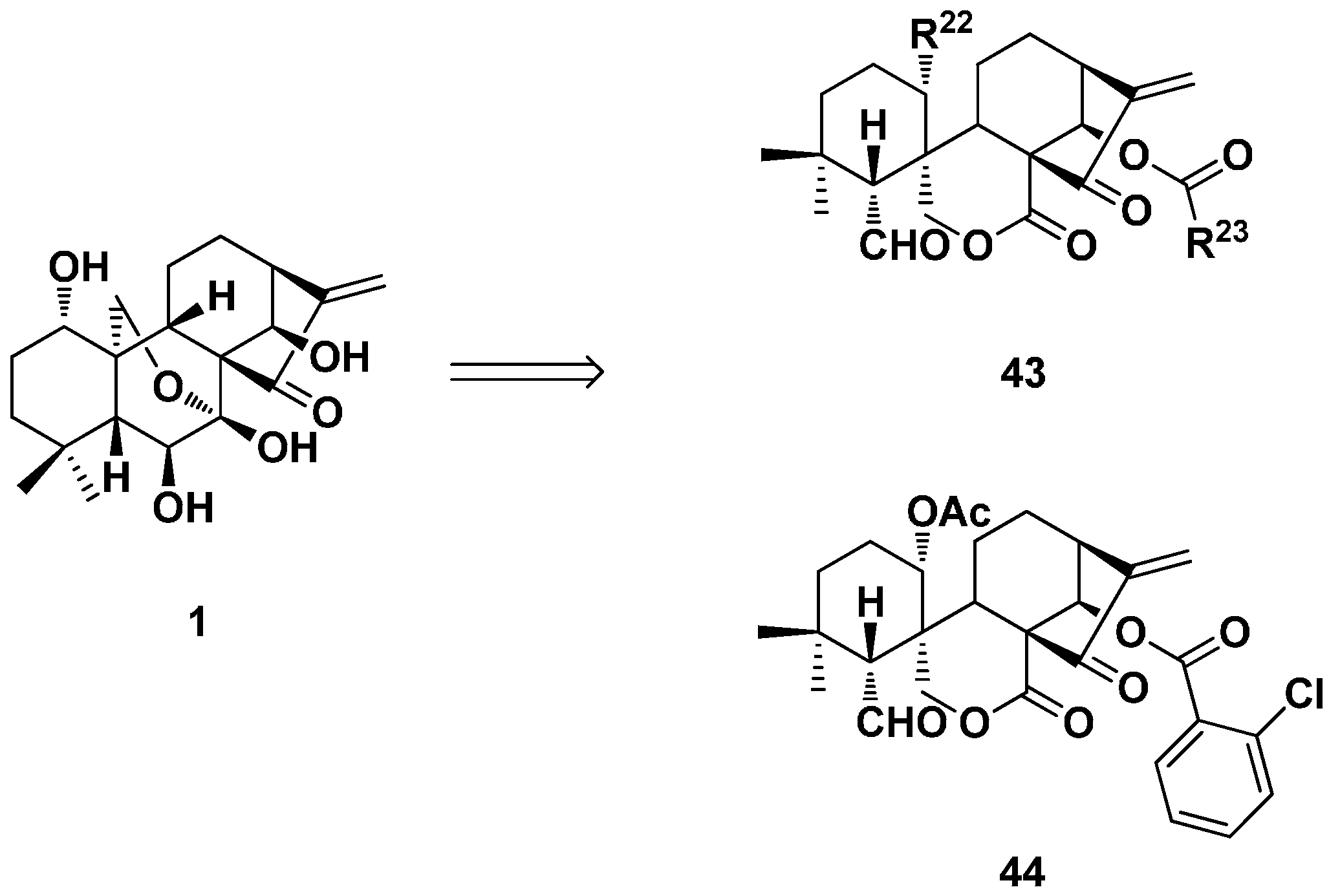
3.2. 6,7-seco-ent-Kaurane Diterpenoid Derivatives
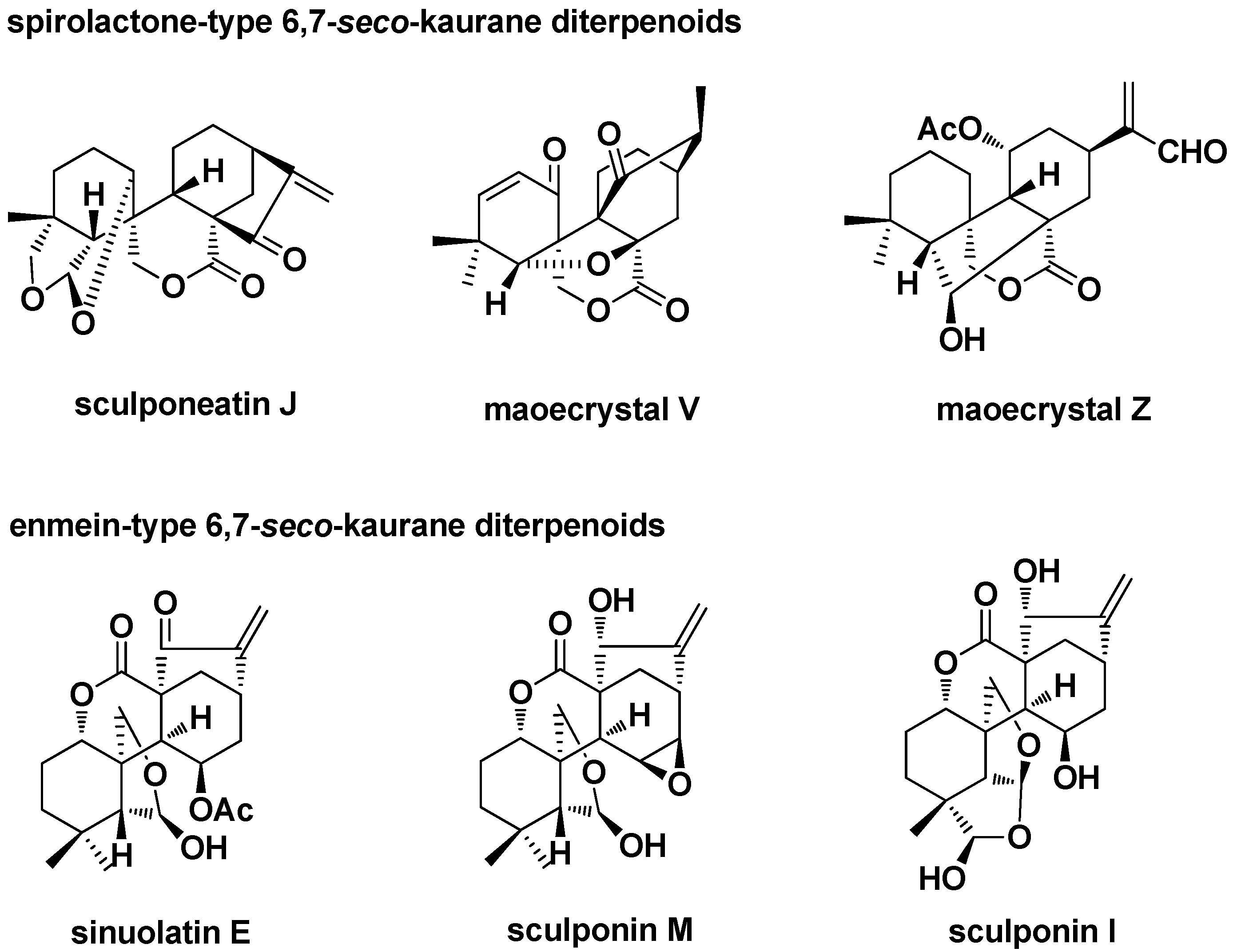
3.2.1. Enmein-Type 6,7-seco-ent-Kaurane Diterpenoid Derivatives
3.2.2. Spirolactone-Type 6,7-seco-ent-Kaurane Diterpenoid Derivatives
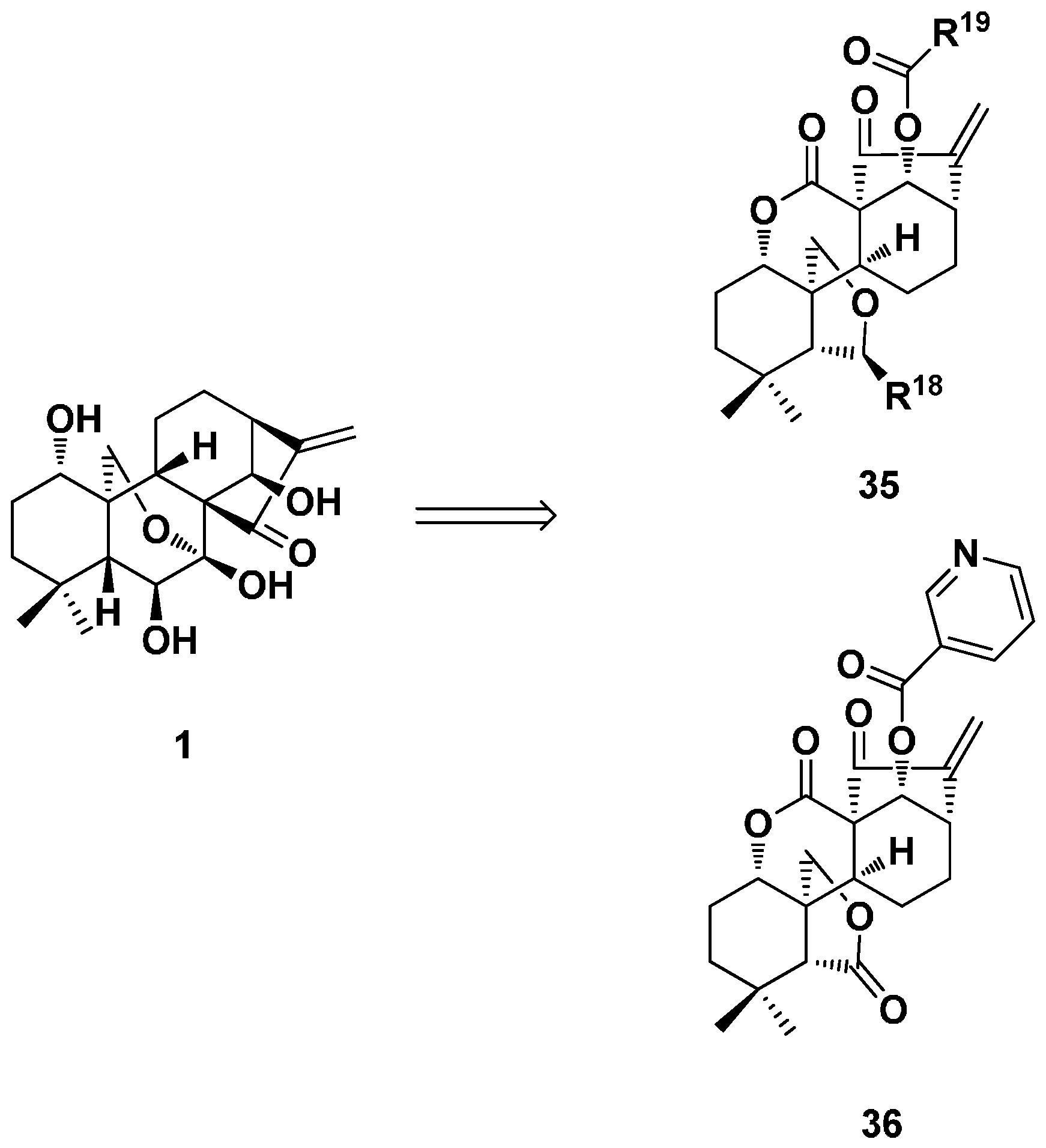
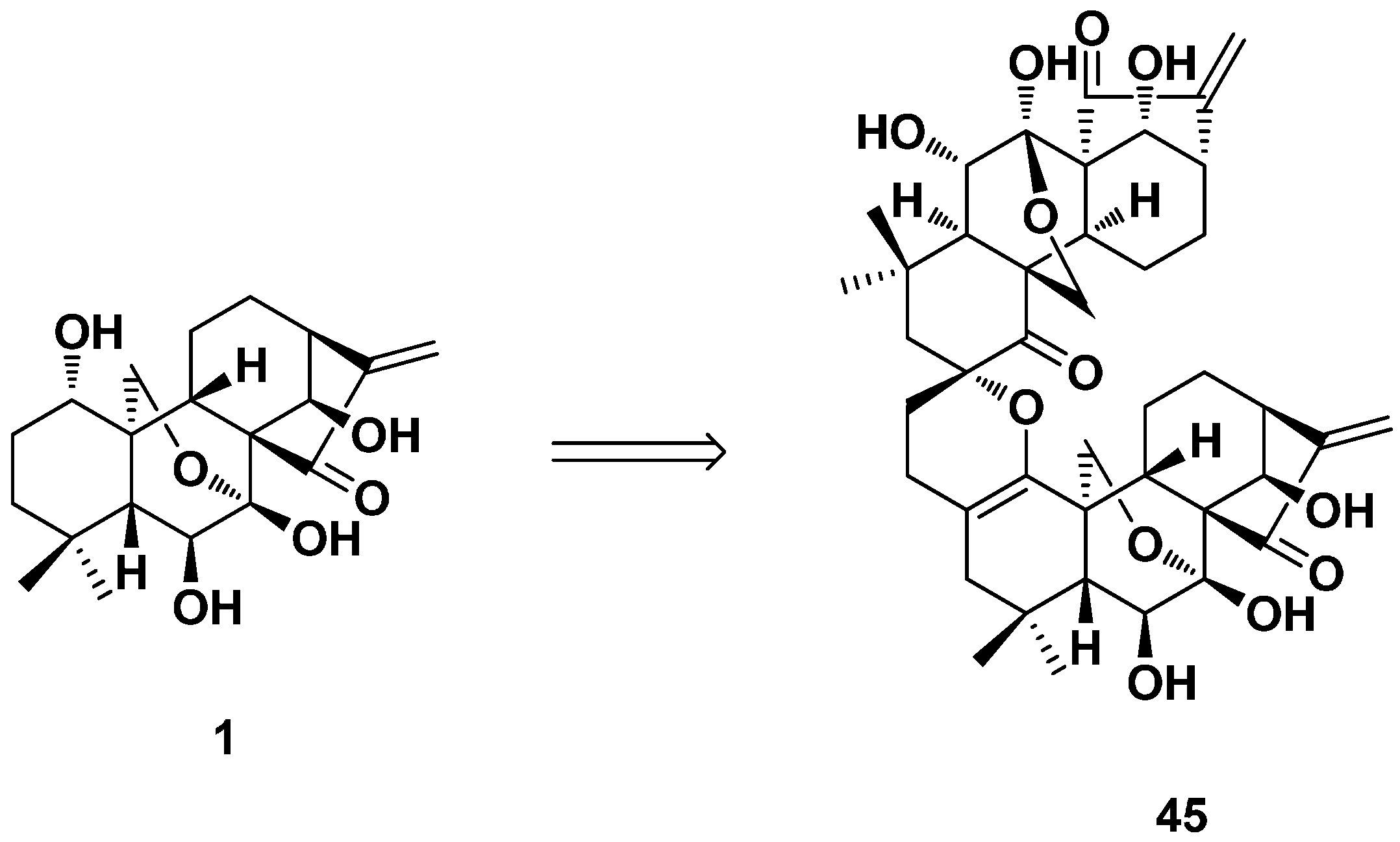
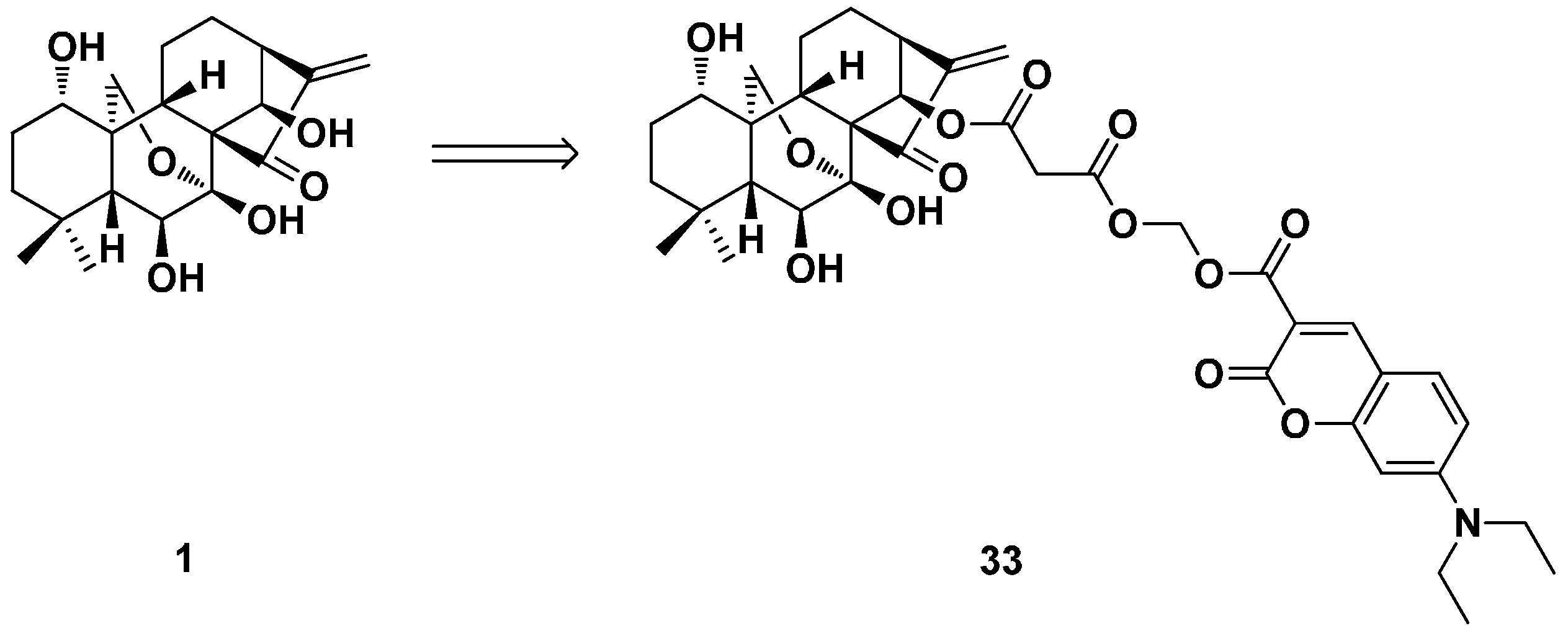
3.3. ent-Kaurane Diterpenoid Dimers
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Mw | molecular weight |
| NP | natural product |
| SAR | structure activity relationship |
| DMAP | 4-dimethylaminopyridine |
| NO | nitric oxide |
| rt | room temperature |
| THF | tetrahydrofuran |
| TEA | triethylamine |
| Ac | acetyl group |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MIC | minimal inhibitory concentration |
| DR5 | death receptor 5 |
| IED | inverse electron demand |
| HDA | hetero-Diels–Alder |
| SI | selectivity index |
References
- Fujita, E.; Fujita, T.; Katayama, H.; Shibuya, M.; Shingu, T. Terpenoids. Part XV. Structure and absolute configuration oridonin isolated from Isodon japonicus and Isodon trichocarpus. J. Chem. Soc. C 1970, 21, 1674–1681. [Google Scholar] [CrossRef]
- Fujita, E.; Nagao, Y.; Node, M.; Kaneko, K.; Nakazawa, S.; Kuroda, H. Antitumor activity of the isodon diterpenoids: Structural requirements for the activity. Cell. Mol. Life Sci. 1976, 32, 203–206. [Google Scholar] [CrossRef]
- Weng, H.; Huang, H.; Dong, B.; Zhao, P.; Zhou, H.; Qu, L. Inhibition of miR-17 and miR-20a by oridonin triggers apoptosis and reverses chemoresistance by derepressing BIM-S. Cancer Res. 2014, 74, 4409–4419. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.B.; Yue, J.Y. Preliminary study on the mechanism of oridonin-induced apoptosis in human squamous cell oesophageal carcinoma cell line EC9706. J. Int. Med. Res. 2014, 42, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Dal Piaz, F.; Cotugno, R.; Lepore, L.; Vassallo, A.; Malafronte, N.; Lauro, G.; Bifulco, G.; Belisario, M.A.; de Tommasi, N. Chemical proteomics reveals HSP70 1A as a target for the anticancer diterpene oridonin in Jurkat cells. J. Proteom. 2013, 82, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, T.; Li, J.; Deng, H.; Song, Y.; Zhai, D.; Peng, Y.; Lu, X.; Liu, M.; Zhao, Y.; et al. Oridonin inhibits tumor growth and metastasis through anti-angiogenesis by blocking the Notch signaling. PLoS ONE 2014, 9, e113830. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.Y.; Li, J.; Qu, X.Y.; Zhu, N.; Ji, Y.B. Downregulation of Cdk1 and cyclinB1 expression contributes to oridonin-induced cell cycle arrest at G2/M phase and growth inhibition in SGC-7901 gastric cancer cells. Asian Pac. J. Cancer Prev. 2014, 15, 6437–6441. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Y.; Xu, B.; Chen, S.F.; Chen, S.S.; Zhang, T.; Ren, J.; Xu, J. Effect of oridonin-mediated hallmark changes on inflammatory pathways in human pancreatic cancer (BxPC-3) cells. World J. Gastroenterol. 2014, 20, 14895–14903. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ye, Y.; Yu, Z.L. Proteomic and functional analyses demonstrate the involvement of oxidative stress in the anticancer activities of oridonin in HepG2 cells. Oncol. Rep. 2014, 31, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Wang, X.; Qi, R.; Wei, L.; Huo, Y.; Ma, Y.; Shi, L.; Chang, Y.; Li, G.; Zhou, L. Oridonin induces apoptosis in uveal melanoma cells by upregulation of Bim and downregulation of fatty acid synthase. Biochem. Biophys. Res. Commun. 2015, 457, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.H.; Chai, K.; Tashiro, S.; Onodera, S.; Ikejima, T. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. J. Pharm. Pharmacol. 2013, 65, 1622–1642. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Huang, R.W.; Lin, D.J.; Wu, X.Y.; Peng, J.; Pan, X.L.; Lin, Q.; Hou, M.; Zhang, M.H.; Chen, F. Antiproliferation effects of oridonin on HPB-ALL cells and its mechanisms of action. Am. J. Hematol. 2006, 81, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Y.; Wu, D.; Tashiro, S.; Onodera, S.; Ikejima, T. NF-κB facilitates oridonin-induced apoptosis and autophagy in HT1080 cells through a p53-mediated pathway. Arch. Biochem. Biophys. 2009, 489, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fan, S.M.; Song, J.K.; Tashiro, S.; Onodera, S.; Ikejima, T. Hydroxyl radical (·OH) played a pivotal role in oridonin-induced apoptosis and autophagy in human epidermoid carcinoma A431 cells. Biol. Pharm. Bull. 2012, 35, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Zhang, J.H.; Qiu, F.; Tashiro, S.; Onodera, S.; Ikejima, T. Inhibition of EGFR signaling augments oridonin-induced apoptosis in human laryngeal cancer cells via enhancing oxidative stress coincident with activation of both the intrinsic and extrinsic apoptotic pathways. Cancer Lett. 2010, 294, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Zhang, J.H.; Qiu, F.; Chen, S.; Tashiro, S.; Onodera, S.; Ikejima, T. Induction of G2/M phase arrest and apoptosis by oridonin in human laryngeal carcinoma cells. J. Nat. Prod. 2010, 73, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.K.; Wang, H.Z.; Xie, L.P.; Chen, D.W.; Liu, X.; Sun, J.; Nie, Y.C.; Zhang, R.Q. The effects of oridonin on cell growth, cell cycle, cell migration and differentiation in melanoma cells. J. Ethnopharmacol. 2006, 103, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Tang, Q.; Zhang, J.; Yang, Y.; Liu, Y.; Pan, Y. Oridonin-induced apoptosis in SW620 human colorectal adenocarcinoma cells. Oncol. Lett. 2011, 2, 1303–1307. [Google Scholar] [PubMed]
- Li, D.; Hu, X.; Han, T.; Xu, S.; Zhou, T.; Wang, Z.; Cheng, K.; Li, Z.; Hua, H.; Xiao, W.; et al. Synthesis, biological activity, and apoptotic properties of NO-Donor/enmein-type ent-kauranoid hybrids. Int. J. Mol. Sci. 2016, 17, 747. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ouyang, L.; Peng, H.; Zhang, W.Z. Oridonin: Targeting programmed cell death pathways as an anti-tumour agent. Cell Prolif. 2012, 45, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Wang, E.Q.; Cheng, Y.; Bao, J.K. Oridonin: An active diterpenoid targeting cell cycle arrest, apoptotic and autophagic pathways for cancer therapeutics. Int. J. Biochem. Cell Biol. 2011, 43, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.H.; Xu, J.; Zhao, J.H.; Feng, N.P.; Liu, Y.; Wei, L. Method for determination of oridonin in rabbits using isopsoralen as an internal standard and its application to pharmacokinetic studies for oridonin-loaded nanoparticles. J. Chromatogr. B 2008, 869, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.F.; Liu, P.W.; Shi, X.W.; Jin, Y.R.; Wang, Q.; Zhang, X.W. A novel analysis method for diterpenoids in rat plasma by liquid chromatography–electrospray ionization mass spectrometry. Anal. Biochem. 2010, 407, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Manly, C.J.; Chandrasekhar, J.; Ochterski, J.W.; Hammer, J.D.; Warfield, B.B. Strategies and tactics for optimizing the Hit-to-Lead process and beyond—a computational chemistry perspective. Drug Discov. Today 2008, 13, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Oprea, T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Luo, S.; Yao, H.; Cai, H.; Miao, X.; Wu, F.; Yang, D.H.; Wu, X.; Xie, W.; Yao, H.; et al. Probing the anticancer action of oridonin with fluorescent analogues: Visualizing subcellular localization to mitochondria. J. Med. Chem. 2016, 59, 5022–5034. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, G.; Lin, Y.; Zhang, Y.; Pei, L.; Yao, H.; Hu, M.; Qiu, Y.; Huang, Z.; Zhang, Y.; et al. Novel anticancer oridonin derivatives possessing a diazen-1-ium-1,2-diolate nitric oxide donor moiety: Design, synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett. 2016, 26, 2795–2800. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Han, T.; Xu, S.; Zhou, T.; Tian, K.; Hu, X.; Cheng, K.; Li, Z.; Hua, H.; Xu, J. Antitumor and antibacterial derivatives of oridonin: A main composition of Dong-Ling-Cao. Molecules 2016, 21, 575. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.T.; Pei, L.L.; Wang, C.Q.; Zhang, Y.K.; Li, D.H.; Yao, H.Q.; Wu, X.M.; Chen, Z.S.; Sun, Y.J.; Xu, J.Y. Novel hybrids of natural oridonin-bearing nitrogen mustards as potential anticancer drug candidates. ACS Med. Chem. Lett. 2014, 5, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pei, L.; Li, D.; Yao, H.; Cai, H.; Yao, H.; Wu, X.; Xu, J. Synthesis and antimycobacterial evaluation of natural oridonin and its enmein-type derivatives. Fitoterapia 2014, 99, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, D.; Pei, L.; Yao, H.; Wang, C.; Cai, H.; Yao, H.; Wu, X.; Xu, J. Design, synthesis and antimycobacterial activity evaluation of natural oridonin derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 2811–2814. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xu, S.; Cai, H.; Pei, L.; Zhang, H.; Wang, L.; Yao, H.; Wu, X.; Jiang, J.; Sun, Y.; et al. Enmein-type diterpenoid analogs from natural kaurene-type oridonin: Synthesis and their antitumor biological evaluation. Eur. J. Med. Chem. 2013, 64, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cai, H.; Jiang, B.; Liu, G.; Wang, Y.; Wang, L.; Yao, H.; Wu, X.; Sun, Y.; Xu, J. Synthesis of spirolactone-type diterpenoid derivatives from kaurene-type oridonin with improved antiproliferative effects and their apoptosis-inducing activity in human hepatoma Bel-7402 cells. Eur. J. Med. Chem. 2013, 59, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.; Wang, L.; Cai, H.; Jiang, B.W.; Zhang, Y.H.; Sun, Y.J.; Xu, J.Y. Synthesis of novel furozan-based nitric oxide-releasing derivatives of 1-oxo-oridonin with anti-proliferative activity. Chin. J. Nat. Med. 2012, 10, 471–476. [Google Scholar] [CrossRef]
- Li, D.; Xu, S.; Cai, H.; Pei, L.; Wang, L.; Wu, X.; Yao, H.; Jiang, J.; Sun, Y.; Xu, J. Library construction and biological evaluation of enmein-type diterpenoid analogues as potential anticancer agents. ChemMedChem 2013, 8, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, D.; Xu, S.; Cai, H.; Yao, H.; Zhang, Y.; Jiang, J.; Xu, J. The conversion of oridonin to spirolactone-type or enmein-type diterpenoid: Synthesis and biological evaluation of ent-6,7-seco-oridonin derivatives as novel potential anticancer agents. Eur. J. Med. Chem. 2012, 52, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, L.; Cai, H.; Zhang, Y.; Xu, J. Synthesis and biological evaluation of novel furozan-based nitric oxide-releasing derivatives of oridonin as potential anti-tumor agents. Molecules 2012, 17, 7556–7568. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, D.H.; Wang, C.L.; Zhang, Y.H.; Xu, J.Y. Recent progress in the development of natural ent-kaurane diterpenoids with anti-tumor activity. Mini Rev. Med. Chem. 2011, 11, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ran, Q.; Li, D.H.; Yao, H.Q.; Zhang, Y.H.; Yuan, S.T.; Zhang, L.Y.; Shen, M.Q.; Xu, J.Y. Synthesis and anti-tumor activity of 14-O-derivatives of natural oridonin. Chin. J. Nat. Med. 2011, 9, 194–198. [Google Scholar]
- Xu, J.Y.; Yang, J.Y.; Ran, Q.; Wang, L.; Liu, J.; Wang, Z.X.; Wu, X.M.; Hua, W.Y.; Yuan, S.T.; Zhang, L.Y.; et al. Synthesis and biological evaluation of novel 1-O- and 14-O-derivatives of oridonin as potential anticancer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 4741–4744. [Google Scholar] [CrossRef] [PubMed]
- Owona, B.A.; Schluesener, H.J. Molecular insight in the multifunctional effects of oridonin. Drugs R D 2015, 15, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, Y. Oridonin, a promising antitumor natural product in the chemotherapy of hematological malignancies. Curr. Pharm. Biotechnol. 2014, 15, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, S.Y. Recent advances in the molecular basis of anti-neoplastic mechanisms of oridonin. Chin. J. Integr. Med. 2013, 19, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ding, C.; Ye, N.; Liu, Z.; Wold, E.A.; Chen, H.; Wild, C.; Shen, Q.; Zhou, J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 2016, 122, 102–117. [Google Scholar] [CrossRef]
- Fujita, E.; Nagao, Y.; Kaneko, K.; Nakazawa, S.; Kuroda, H. The antitumor and antibacterial activity of isodon diterpenoid. Chem. Pharm. Bull. 1976, 24, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Fujita, E.; Nagao, Y.; Kohno, T.; Matsuda, M.; Ozaki, M. Antitumor activity of acylated oridonin. Chem. Pharm. Bull. 1981, 29, 3208–3213. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Fujita, E.; Kohno, T.; Yagi, M. An efficient method for selective acetylation of alcoholic hydroxyl groups. Chem. Pharm. Bull. 1981, 29, 3202–3207. [Google Scholar] [CrossRef]
- Zhou, W.S.; Cheng, Y.X. The chemical selective synthesis of eriocalyxin B and its analogues. Acta Chim. Sin. 1990, 48, 1185–1190. [Google Scholar]
- Yan, X.B.; Lei, M.; Zhang, J.Y.; Liu, H.M. Synthesis of oridonin glucopyranoside. Chin. J. Org. Chem. 2005, 25, 222–224. [Google Scholar]
- Yan, X.B.; Li, W.; Liu, H.M. Study on the synthesis and activity of lasiokaurin (in Chinese). J. Zhengzhou Univ. Eng. Sci. 2006, 27, 113–115. [Google Scholar]
- Yan, X.B.; Zhang, J.Y.; Ke, Y.; Zhou, X.; Liu, H.M. Synthesis of amino-derivatives of oridonin and their antitumor activity (in Chinese). J. Zhengzhou Univ. Med. Sci. 2007, 42, 39–41. [Google Scholar]
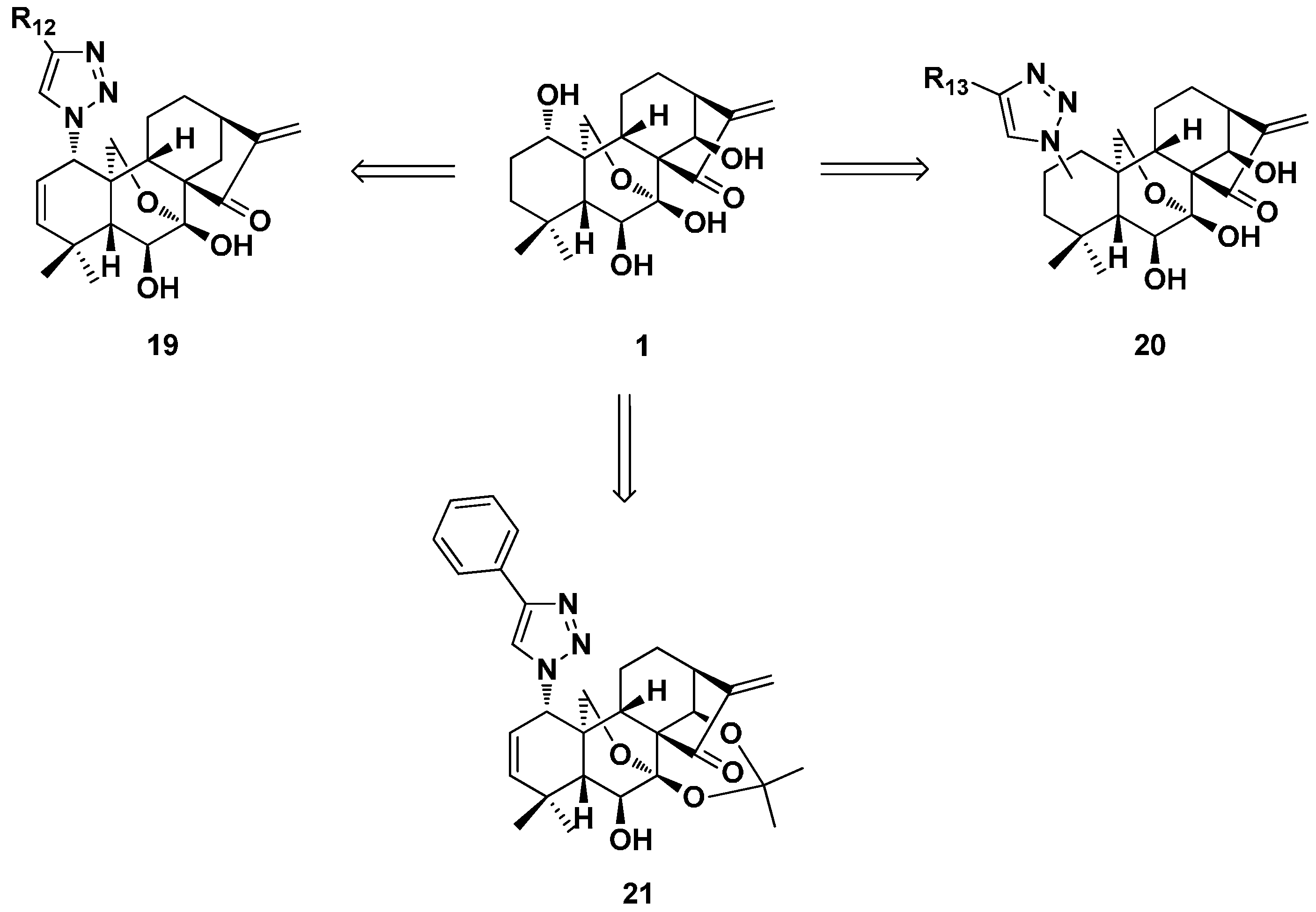
- Ding, C.; Zhang, Y.; Chen, H.; Wild, C.; Wang, T.; White, M.A.; Shen, Q.; Zhou, J. Overcoming synthetic challenges of oridonin A-ring structural diversification: Regio- and stereoselective installation of azides and 1,2,3-triazoles at the C-1, C-2, or C-3 position. Org. Lett. 2013, 15, 3718–3721. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Zhang, Y.; Chen, H.; Yang, Z.; Wild, C.; Chu, L.; Liu, H.; Shen, Q.; Zhou, J. Novel nitrogen-enriched oridonin analogues with thiazole-fused A-ring: Protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J. Med. Chem. 2013, 56, 5048–5058. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ding, Y.; Chen, C.H.; Zhou, Z.; Ding, C.; Chen, H.; Zhou, J.; Chen, C. A new oridonin analog suppresses triple-negative breast cancer cells and tumor growth via the induction of death receptor 5. Cancer Lett. 2016, 380, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Zhang, Y.; Chen, H.; Yang, Z.; Wild, C.; Ye, N.; Ester, C.D.; Xiong, A.; White, M.A.; Shen, Q.; et al. Oridonin ring A-based diverse constructions of enone functionality: Identification of novel dienone analogues effective for highly aggressive breast cancer by inducing apoptosis. J. Med. Chem. 2013, 56, 8814–8825. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Zhang, Y.; Yang, S.; Wang, X.; Yu, B.; Yang, H. Oridonin: A small molecule inhibitor of cystic fibrosis transmembrane conductance regulator (CFTR) isolated from traditional Chinese medicine. Fitoterapia 2015, 100, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Bohanon, F.J.; Wang, X.; Ding, C.; Ding, Y.; Radhakrishnan, G.L.; Rastellini, C.; Zhou, J.; Radhakrishnan, R.S. Oridonin inhibits hepatic stellate cell proliferation and fibrogenesis. J. Surg. Res. 2014, 190, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.M.; Kuo, C.Y.; Lin, C.Y.; Hung, M.F.; Shen, J.J.; Hwang, T.L. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules 2014, 19, 3327–3344. [Google Scholar] [CrossRef] [PubMed]
- Bohanon, F.J.; Wang, X.; Graham, B.M.; Ding, C.; Ding, Y.; Radhakrishnan, G.L.; Rastellini, C.; Zhou, J.; Radhakrishnan, R.S. Enhanced effects of novel oridonin analog CYD0682 for hepatic fibrosis. J. Surg. Res. 2015, 199, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Bohanon, F.J.; Wang, X.; Graham, B.M.; Prasai, A.; Vasudevan, S.J.; Ding, C.; Ding, Y.; Radhakrishnan, G.L.; Rastellini, C.; Zhou, J.; et al. Enhanced anti-fibrogenic effects of novel oridonin derivative CYD0692 in hepatic stellate cells. Mol. Cell. Biochem. 2015, 410, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Wang, L.; Chen, H.; Wild, C.; Ye, N.; Ding, Y.; Wang, T.; White, M.A.; Shen, Q.; Zhou, J. ent-Kaurane-based regio- and stereoselective inverse electron demand hetero-Diels-Alder reactions: Synthesis of dihydropyran-fused diterpenoids. Org. Biomol. Chem. 2014, 12, 8442–8452. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.D.; Huang, S.X.; Han, Q.B. Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep. 2006, 23, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, W.G.; Wu, H.Y.; Du, X.; Li, X.N.; Li, Y.; Pu, J.X.; Sun, H.D. Bioactive enmein-type ent-kaurane diterpenoids from Isodon phyllostachys. J. Nat. Prod. 2016, 79, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.G.; Du, X.; Li, X.N.; Wu, H.Y.; Liu, X.; Shang, S.Z.; Zhan, R.; Liang, C.Q.; Kong, L.M.; Li, Y.; et al. New bicyclo[3.1.0]hexane unit ent-kaurane diterpene and its seco-derivative from Isodon eriocalyx var. laxiflora. Org. Lett. 2012, 14, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Pan, L.; Li, Q.; Zhao, J.; Pu, J.; Yao, P.; Gong, N.; Lu, Y.; Kondratyuk, T.P.; Pezzuto, J.M.; et al. Rubesanolides A and B: Diterpenoids from Isodon rubescens. Org. Lett. 2011, 13, 1406–1409. [Google Scholar] [CrossRef] [PubMed]
- Brucoli, F.; Borrello, M.T.; Stapleton, P.; Parkinson, G.N.; Gibbons, S. Structural characterization and antimicrobial evaluation of atractyloside, atractyligenin, and 15-didehydroatractyligenin methyl ester. J. Nat. Prod. 2012, 75, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.G.; Li, X.N.; Du, X.; Wu, H.Y.; Liu, X.; Su, J.; Li, Y.; Pu, J.X.; Sun, H.D. Laxiflorolides A and B, epimeric bishomoditerpene lactones from Isodon eriocalyx. J. Nat. Prod. 2012, 75, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Wijeratne, E.M.; Bashyal, B.P.; Liu, M.X.; Rocha, D.D.; Gunaherath, G.M.; U’Ren, J.M.; Gunatilaka, M.K.; Arnold, A.E.; Whitesell, L.; Gunatilaka, A.A. Geopyxins A-E, ent-kaurane diterpenoids from endolichenic fungal strains Geopyxis aff. majalis and Geopyxis sp. AZ0066: Structure-activity relationships of geopyxins and their analogues. J. Nat. Prod. 2012, 75, 361–369. [Google Scholar] [PubMed]
- Gong, J.X.; Lin, G.A.; Sun, W.B.; Li, C.C.; Yang, Z. Total synthesis of (±) maoecrystal, V. J. Am. Chem. Soc. 2010, 132, 16745–16746. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Yeoman, J.T.S.; Reisman, S.E. A concise total synthesis of (−)-maoecrystal, Z. J. Am. Chem. Soc. 2011, 133, 14964–14967. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Zheng, C.; Wang, H.; Chen, Y.; Li, Y.; Cheng, B.; Zhai, H. Total synthesis of (±)-sculponeatin, N. Org. Lett. 2014, 16, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.; Lu, W.; Nan, F.J. Synthesis and revision of stereochemistry of rubescensin, S. Org. Biomol. Chem. 2011, 9, 4436–4439. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Wang, J.; Niu, X.M.; Shen, Y.H.; Zhang, H.J.; Sun, H.D.; Li, M.L.; Tian, Q.E.; Lu, Y.; Cao, P.; et al. Maoecrystal V, cytotoxic diterpenoid with a novel C19 skeleton from Isodon eriocalyx (Dunn.) hara. Org. Lett. 2004, 6, 4327–4330. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wang, W.G.; Zhou, M.; Wu, H.Y.; Zhan, R.; Li, X.N.; Du, X.; Li, Y.; Pu, J.X.; Sun, H.D. Diterpenoids from Isodon sculponeatus. Fitoterapia 2014, 93, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.B.; Cheung, S.; Tai, J.; Qiao, C.F.; Song, J.Z.; Tso, T.F.; Sun, H.D.; Xu, H.X. Maoecrystal Z, a cytotoxic diterpene from Isodon eriocalyx with a unique skeleton. Org. Lett. 2006, 8, 4727–4730. [Google Scholar] [CrossRef] [PubMed]
- Li, L.M.; Li, G.Y.; Pu, J.X.; Xiao, W.L.; Ding, L.S.; Sun, H.D. ent-Kaurane and cembrane diterpenoids from Isodon sculponeatus and their cytotoxicity. J. Nat. Prod. 2009, 72, 1851–1856. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wang, W.G.; Zhou, M.; Wu, H.Y.; Zhan, R.; Li, X.N.; Du, X.; Li, Y.; Pu, J.X.; Sun, H.D. Enmein-type 6,7-seco-ent-kauranoids from Isodon sculponeatus. J. Nat. Prod. 2009, 72, 1851–1856. [Google Scholar]
- He, F.; Xiao, W.L.; Pu, J.X.; Wu, Y.L.; Zhang, H.B.; Li, X.N.; Zhao, Y.; Yang, L.B.; Chen, G.Q.; Sun, H.D. Cytotoxic ent-kaurane diterpenoids from Isodon sinuolata. Phytochemistry 2009, 70, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Thuong, P.T.; Pham, T.H.; Le, T.V.; Dao, T.T.; Dang, T.T.; Nguyen, Q.T.; Oh, W.K. Symmetric dimers of ent-kaurane diterpenoids with cytotoxic activity from Croton tonkinensis. Bioorg. Med. Chem. Lett. 2012, 22, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.P.; Wei, Q.Y.; Jin, X.L.; Wu, Q.X.; Yang, L. Two novel ent-kauranoid diterpenoids from Isodon japonica leaves. Planta Med. 2005, 71, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Braca, A.; Abdel-Razik, A.F.; Mendez, J.; Morelli, I. A new kaurane diterpene dimer from Parinari campestris. Fitoterapia 2005, 76, 614–619. [Google Scholar] [CrossRef] [PubMed]









© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Han, T.; Liao, J.; Hu, X.; Xu, S.; Tian, K.; Gu, X.; Cheng, K.; Li, Z.; Hua, H.; et al. Oridonin, a Promising ent-Kaurane Diterpenoid Lead Compound. Int. J. Mol. Sci. 2016, 17, 1395. https://doi.org/10.3390/ijms17091395
Li D, Han T, Liao J, Hu X, Xu S, Tian K, Gu X, Cheng K, Li Z, Hua H, et al. Oridonin, a Promising ent-Kaurane Diterpenoid Lead Compound. International Journal of Molecular Sciences. 2016; 17(9):1395. https://doi.org/10.3390/ijms17091395
Chicago/Turabian StyleLi, Dahong, Tong Han, Jie Liao, Xu Hu, Shengtao Xu, Kangtao Tian, Xiaoke Gu, Keguang Cheng, Zhanlin Li, Huiming Hua, and et al. 2016. "Oridonin, a Promising ent-Kaurane Diterpenoid Lead Compound" International Journal of Molecular Sciences 17, no. 9: 1395. https://doi.org/10.3390/ijms17091395