
Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
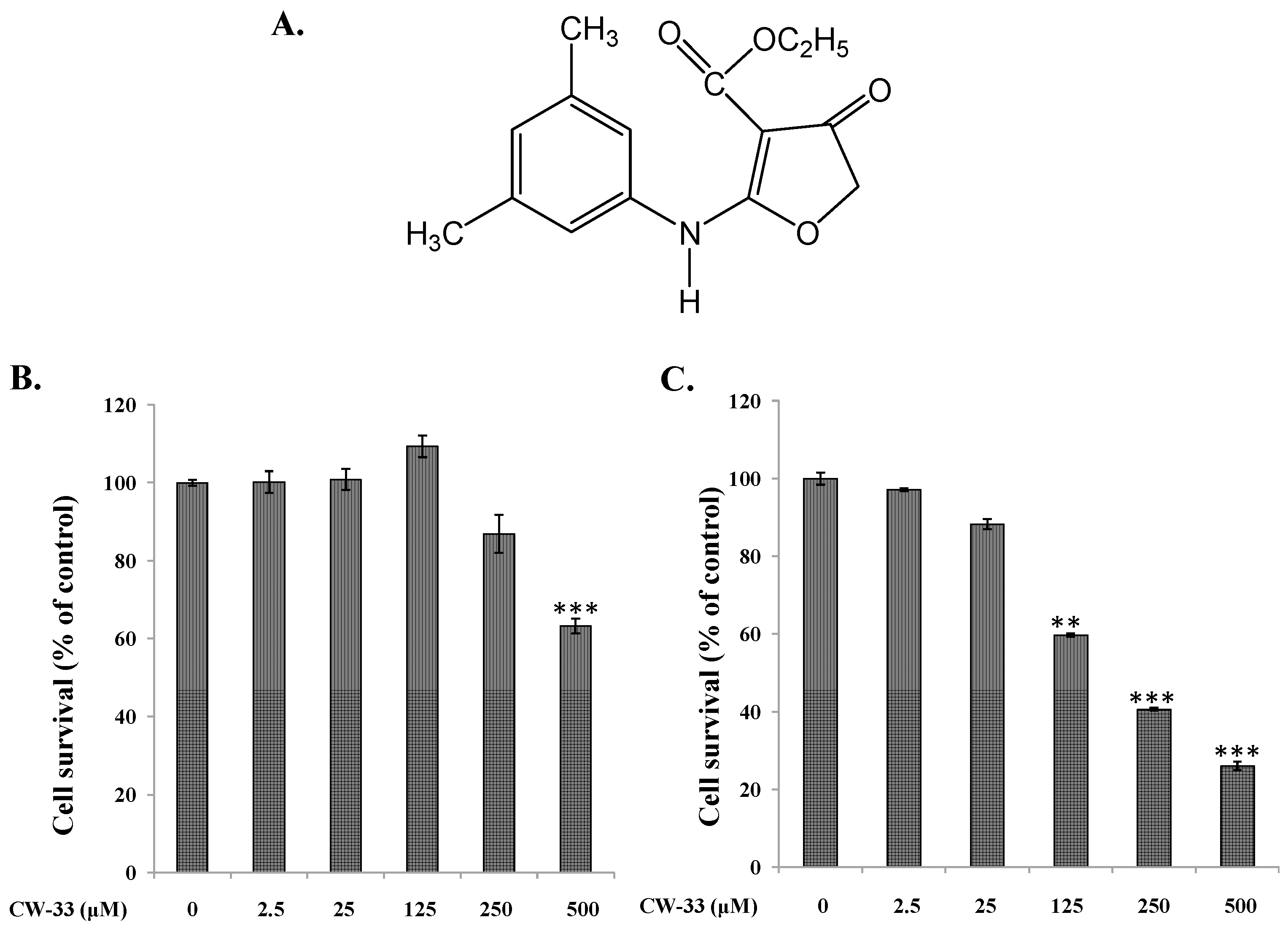
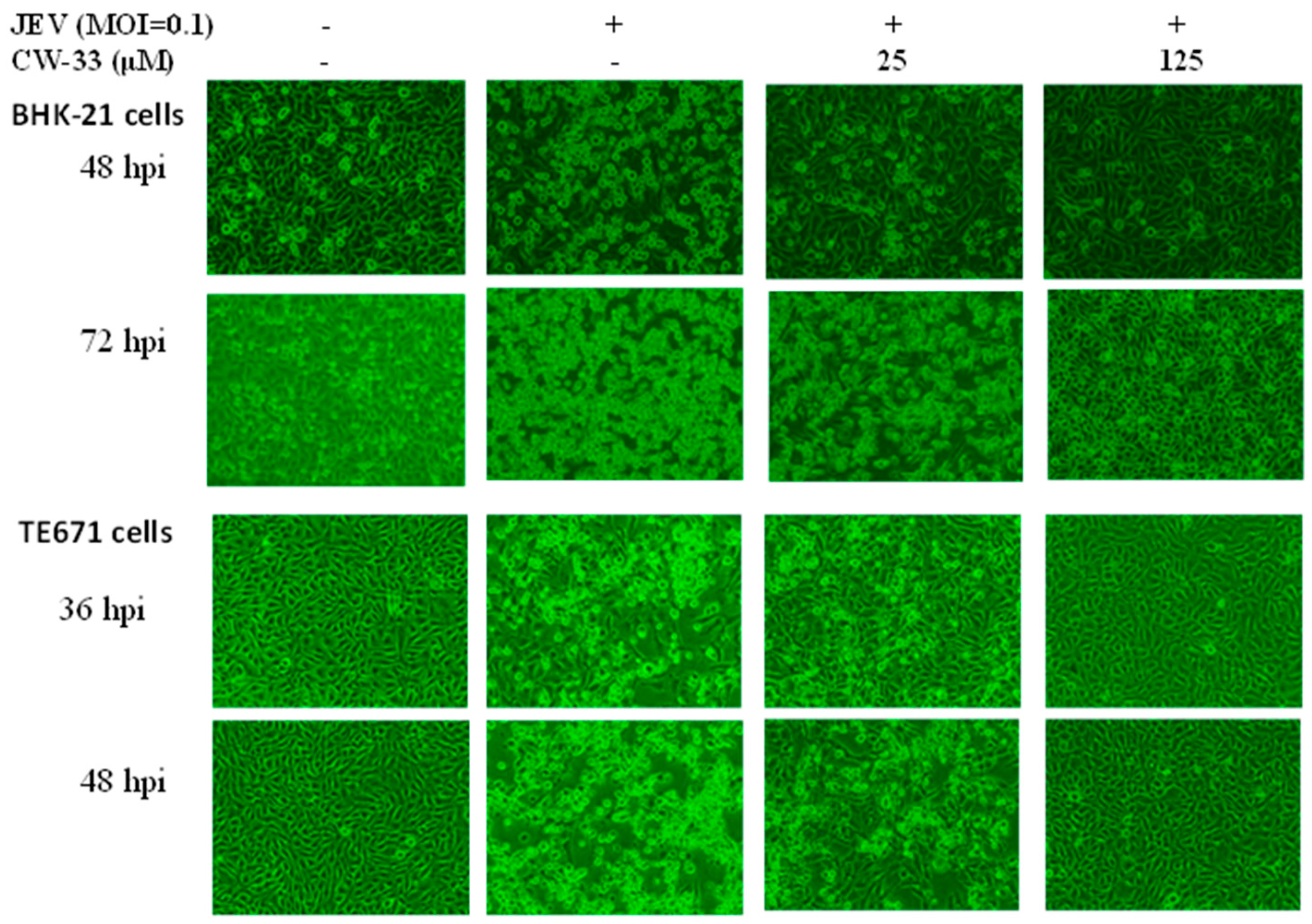
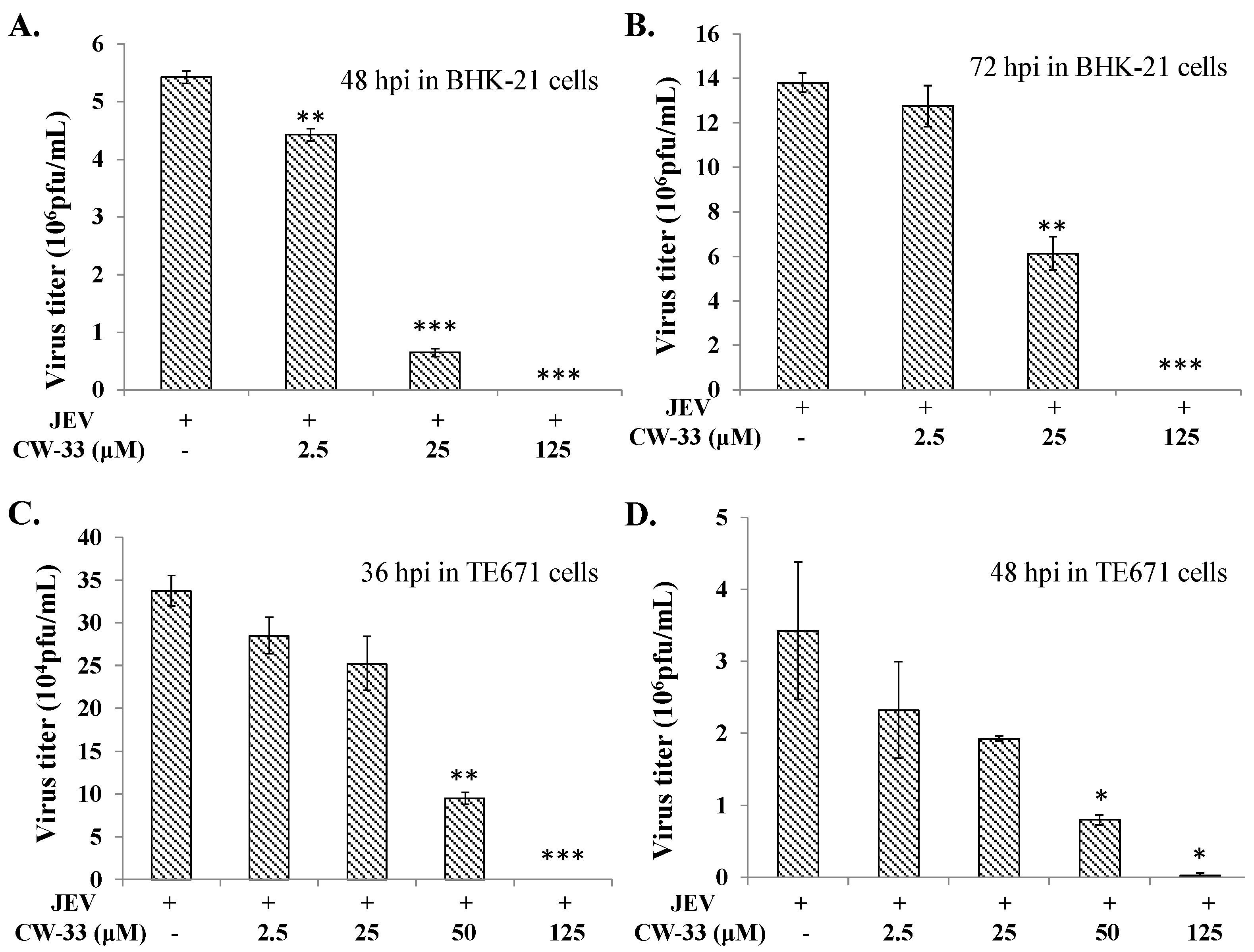
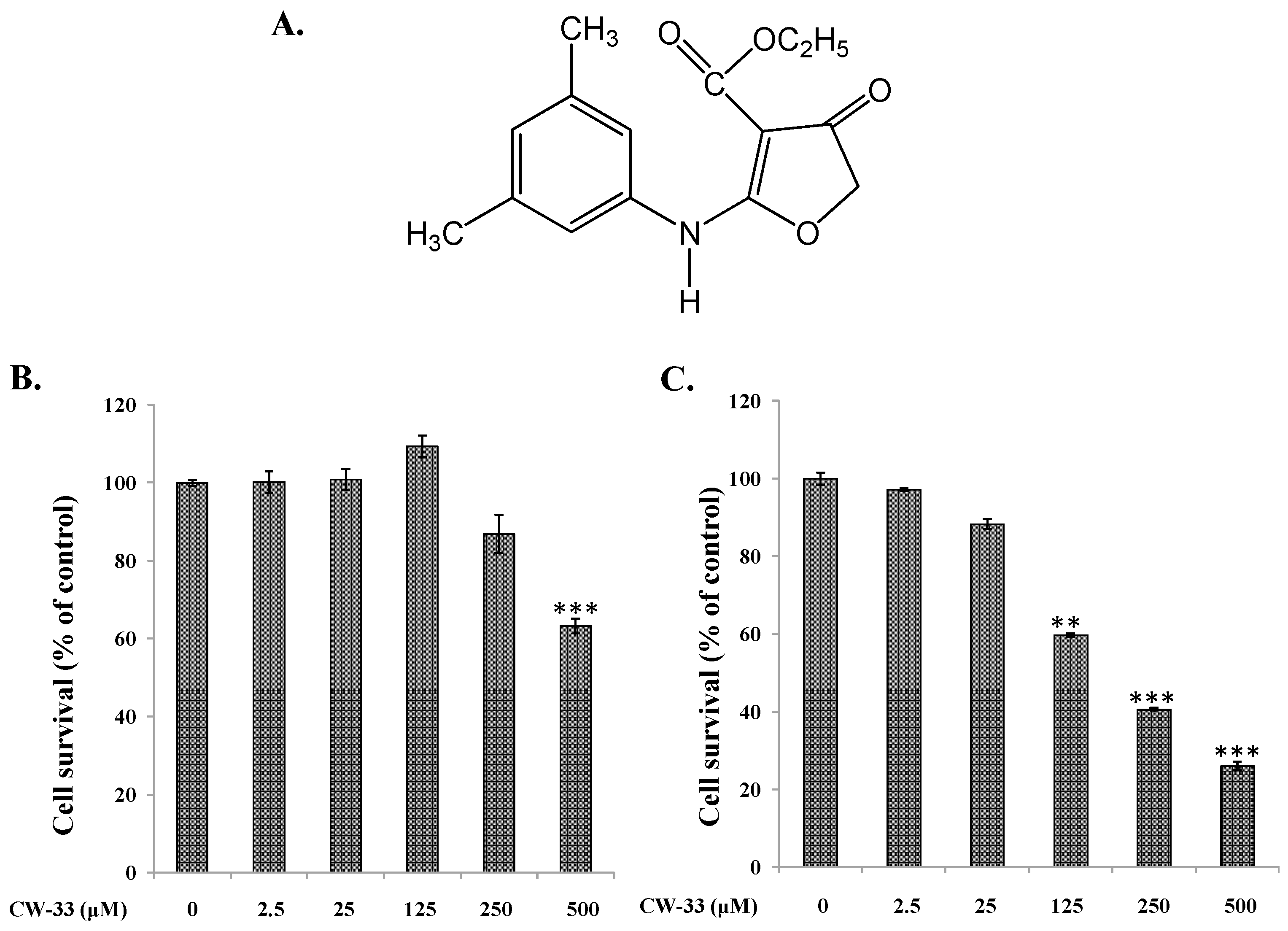
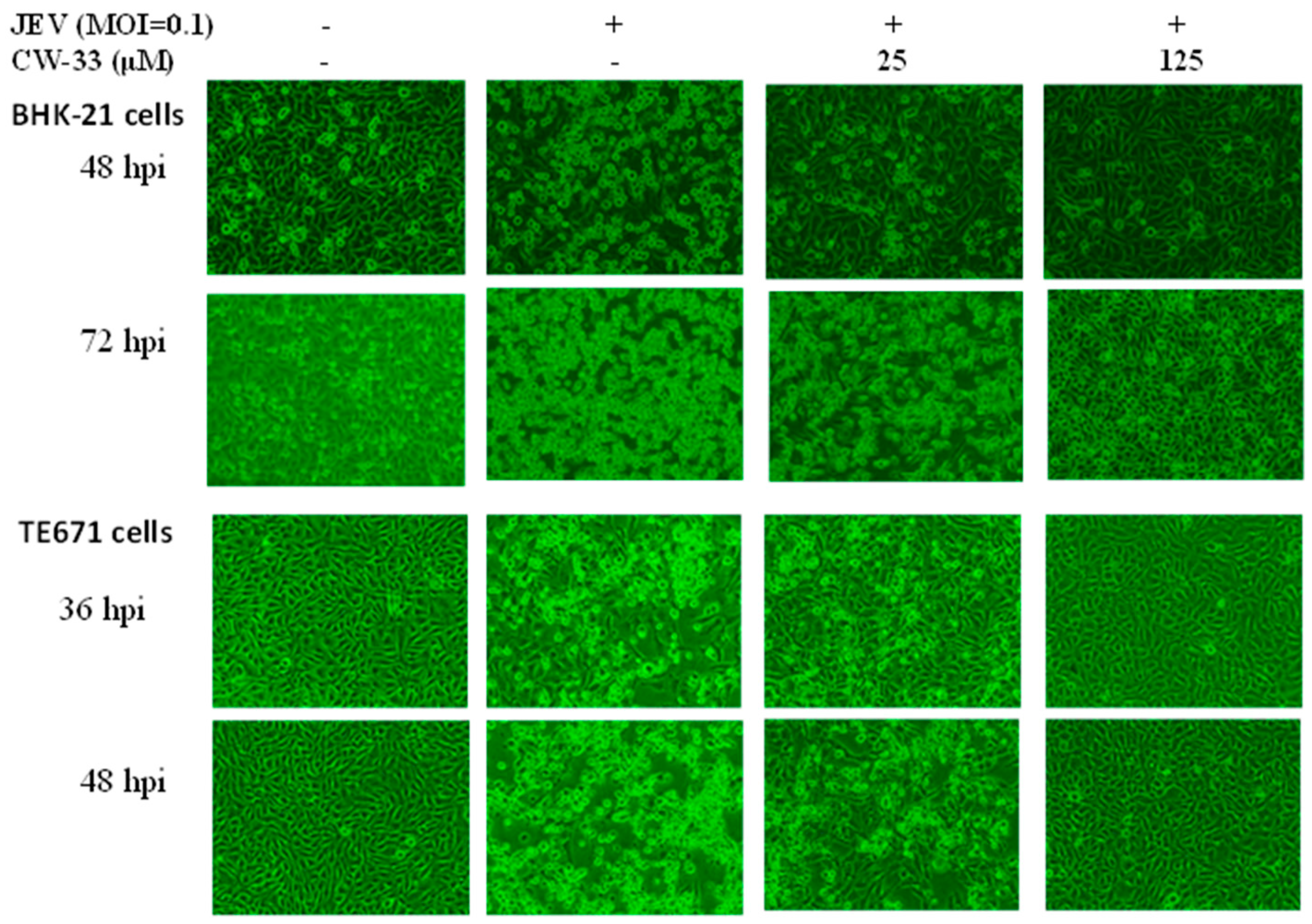
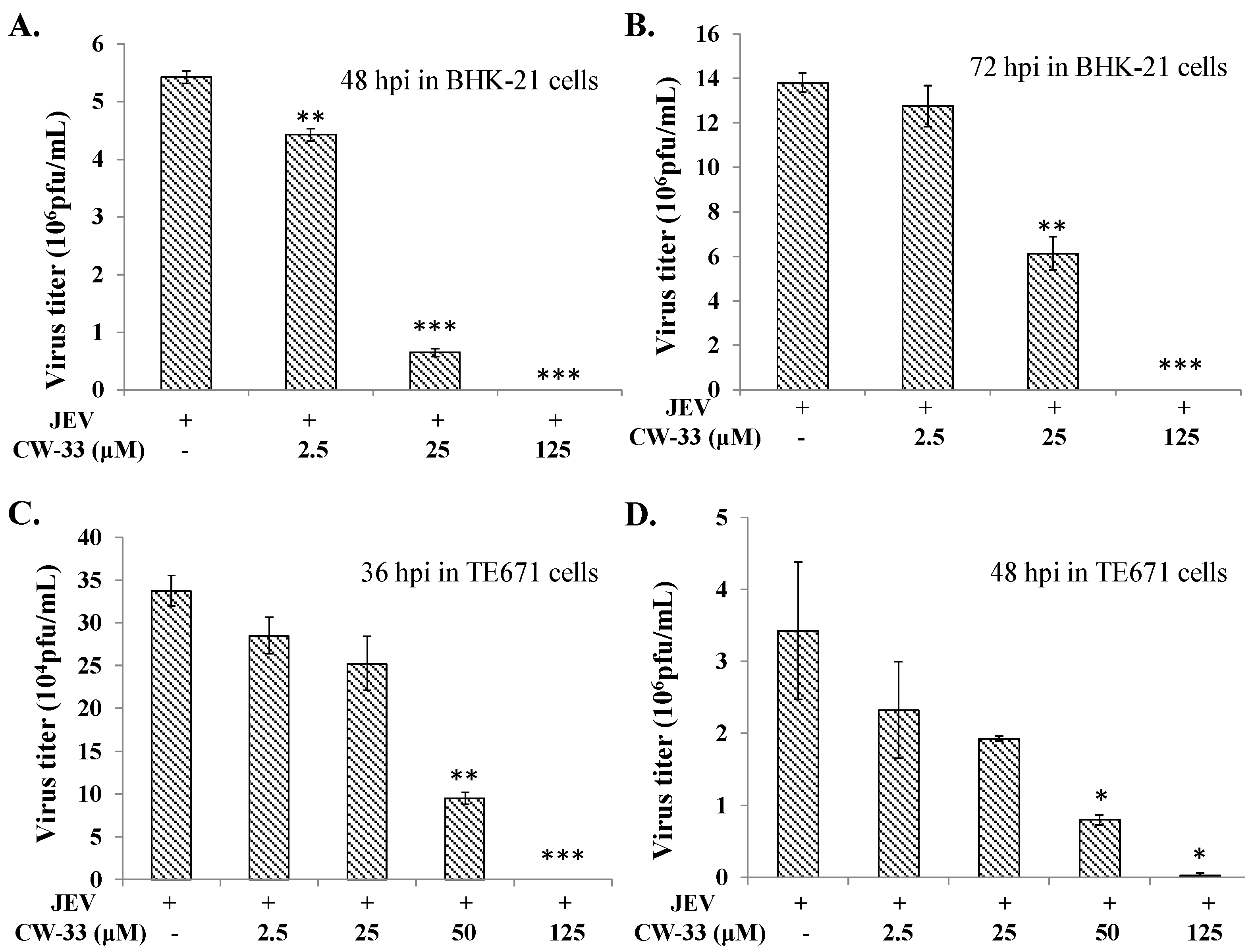
2.1. Antiviral Activity of CW-33 against JEV (Japanese Encephalitis Virus)
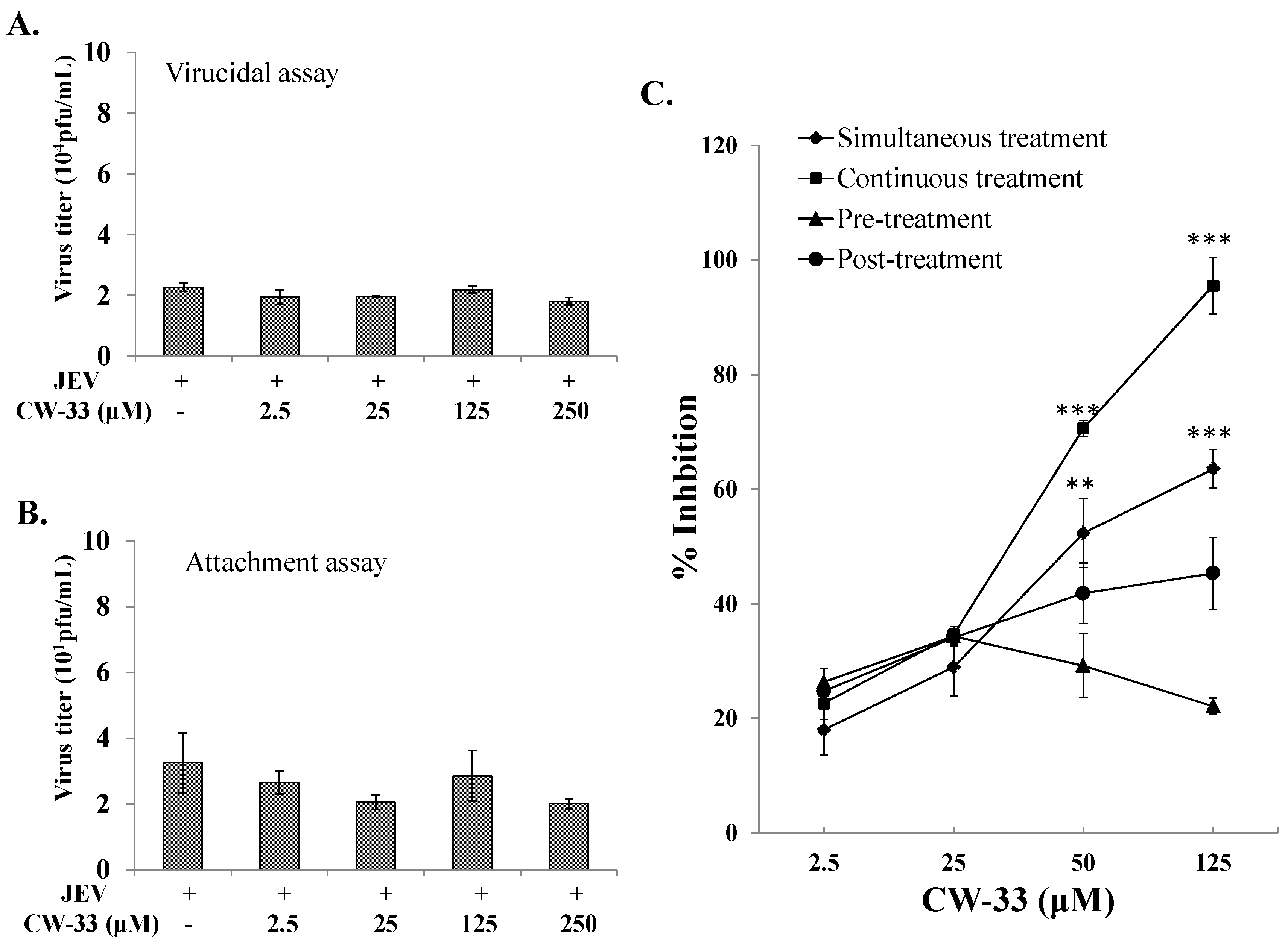
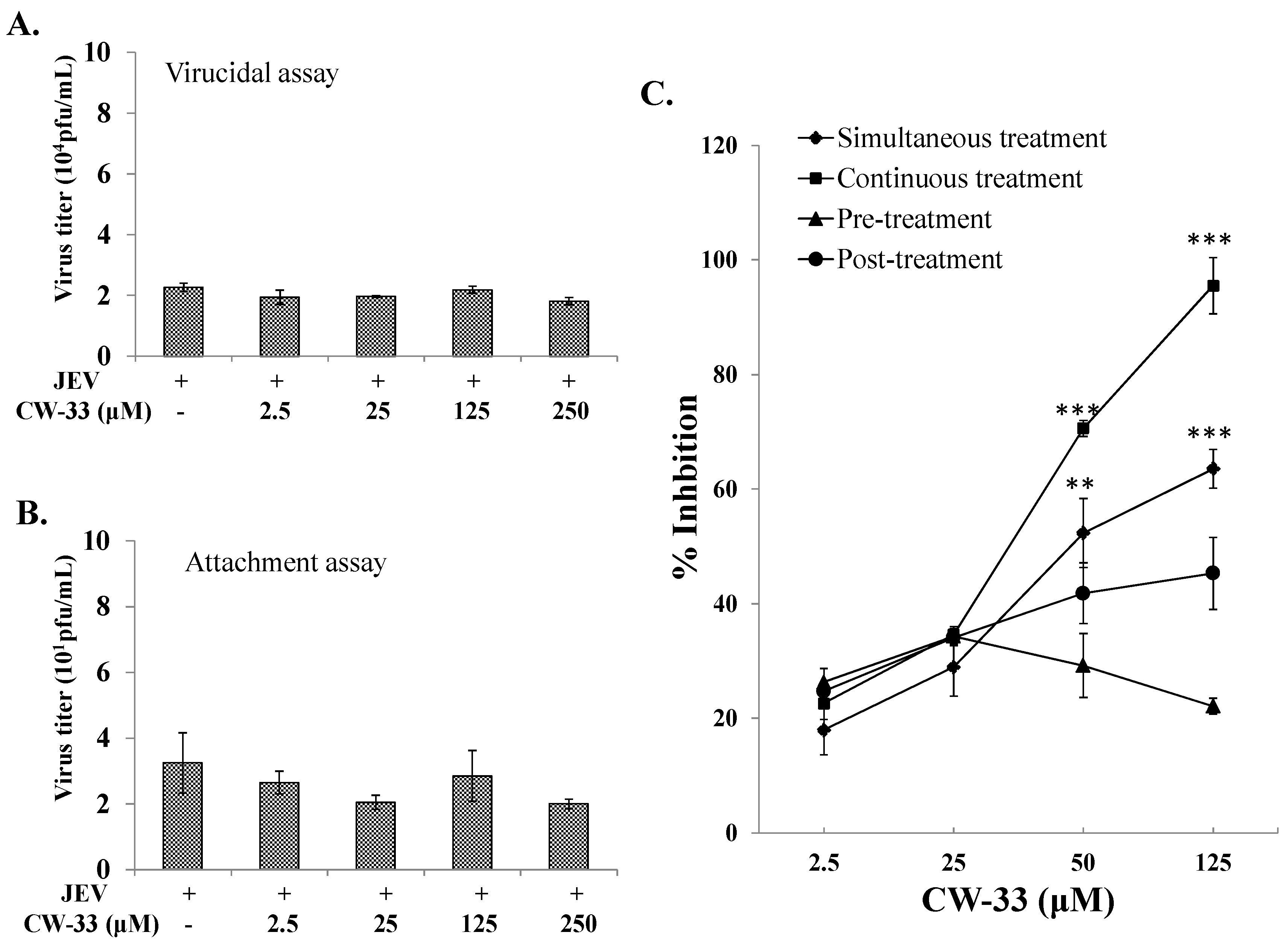
2.2. No Virucidal Activity and Attachment Inhibition by CW-33
2.3. Time-of-Addition Effect of CW-33 against JEV
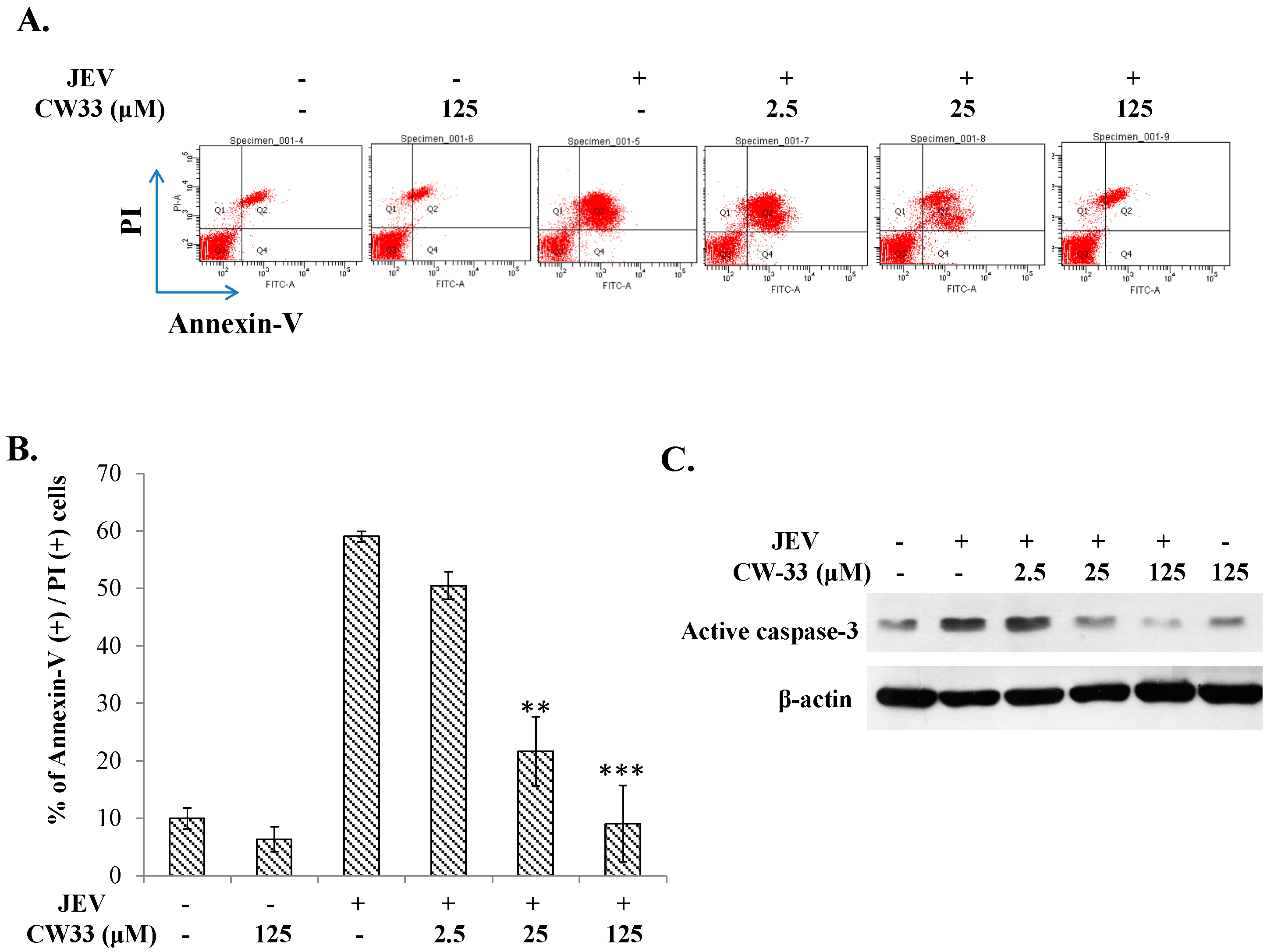
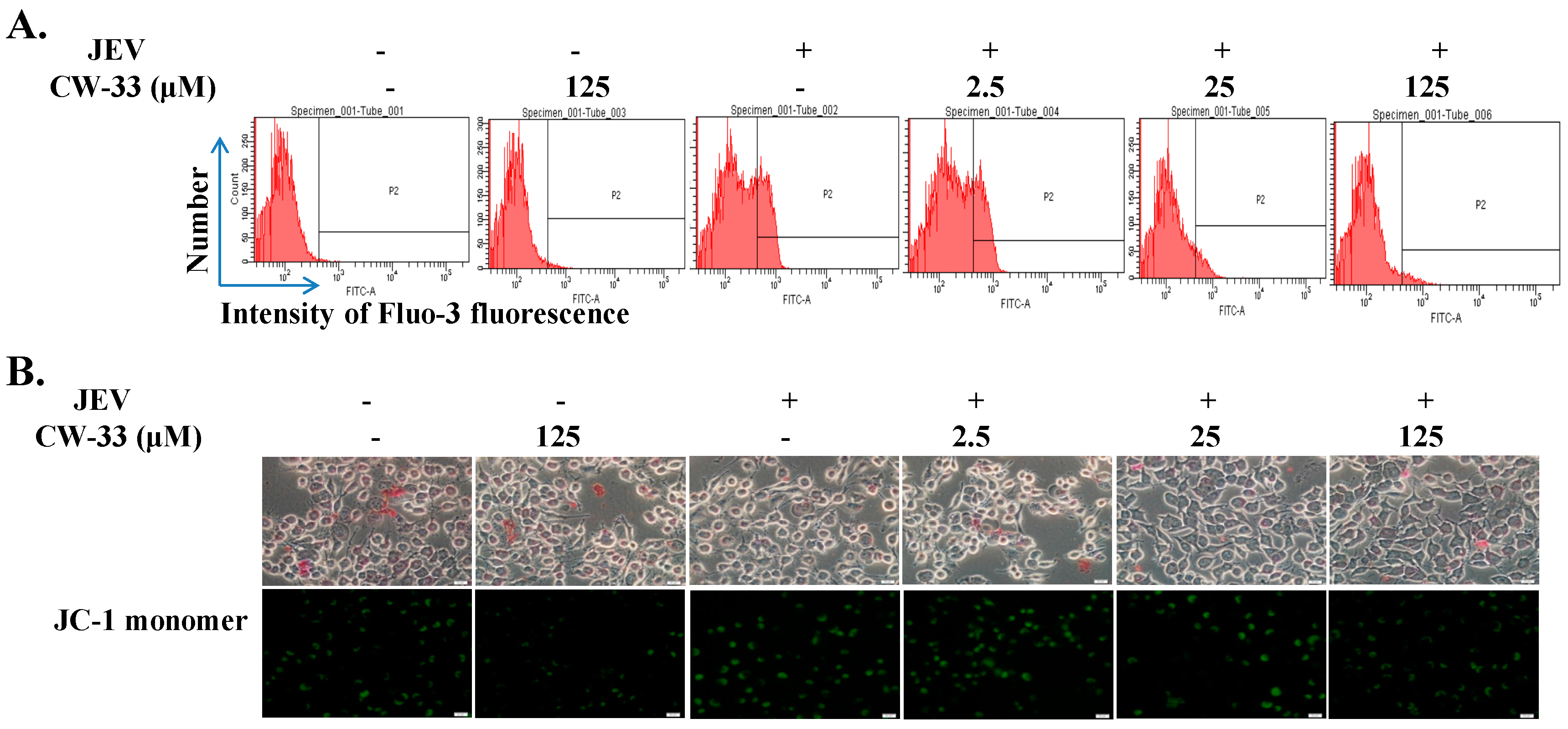
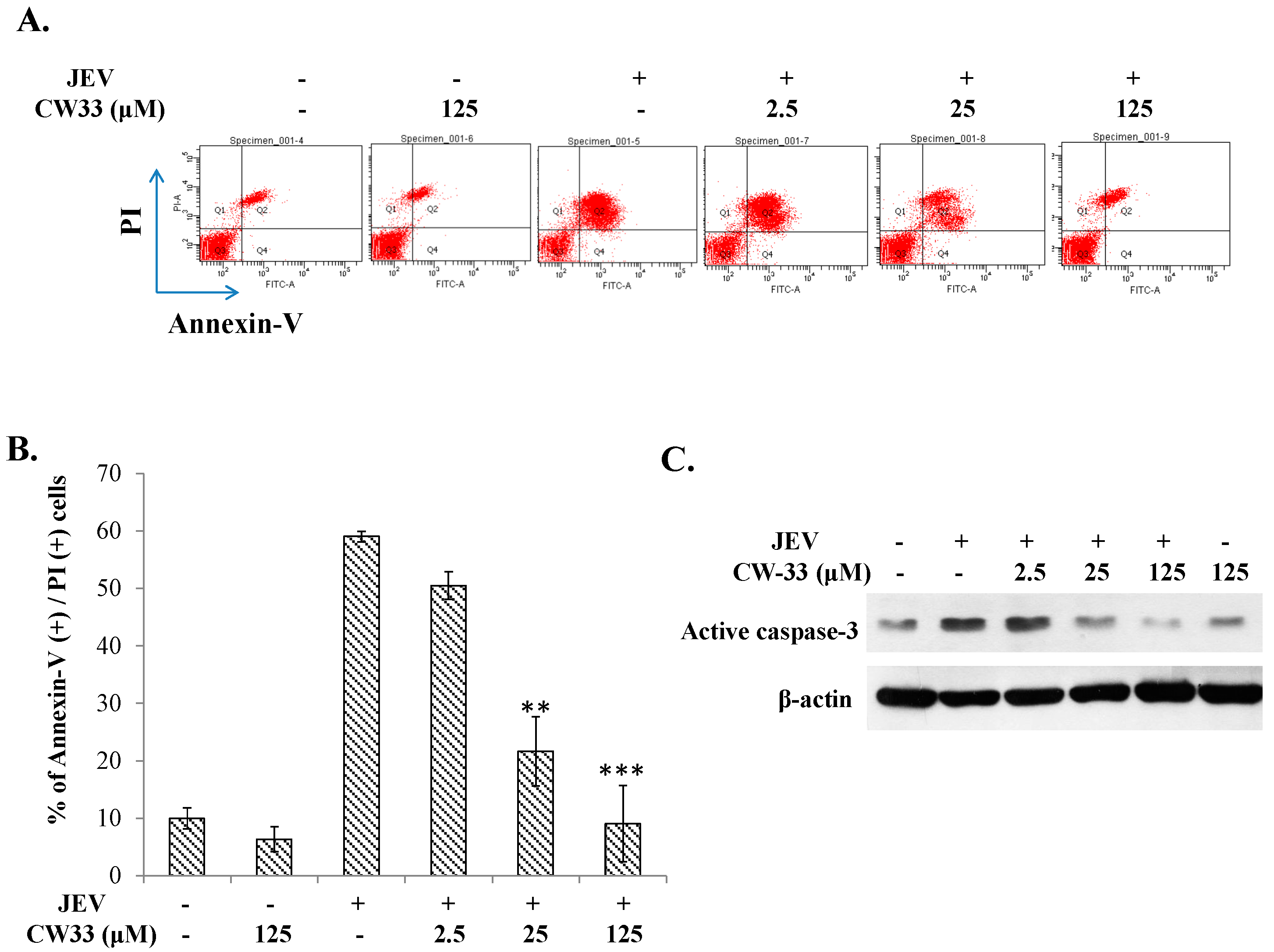
2.4. Reduction of JEV-Induced Calcium Overload by CW-33
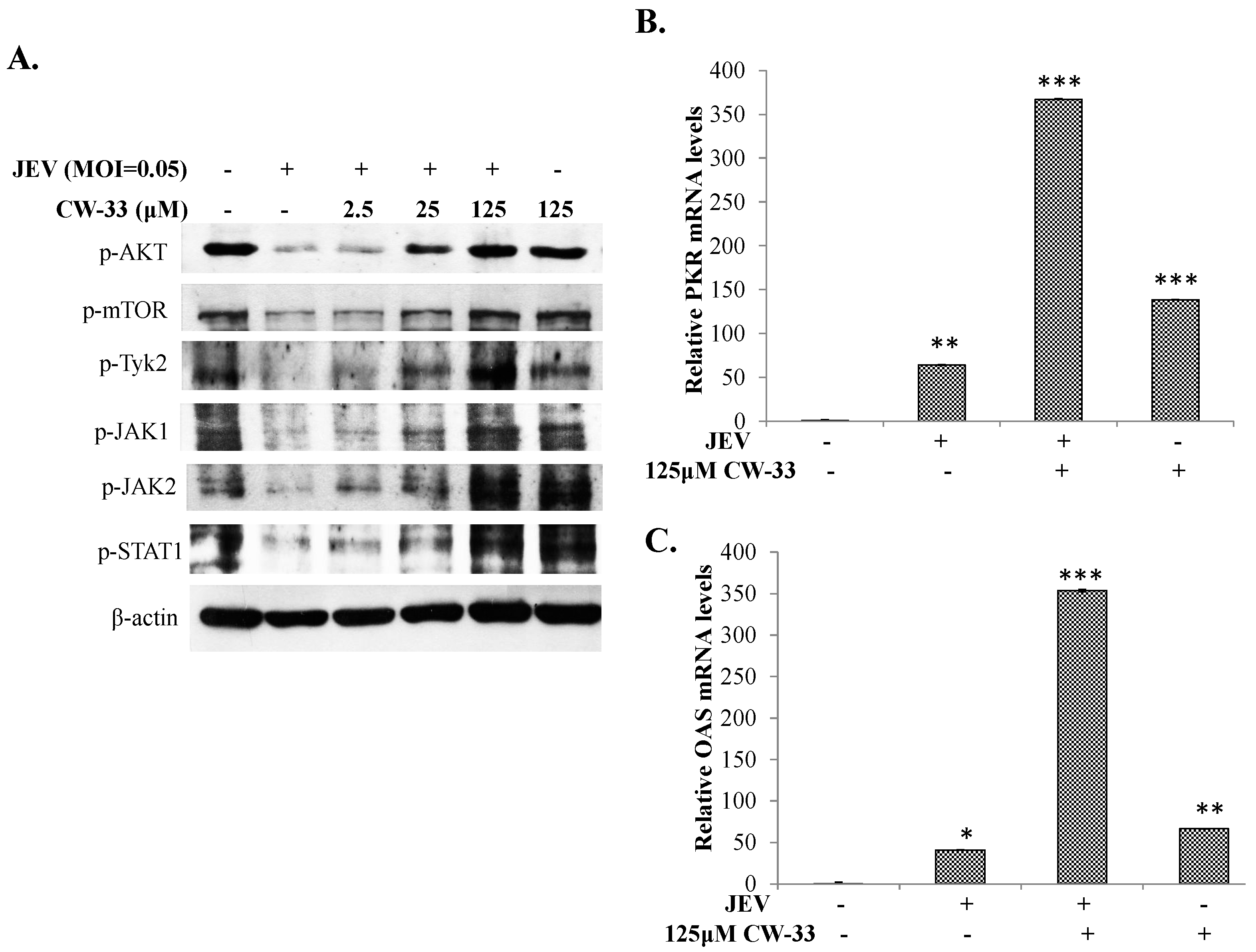
2.5. Activation of Akt/mTOR and Jak/STAT Pathway in JEV-Infected Cells by CW-33
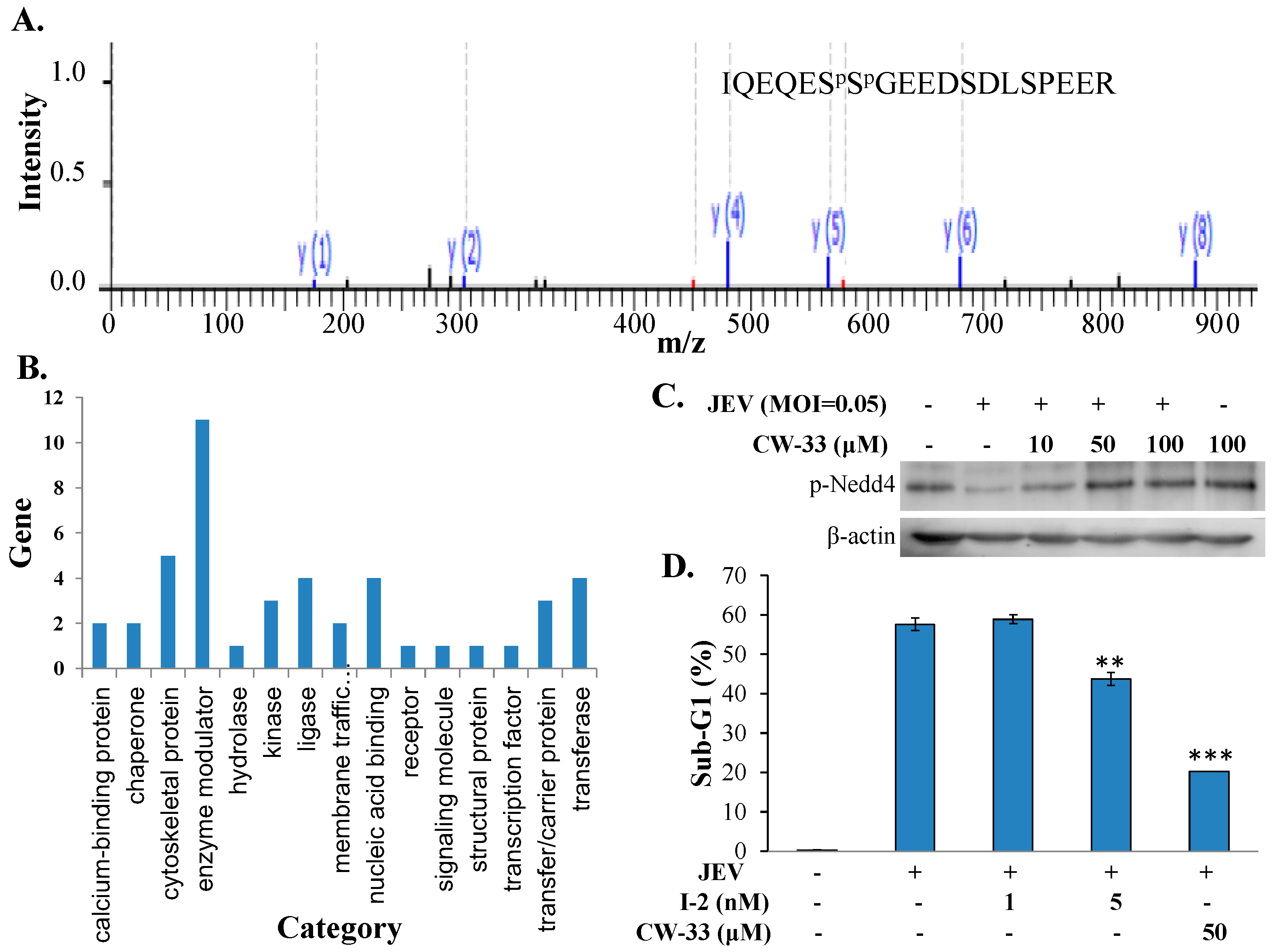
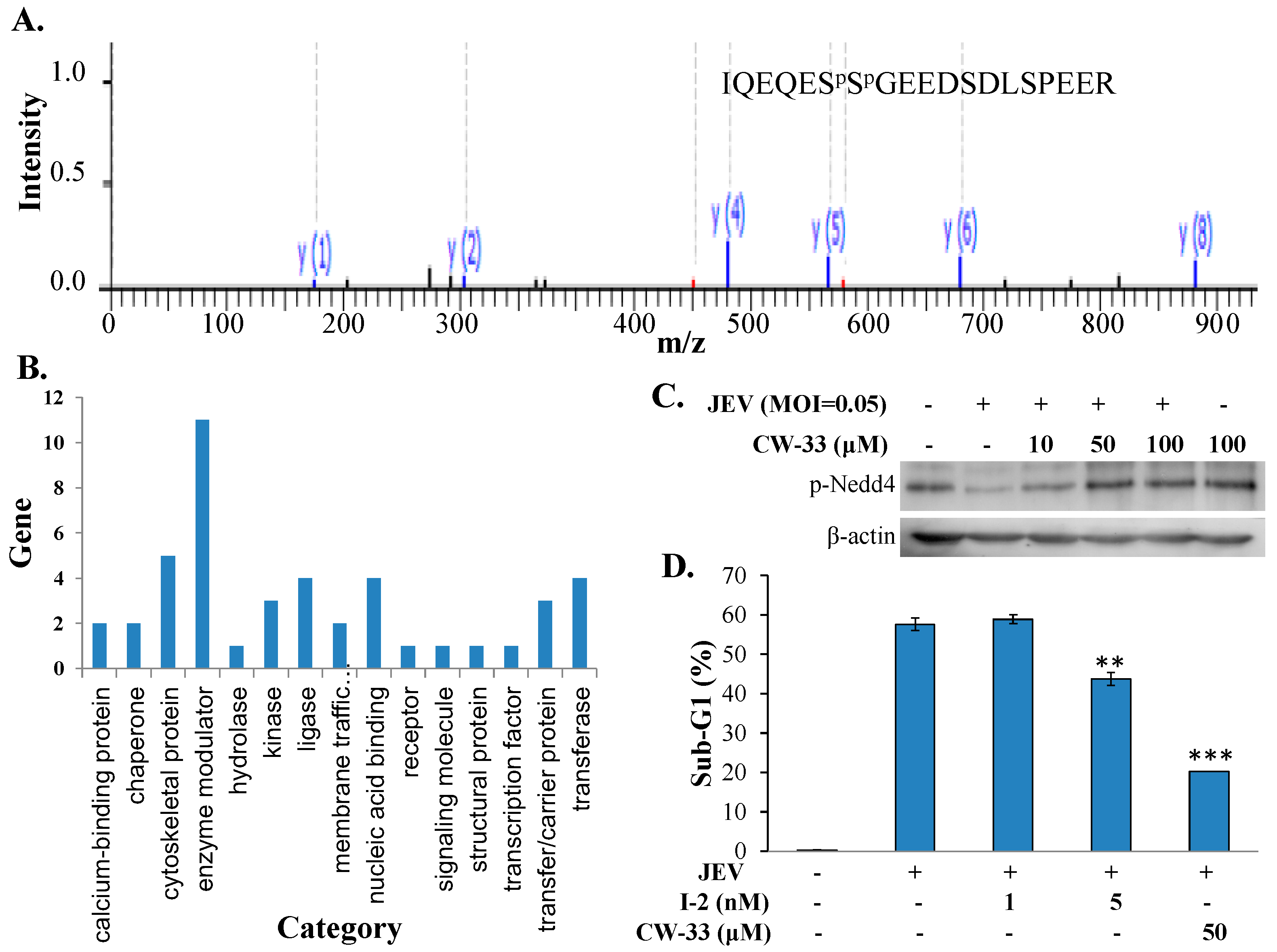
2.6. Regulation of Protein Phosphatase-1 Inhibitor-2 (I-2) in JEV-Infected Cells by CW-33
3. Discussion
4. Materials and Methods
4.1. Cells and Viruses
4.2. Synthesis of Compound CW-33
4.3. MTT Cytotoxicity Test
4.4. Cytopathic Effect Inhibition
4.5. Western Blotting Analysis of Signaling Pathways
4.6. Virus Yield Reduction Assays
4.7. Virucidal and Attachment Assays
4.8. Time-of-Addition Assay
4.9. Detecting Intracellular Free Ca2+
4.10. Mitochondrial Membrane Potential Assay with JC-1 Dye
4.11. Quantification of Gene Expression Using Real Time RT-PCR
4.12. Phosphopeptide Analysis of JEV-Infected Cells Treated with or without CW-33
4.13. Inhibition of JEV-Induced Apoptosis by Protein Phosphatase 1 Inhibitor-2 (I-2)
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coffey, L.L.; Forrester, N.; Tsetsarkin, K.; Vasilakis, N.; Weaver, S.C. Factors shaping the adaptive landscape for arboviruses: Implications for the emergence of disease. Future Microbiol. 2013, 8, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. Zika virus: New clinical syndromes and its emergence in the Western Hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Reisen, W.K. Present and future arboviral threats. Antivir. Res. 2010, 85, 328–345. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Ni, H.; Beasley, D.W.; Ekkelenkamp, M.; Cardosa, M.J.; Barrett, A.D. Origin and evolution of Japanese encephalitis virus in southeast Asia. J. Virol. 2003, 77, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Fan, Y.C.; Tu, W.C.; Chang, R.Y.; Shih, C.C.; Lu, I.H.; Chien, M.S.; Lee, W.C.; Chen, T.H.; Chang, G.J.; et al. Japanese encephalitis virus genotype replacement, Taiwan, 2009–2010. Emerg. Infect. Dis. 2011, 17, 2354–2356. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.M.; Cho, J.E.; Ju, Y.R.; Kim, S.Y.; Ryou, J.; Han, M.G.; Choi, W.Y.; Jeong, Y.E. Molecular epidemiology of Japanese encephalitis virus circulating in South Korea, 1983–2005. Virol. J. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, J.F.; Ståhl, K.; Chirico, J.; Boqvist, S.; Thu, H.T.; Magnusson, U. Circulation of Japanese encephalitis virus in pigs and mosquito vectors within Can Tho city, Vietnam. PLoS Negl. Trop. Dis. 2013, 7, e2153. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.C.; Chen, J.M.; Chiu, H.C.; Chen, Y.Y.; Lin, J.W.; Shih, C.C.; Chen, C.M.; Chang, C.C.; Chang, G.J.; Chiou, S.S. Partially neutralizing potency against emerging genotype I virus among children received formalin-inactivated Japanese encephalitis virus vaccine. PLoS Negl. Trop. Dis. 2012, 6, e1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.J.; Kim, H.C.; Klein, T.A.; Ramey, A.M.; Lee, J.H.; Kyung, S.G.; Park, J.Y.; Cho, Y.S.; Cho, I.S.; Yeh, J.Y. Molecular detection and genotyping of Japanese encephalitis virus in mosquitoes during a 2010 outbreak in the Republic of Korea. PLoS ONE 2013, 8, e55165. [Google Scholar]
- Kuo, S.C.; Huang, S.C.; Hung, L.J.; Cheng, H.E.; Lin, T.P.; Wu, C.H.; Ishii, K.; Nakamura, H. Studies of heterocyclic compounds. VIII. Synthesis, anti-inflammatory and antiallergic activities of n-alkyl-2,3,4,9-tetrahydrofuro[2,3-b]quinoline-3,4-diones and related compounds. J. Heterocycl. Chem. 1991, 28, 955–963. [Google Scholar] [CrossRef]
- Su, M.J.; Chang, G.J.; Wu, M.H.; Kuo, S.C. Electrophysiological basis for the antiarrhythmic action and positive inotropy of HA-7, a furoquinoline alkaloid derivative, in rat heart. Br. J. Pharmacol. 1997, 122, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wolfender, J.L.; Hostettmann, K.; Xu, R.; Qin, G. Antifungal alkaloids and limonoid derivatives from Dictamnus dasycarpus. Phytochemistry 1998, 47, 7–11. [Google Scholar] [CrossRef]
- Severino, V.G.; da Silva, M.F.; Lucarini, R.; Montanari, L.B.; Cunha, W.R.; Vinholis, A.H.; Martins, C.H. Determination of the antibacterial activity of crude extracts and compounds isolated from Hortia oreadica (Rutaceae) against oral pathogens. Braz. J. Microbiol. 2009, 40, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Wansi, J.D.; Mesaik, M.A.; Chiozem, D.D.; Devkota, K.P.; Gaboriaud-Kolar, N.; Lallemand, M.C.; Wandji, J.; Choudhary, M.I.; Sewald, N. Oxidative burst inhibitory and cytotoxic indoloquinazoline and furoquinoline alkaloids from Oricia suaveolens. J. Nat. Prod. 2008, 71, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Huang, A.C.; Hour, M.J.; Huang, S.H.; Kung, S.H.; Chen, C.H.; Chen, I.C.; Chang, Y.S.; Lien, J.C.; Lin, C.W. Antiviral Potential of a Novel Compound CW-33 against Enterovirus A71 via Inhibition of Viral 2A Protease. Viruses 2015, 7, 3155–3171. [Google Scholar] [CrossRef] [PubMed]
- Takegami, T.; Simamura, E.; Hirai, K.; Koyama, J. Inhibitory effect of furanonaphthoquinone derivatives on the replication of Japanese encephalitis virus. Antivir. Res. 1998, 37, 37–45. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Kondo, T.; Zhao, Q.L.; Feril, L.B., Jr.; Kitagawa, H. Role of intracellular calcium ions and reactive oxygen species in apoptosis induced by ultrasound. Ultrasound Med. Biol. 2004, 30, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Rimessi, A.; Giorgi, C.; Baldini, C.; Ferroni, L.; Rizzuto, R.; Pinton, P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem. Biophys. Res. Commun. 2008, 375, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef]
- Ding, W.; Albrecht, B.; Kelley, R.E.; Muthusamy, N.; Kim, S.J.; Altschuld, R.A.; Lairmore, M.D. Human T cell lymphotropic virus type 1 p12(I) expression increases cytoplasmic calcium to enhance the activation of nuclear factor of activated T cells. J. Virol. 2002, 76, 10374–10382. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, B.; D’Souza, C.D.; Ding, W.; Tridandapani, S.; Coggeshall, K.M.; Lair-more, M.D. Activation of nuclear factor of activated T cells by human T-lymphotropic virus type 1 accessory protein p12(I). J. Virol. 2002, 76, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, A.; Sundstrom, S.; Dimberg, L.Y.; Gylfe, E.; Masucci, M.G. The hepatitis C virus core protein modulates T cell responses by inducing spontaneous and altering T-cell receptor-triggered Ca2+ oscillations. J. Biol. Chem. 2003, 278, 18877–18883. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, A.; Rice, C.M. Transcriptional activation of the interleukin-2 promoter by hepatitis C virus core protein. J. Virol. 2001, 75, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, S.H.; Feng, P.; Chang, H.; Cho, N.H.; Jung, J.U. Characterization of the Kaposi’s sarcoma associated herpesvirus K1 signalosome. J. Virol. 2005, 79, 12173–12184. [Google Scholar] [CrossRef] [PubMed]
- Van Kuppeveld, F.J.; de Jong, A.S.; Melchers, W.J.; Willems, P.H. Enterovirus protein 2B po(u)res out the calcium: A viral strategy to survive? Trends Microbiol. 2005, 13, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Frey, T.K.; Yang, J.J. Viral calciomics: Interplays between Ca2+ and virus. Cell Calcium 2009, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Gong, L.L.; Yuan, D.; Deng, M.; Zeng, X.M.; Chen, L.L.; Zhang, L.; Yan, Q.; Liu, J.P.; Hu, X.H.; et al. Protein phosphatase-1 regulates Akt1 signal transduction pathway to control gene expression, cell survival and differentiation. Cell Death Differ. 2010, 17, 1448–1462. [Google Scholar] [CrossRef] [PubMed]
- Thayyullathil, F.; Chathoth, S.; Shahin, A.; Kizhakkayil, J.; Hago, A.; Patel, M.; Galadari, S. Protein phosphatase 1-dependent dephosphorylation of Akt is the prime signaling event in sphingosine-induced apoptosis in Jurkat cells. J. Cell. Biochem. 2011, 112, 1138–1153. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Nishimoto, H.; Kitaura, J.; Maeda-Yamamoto, M.; Kato, R.M.; Littman, D.R.; Leitges, M.; Rawlings, D.J.; Kawakami, T. Protein kinase C betaII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J. Biol. Chem. 2004, 279, 47720–47725. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Liao, C.L.; Lin, E.; Lin, Y.L. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J. Virol. 2004, 78, 9285–9294. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Chang, B.L.; Yu, H.P.; Liao, C.L.; Lin, Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef]
- Yang, T.C.; Li, S.W.; Lai, C.C.; Lu, K.Z.; Chiu, M.T.; Hsieh, T.H.; Wan, L.; Lin, C.W. Proteomic analysis for Type I interferon antagonism of Japanese encephalitis virus NS5 protein. Proteomics 2013, 13, 3442–3456. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Briguglio, I.; Piras, S.; Corona, P.; Boatto, G.; Nieddu, M.; Giunchedi, P.; Marongiu, M.E.; Giliberti, G.; Iuliano, F.; et al. Quinoline tricyclic derivatives. Design, synthesis and evaluation of the antiviral activity of three new classes of RNA-dependent RNA polymerase inhibitors. Bioorg. Med. Chem. 2011, 19, 7070–7084. [Google Scholar] [CrossRef] [PubMed]
- Briguglio, I.; Loddo, R.; Laurini, E.; Fermeglia, M.; Piras, S.; Corona, P.; Giunchedi, P.; Gavini, E.; Sanna, G.; Giliberti, G.; et al. Synthesis, cytotoxicity and antiviral evaluation of new series of imidazo[4,5-g]quinoline and pyrido[2,3-g]quinoxalinone derivatives. Eur. J. Med. Chem. 2015, 105, 63–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, S.; Duhamel, F.; Coulombe, P.; Popoff, M.R.; Meloche, S. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol. Cell. Biol. 2003, 23, 1316–1333. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Jin, H.; Cao, Z.; Feng, N.; Zhao, Y.; Zheng, X.; Wang, J.; Li, Q.; Zhao, G.; et al. Interferon-inducible GTPase: A novel viral response protein involved in rabies virus infection. Arch. Virol. 2016, 161, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.; Graf, L.; Olal, D.; von der Malsburg, A.; Gao, S.; Kochs, G.; Daumke, O. Role of nucleotide binding and GTPase domain dimerization in dynamin-like myxovirus resistance protein A for GTPase activation and antiviral activity. J. Biol. Chem. 2015, 290, 12779–12792. [Google Scholar] [CrossRef] [PubMed]
- Brisac, C.; Salloum, S.; Yang, V.; Schaefer, E.A.; Holmes, J.A.; Chevaliez, S.; Hong, J.; Carlton-Smith, C.; Alatrakchi, N.; Kruger, A.; et al. IQGAP2 is a novel interferon-alpha antiviral effector gene acting nonconventionally through the NF-κB pathway. J. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Chang, Y.C.; Lu, K.Z.; Tsou, Y.Y.; Lin, C.W. Antiviral Activity of Isatis indigotica Extract and Its Derived Indirubin against Japanese Encephalitis Virus. Evid. Based Complement. Altern. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Huang, S.C.; Zhang, Y.; Lai, Z.R.; Kung, S.H.; Chang, Y.S.; Lin, C.W. Antiviral Ability of Kalanchoe gracilis Leaf Extract against Enterovirus 71 and Coxsackievirus A16. Evid Based Complement. Altern. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Jou, Y.J.; Chen, C.J.; Liu, Y.C.; Way, T.D.; Lai, C.H.; Hua, C.H.; Wang, C.Y.; Huang, S.H.; Kao, J.Y.; Lin, C.W. Quantitative phosphoproteomic analysis reveals γ-bisabolene inducing p53-mediated apoptosis of human oral squamous cell carcinoma via HDAC2 inhibition and ERK1/2 activation. Proteomics 2015, 15, 3296–3309. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.-H.; Lien, J.-C.; Chen, C.-J.; Liu, Y.-C.; Wang, C.-Y.; Ping, C.-F.; Lin, Y.-F.; Huang, A.-C.; Lin, C.-W. Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload. Int. J. Mol. Sci. 2016, 17, 1386. https://doi.org/10.3390/ijms17091386
Huang S-H, Lien J-C, Chen C-J, Liu Y-C, Wang C-Y, Ping C-F, Lin Y-F, Huang A-C, Lin C-W. Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload. International Journal of Molecular Sciences. 2016; 17(9):1386. https://doi.org/10.3390/ijms17091386
Chicago/Turabian StyleHuang, Su-Hua, Jin-Cherng Lien, Chao-Jung Chen, Yu-Ching Liu, Ching-Ying Wang, Chia-Fong Ping, Yu-Fong Lin, An-Cheng Huang, and Cheng-Wen Lin. 2016. "Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload" International Journal of Molecular Sciences 17, no. 9: 1386. https://doi.org/10.3390/ijms17091386