
Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Riemerella anatipestifer

 ,
,
Abstract
:
1. Introduction
2. Results
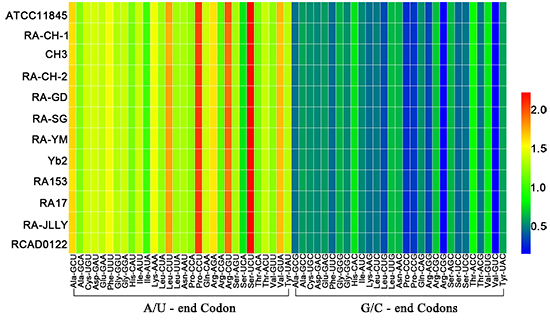
2.1. The Codon Usage Pattern between Riemerella anatipestifer (RA)
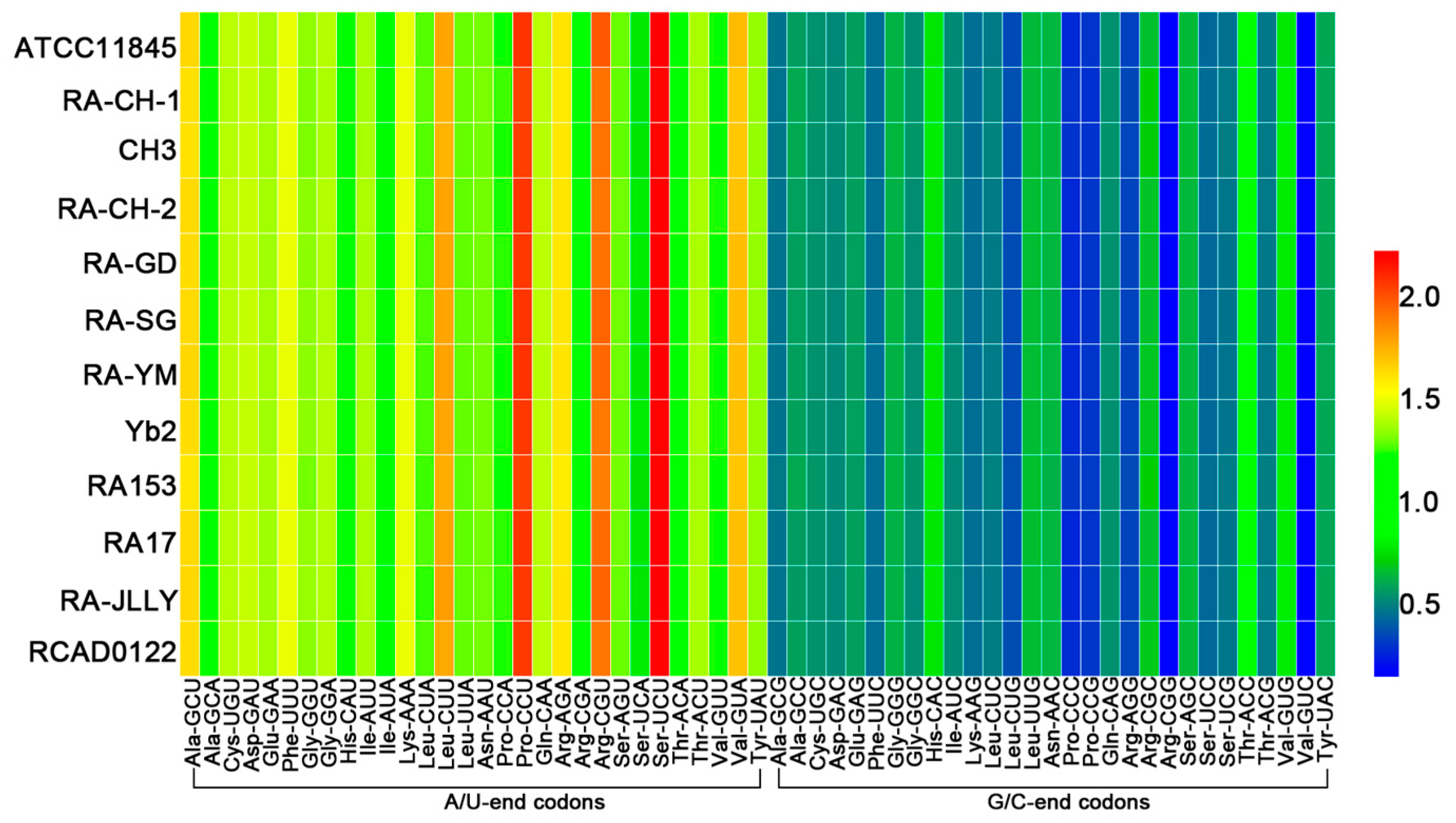
2.2. The Codon Usage Bias of RA not Affected by Mutation Bias
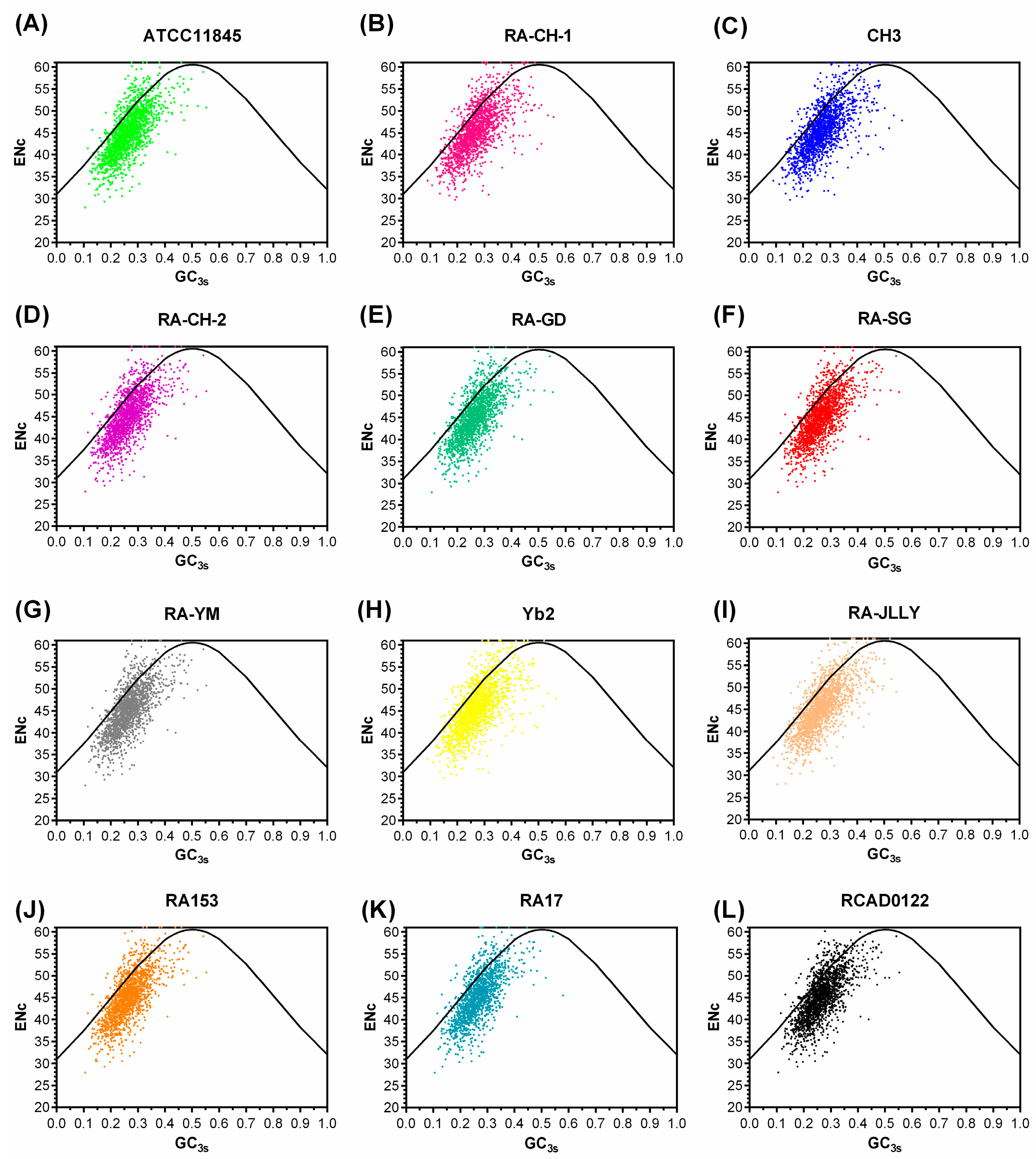
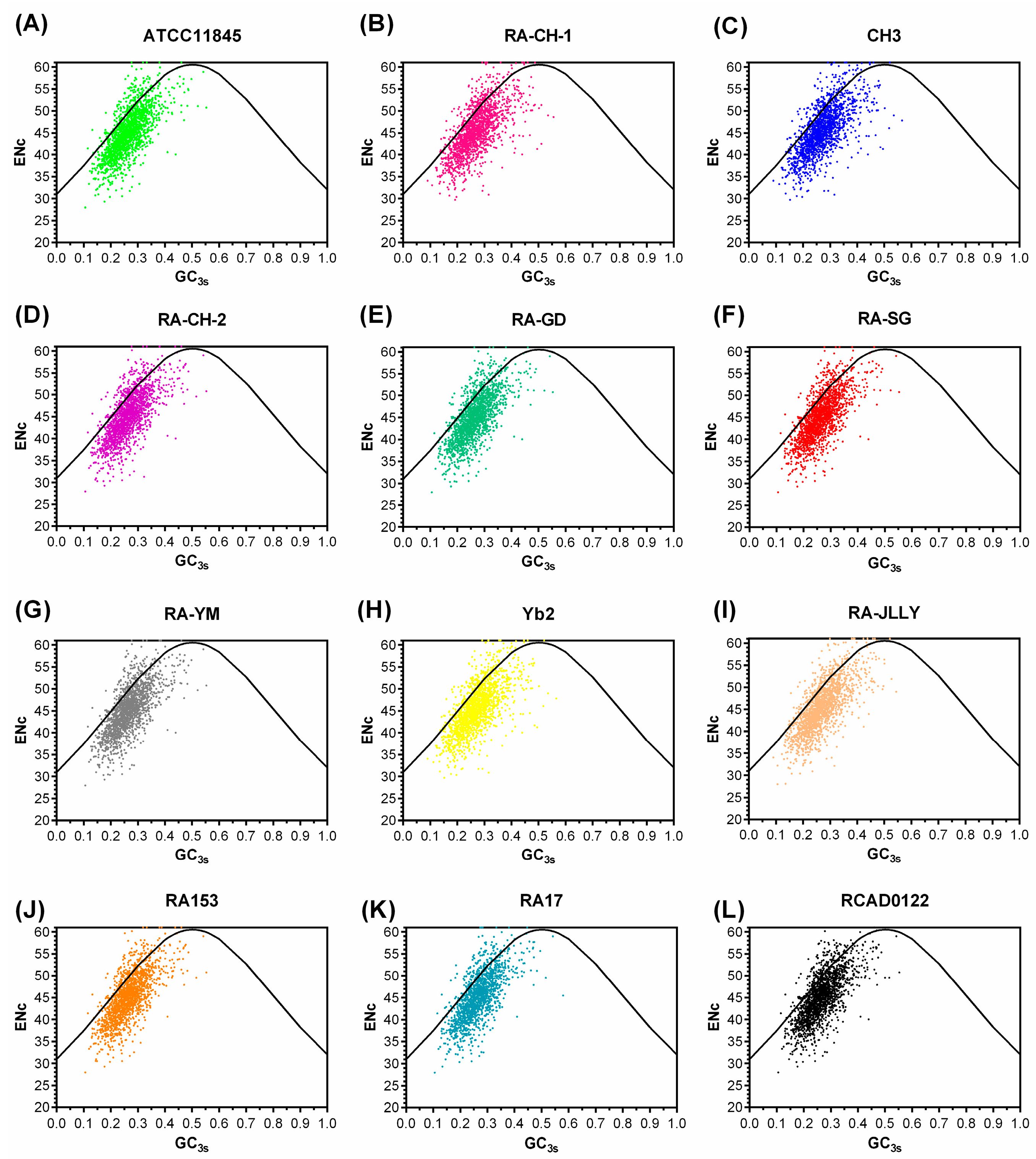
2.3. Correspondence Analysis (COA)
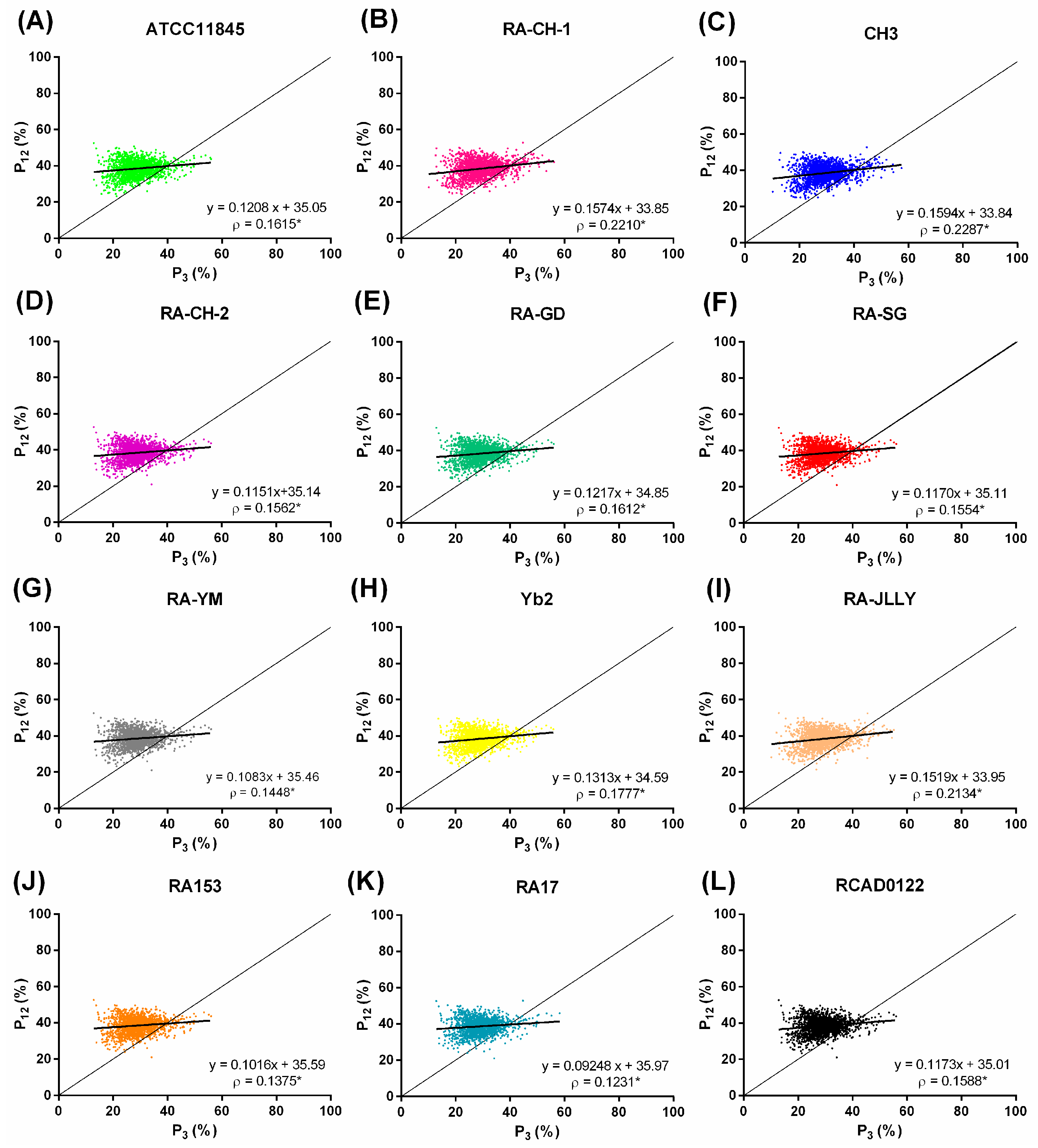
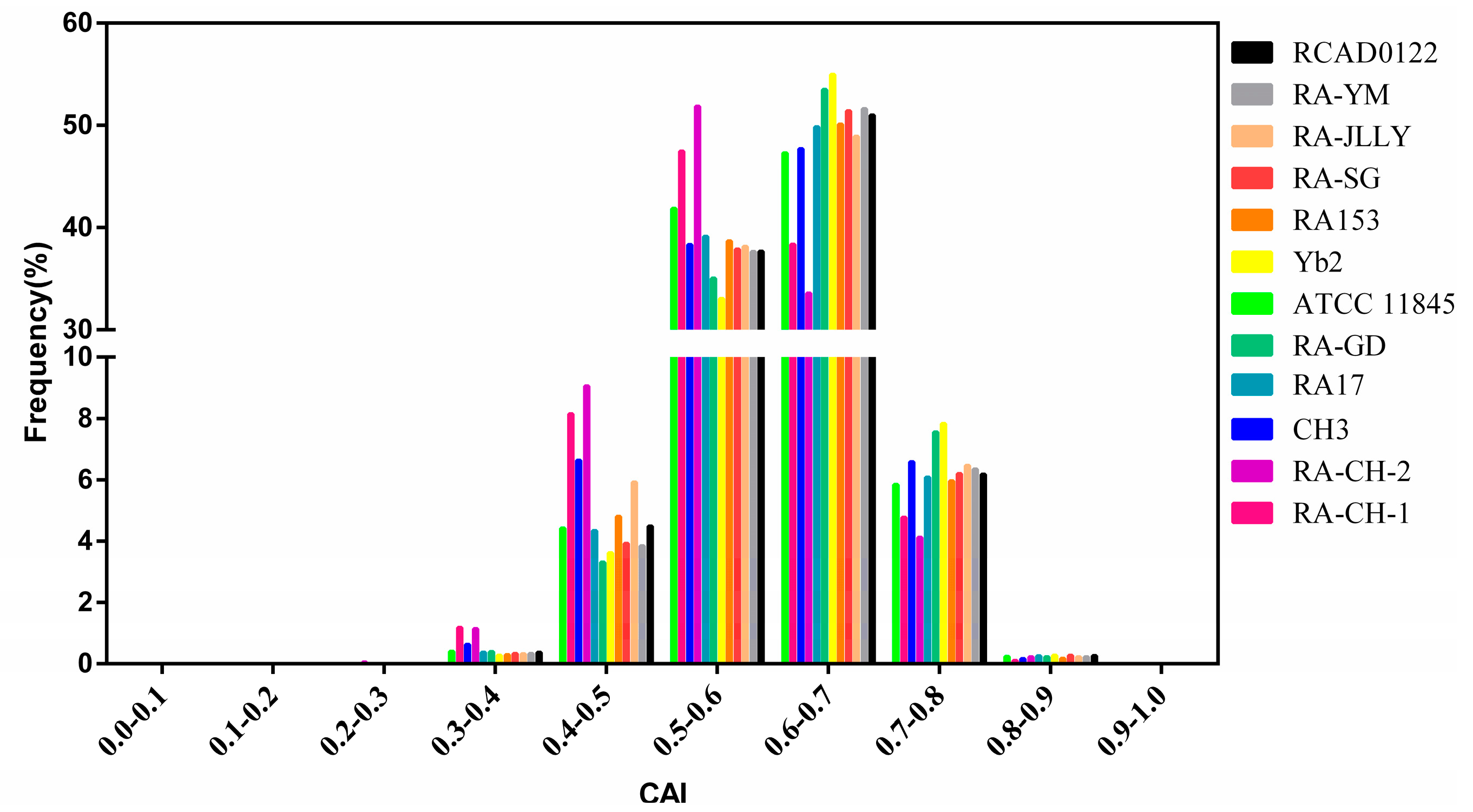
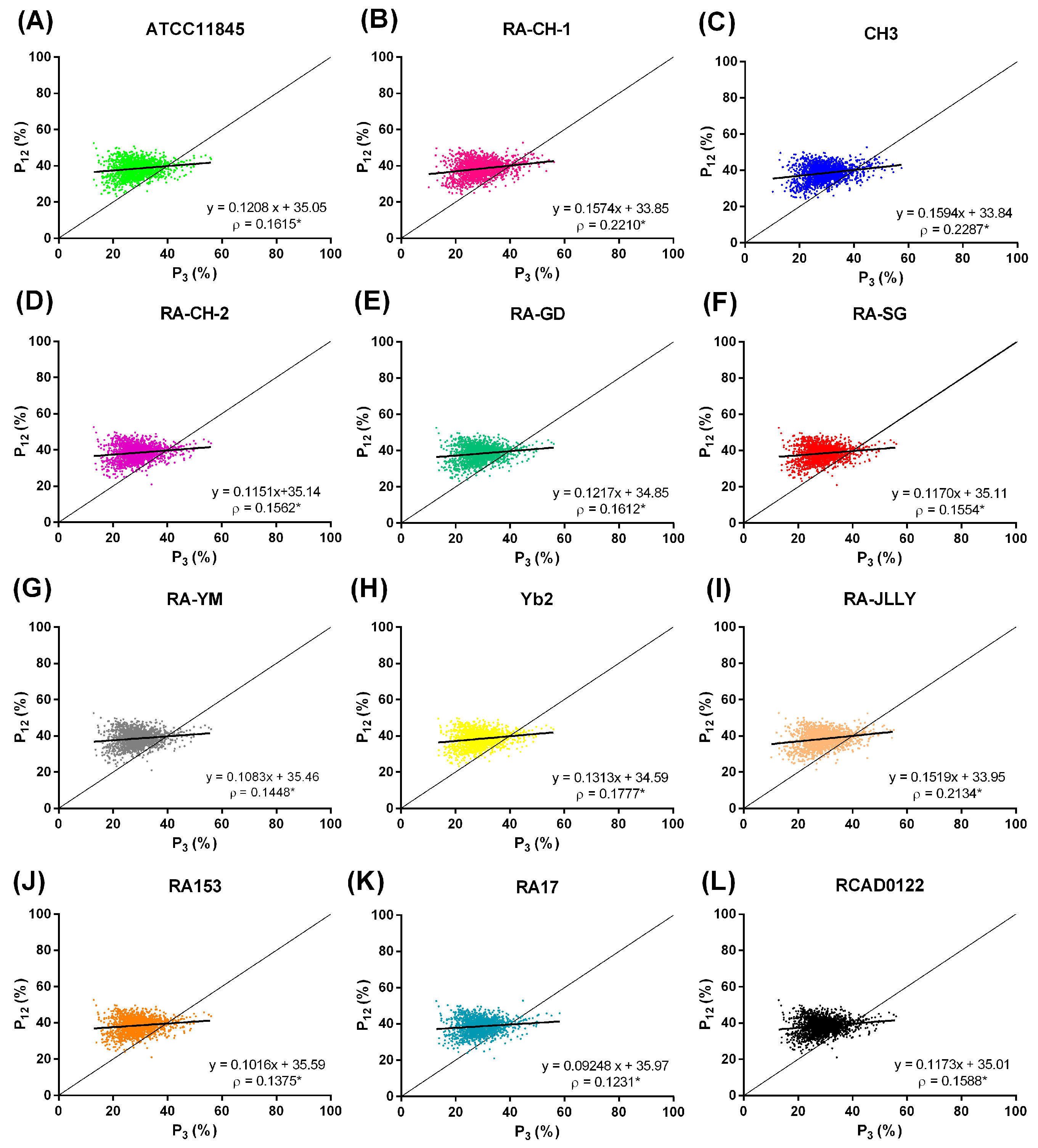
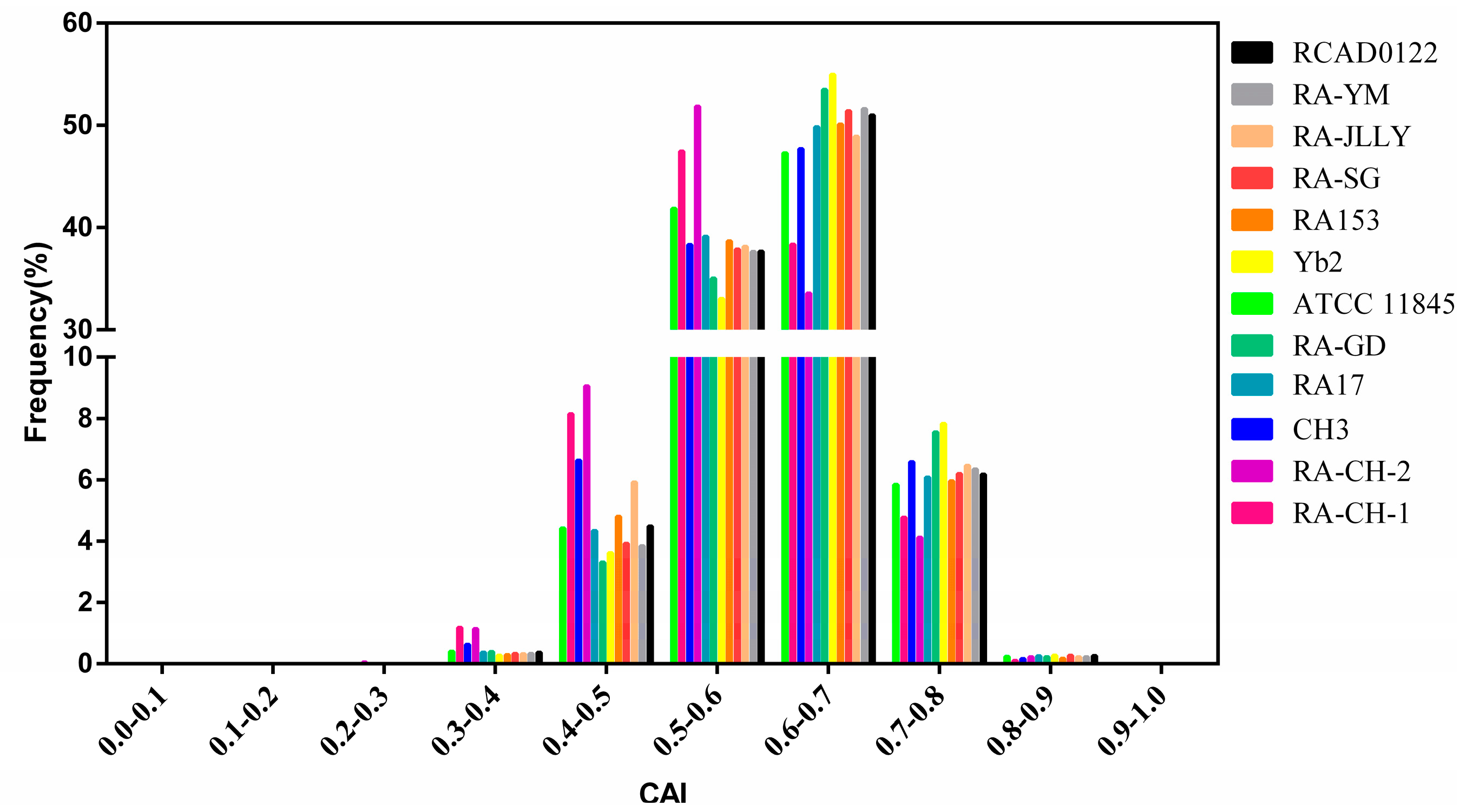
2.4. Natural Selection Influences the Codon Bias of RA
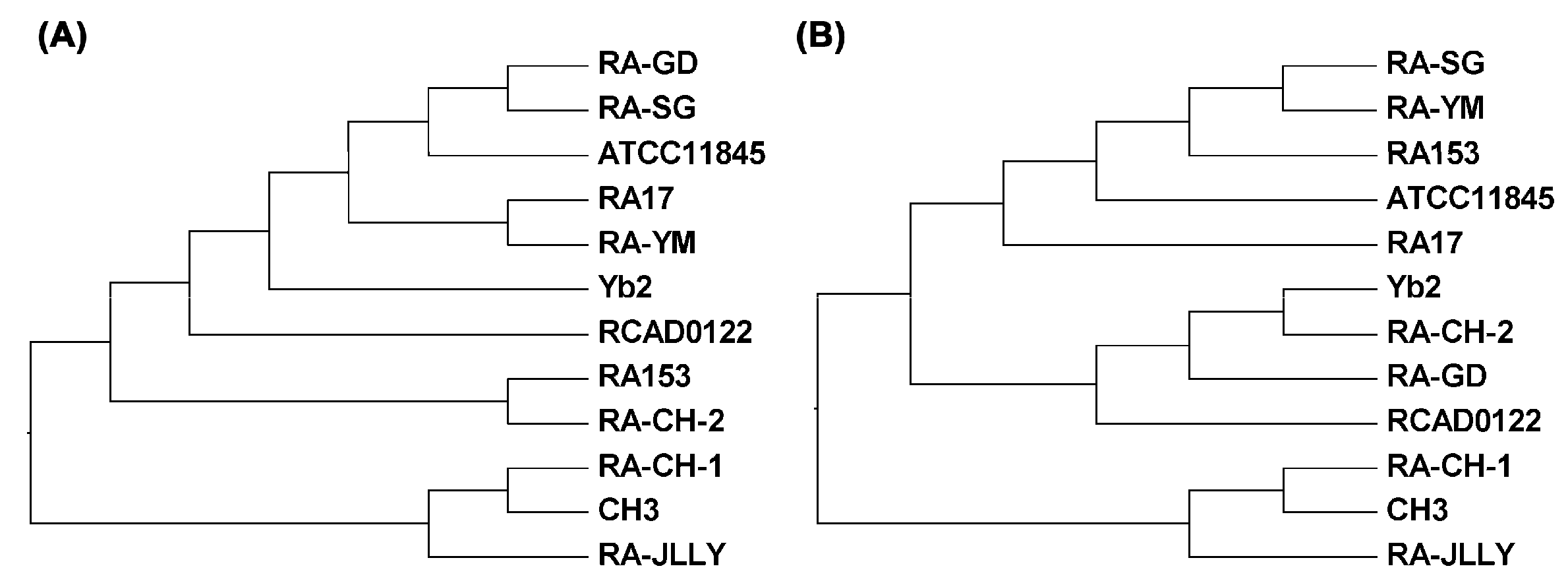
2.5. Cluster Analysis
2.6. Understanding Pathway Level Functions in RA through CUB
3. Discussion
4. Materials and Methods
4.1. Sequences Data
4.2. Measurement Indices of Codon Usage Bias
4.3. Correspondence Analysis and Cluster Analysis
4.4. Software and Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Subramaniam, S.; Chua, K.L.; Tan, H.M.; Loh, H.; Kuhnert, P.; Frey, J. Phylogenetic position of Riemerella anatipestifer based on 16S rRNA gene sequences. Int. J. Syst. Bacteriol. 1997, 47, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Swayne, D.E.; Glisson, J.R.; McDougald, L.R. Diseases of Poultry; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 811–813. [Google Scholar]
- Chang, C.F.; Lin, W.H.; Yeh, T.M.; Chiang, T.S.; Chang, Y.F. Antimicrobial susceptibility of Riemerella anatipestifer isolated from ducks and the efficacy of ceftiofur treatment. J. Vet. Diagn. Investig. 2003, 15, 26–29. [Google Scholar] [CrossRef]
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pave, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, r49–r62. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R.; Gautier, C.; Gouy, M.; Jacobzone, M.; Mercier, R. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res. 1981, 9, r43–r74. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wu, J.; Yang, H.; Bao, Q. Codon usage patterns in Corynebacterium glutamicum: Mutational bias, natural selection and amino acid conservation. Comp. Funct. Genom. 2010, 2010, 1–7. [Google Scholar]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Venton, D. Highlight: Tiny bacterial genome opens a huge mystery: At mutational bias in Hodgkinia. Genome Biol. Evol. 2012, 4, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in Aeropyrum pernix K1 and other Crenarchaeota microorganisms. J. Genet. Genom. 2007, 34, 275–284. [Google Scholar] [CrossRef]
- Peixoto, L.; Zavala, A.; Romero, H.; Musto, H. The strength of translational selection for codon usage varies in the three replicons of Sinorhizobium meliloti. Gene 2003, 320, 109–116. [Google Scholar] [CrossRef]
- Nayak, K.C. Comparative study on factors influencing the codon and amino acid usage in Lactobacillus sakei 23K and 13 other lactobacilli. Mol. Biol. Rep. 2012, 39, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Tong, Z.; Ma, J.; Xiao, S.; Lu, Z. The relationship between synonymous codon usage and protein structure in Escherichia coli and Homo sapiens. BioSystems 2004, 73, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhou, T.; Gu, W.; Sun, X.; Lu, Z. Cluster analysis of the codon use frequency of MHC genes from different species. BioSystems 2002, 65, 199–207. [Google Scholar] [CrossRef]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [PubMed]
- Cai, M.S.; Cheng, A.C.; Wang, M.S.; Zhao, L.C.; Zhu, D.K.; Luo, Q.H.; Liu, F.; Chen, X.Y. Characterization of synonymous codon usage bias in the duck plague virus UL35 gene. Intervirology 2009, 52, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Qin, Z.; Chen, Z.; Zhao, Y.; Zhong, J. The factors shaping synonymous codon usage in the genome of Burkholderia mallei. J. Genet. Genom. 2007, 34, 362–372. [Google Scholar]
- Ma, X.X.; Feng, Y.P.; Bai, J.L.; Zhang, D.R.; Lin, X.S.; Ma, Z.R. Nucleotide composition bias and codon usage trends of gene populations in Mycoplasma capricolum subsp. Capricolum and M. Agalactiae. J. Genet. 2015, 94, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Landan, G.; Graur, D. Can GC content at third-codon positions be used as a proxy for isochore composition? Mol. Biol. Evol. 2009, 26, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The “effective number of codons” used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Palacios, C.; Wernegreen, J.J. A strong effect of at mutational bias on amino acid usage in Buchnera is mitigated at high-expression genes. Mol. Biol. Evol. 2002, 19, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Rispe, C.; Delmotte, F.; van Ham, R.C.; Moya, A. Mutational and selective pressures on codon and amino acid usage in Buchnera, endosymbiotic bacteria of aphids. Genome Res. 2004, 14, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, S.; Muto, A.; Kawauchi, Y.; Yamao, F.; Osawa, S. The ribosomal protein gene cluster of Mycoplasma capricolum. Mol. Gen. Genet. 1987, 210, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.; Bibb, M.J. Codon usage in the G + C-rich Streptomyces genome. Gene 1992, 113, 55–65. [Google Scholar] [CrossRef]
- Shields, D.C. Switches in species-specific codon preferences: The influence of mutation biases. J. Mol. Evol. 1990, 31, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Two aspects of DNA base composition: G + C content and translation-coupled deviation from intra-strand rule of A = T and G = C. J. Mol. Evol. 1999, 49, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Kepes, F.; Zinovyev, A. Codon bias signatures, organization of microorganisms in codon space, and lifestyle. Mol. Biol. Evol. 2005, 22, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Jon, B.; Ola, B.; Tammi, V.; Eystein, S.; Ussery, D.W. Amino acid usage is asymmetrically biased in AT- and GC-rich microbial genomes. PLoS ONE 2013, 8, e69878. [Google Scholar]
- Muto, A.; Osawa, S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc. Natl. Acad. Sci. USA 1987, 84, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Evidence that mutation is universally biased towards at in bacteria. PLoS Genet. 2010, 6, e1001115. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, F.; Meyer, A.; Eyre-Walker, A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010, 6, e1001107. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. Predicted highly expressed genes in the genomes of Streptomyces coelicolor and Streptomyces avermitilis and the implications for their metabolism. Microbiology 2005, 151, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Nie, L.; Zhang, W. Predicted highly expressed genes in Nocardia farcinica and the implication for its primary metabolism and nocardial virulence. Antonie Van Leeuwenhoek 2006, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Willenbrock, H.; Friis, C.; Juncker, A.S.; Ussery, D.W. An environmental signature for 323 microbial genomes based on codon adaptation indices. Genome Biol. 2006, 7, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristina, J.; Moreno, P.; Moratorio, G.; Musto, H. Genome-wide analysis of codon usage bias in Ebolavirus. Virus Res. 2015, 196, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Sarkar, R.R. Comparison of codon usage bias across Leishmania and Trypanosomatids to understand mRNA secondary structure, relative protein abundance and pathway functions. Genomics 2015, 106, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.P.; Li, C.; Wang, J.; Wang, Y.; Ding, Z.T. Analysis of synonymous codon usage in FAD7 genes from different plant species. Genet. Mol. Res. 2015, 14, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Singha, H.S.; Chakraborty, S.; Deka, H. Stress induced MAPK genes show distinct pattern of codon usage in Arabidopsis thaliana, Glycine max and Oryza sativa. Bioinformation 2014, 10, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Mavromatis, K.; Lu, M.; Misra, M.; Lapidus, A.; Nolan, M.; Lucas, S.; Hammon, N.; Deshpande, S.; Cheng, J.F.; Tapia, R.; et al. Complete genome sequence of Riemerella anatipestifer type strain (ATCC 11845). Stand. Genom. Sci. 2011, 4, 144–153. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, D.; Wang, M.; Cheng, A.; Jia, R.; Zhou, Y.; Chen, Z.; Luo, Q.; Liu, F.; Wang, Y.; et al. Complete genome sequence of Riemerella anatipestifer reference strain. J. Bacteriol. 2012, 194, 3270–3271. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ding, C.; Han, X.; Wang, S.; Yue, J.; Hou, W.; Cao, S.; Zou, J.; Yu, S. Complete genome sequence of Riemerella anatipestifer serotype 1 strain CH3. Genome Announc. 2015, 3, e01594-14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Peng, X.; Xiao, Y.; Wang, X.; Guo, Z.; Zhu, L.; Liu, M.; Jin, H.; Bi, D.; Li, Z.; et al. Genome sequence of poultry pathogen Riemerella anatipestifer strain RA-YM. J. Bacteriol. 2011, 193, 1284–1285. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, W.; Zhu, D.; Yang, L.; Liu, M.; Yin, S.; Wang, M.; Jia, R.; Chen, S.; Sun, K.; et al. Comparative genomics of Riemerella anatipestifer reveals genetic diversity. BMC Genom. 2014, 15, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, W.; Sun, M.; Song, S.; Cai, J.; Hu, S. Complete genome sequence of the pathogenic bacterium Riemerella anatipestifer strain RA-GD. J. Bacteriol. 2011, 193, 2896–2897. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ding, C.; Wang, S.; Han, X.; Yu, S. Whole-Genome sequence analysis and Genome-Wide virulence gene identification of Riemerella anatipestifer strain Yb2. Appl. Environ. Microbiol. 2015, 81, 5093–5102. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Li, L.; Sun, M.; Dong, J.; Hu, Q. Genome sequence of avirulent Riemerella anatipestifer strain RA-SG. Genome Announc. 2013, 1, e21812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, R.; Luo, Q.; Wen, G.; Ai, D.; Wang, H.; Luo, L.; Wang, H.; Shao, H. Genome sequence of avirulent Riemerella anatipestifer strain RA-JLLY. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Song, X.H.; Zhou, W.S.; Wang, J.B.; Liu, M.F.; Wang, M.S.; Cheng, A.C.; Jia, R.Y.; Chen, S.; Sun, K.F.; Yang, Q.; et al. Genome sequence of Riemerella anatipestifer strain RCAD0122, a multidrug-resistant isolate from ducks. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Translation-coupled violation of parity rule 2 in human genes is not the cause of heterogeneity of the DNA G + C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Greenacre, M.J. Theory and Applications of Correspondence Analysis; Academic Press: Pittsburgh, PA, USA, 1984; pp. 326–339. [Google Scholar]
- Lu, H.; Zhao, W.M.; Zheng, Y.; Wang, H.; Qi, M.; Yu, X.P. Analysis of synonymous codon usage bias in Chlamydia. Acta Biochim. Biophys. Sin. 2005, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Codon, W. Available online: http://codonw.sourceforge.net/ (accessed on 9 August 2016).
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A toolkit for illustrating heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef] [PubMed]
- CAIcal Sever. Available online: http://genomes.urv.es/CAIcal/intro.php (accessed on 9 August 2016).
- BlastKOALA. Available online: http://www.kegg.jp/blastkoala/ (accessed on 9 August 2016).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acids | Codon | RSCU 1 | Amino Acids | Codon | RSCU 1 |
|---|---|---|---|---|---|
| Ala | GCG | 0.49 | Pro | CCC | 0.31 |
| GCC | 0.61 | CCG | 0.33 | ||
| GCU * | 1.66 | CCA * | 1.27 | ||
| GCA * | 1.24 | CCU * | 2.10 | ||
| Cys | UGC | 0.57 | Arg | AGG | 0.37 |
| UGU * | 1.43 | CGC | 0.71 | ||
| Asp | GAC | 0.55 | CGG | 0.19 | |
| GAU * | 1.45 | AGA * | 1.63 | ||
| Glu | GAG | 0.60 | CGA | 1.14 | |
| GAA | 1.40 | CGU * | 1.96 | ||
| Phe | UUC | 0.47 | Ser | AGC | 0.68 |
| UUU * | 1.53 | UCC | 0.46 | ||
| Gly | GGG | 0.66 | UCG | 0.49 | |
| GGC | 0.56 | AGU * | 1.32 | ||
| GGU * | 1.36 | UCA | 0.80 | ||
| GGA * | 1.42 | UCU * | 2.24 | ||
| His | CAC | 0.80 | Thr | ACC | 0.89 |
| CAU * | 1.20 | ACG | 0.50 | ||
| Ile | AUC | 0.55 | ACA * | 1.20 | |
| AUU * | 1.41 | ACU * | 1.41 | ||
| AUA | 1.04 | Val | GUG | 0.82 | |
| Lys | AAG | 0.47 | GUC | 0.18 | |
| AAA * | 1.53 | GUU * | 1.26 | ||
| Leu | CUC | 0.51 | GUA * | 1.74 | |
| CUG | 0.39 | Try | UAC | 0.62 | |
| UUG | 0.68 | UAU * | 1.38 | ||
| CUA * | 1.30 | Gln | CAG | 0.57 | |
| CUU * | 1.81 | CAA * | 1.43 | ||
| UUA * | 1.32 | Stop | UGA | 0.20 | |
| Asn | AAC | 0.66 | UAG | 0.61 | |
| AAU * | 1.34 | UAA * | 2.19 | ||
| Met | AUG | 1.00 |
| RA Strain | GC% | GC3s% | ENc | CAI |
|---|---|---|---|---|
| ATCC11845 | 35.42 ± 3.63 | 26.50 ± 5.75 | 45.04 ± 5.13 | 0.616 ± 0.062 |
| RA-CH-1 | 35.51 ± 3.85 | 27.05 ± 6.28 | 45.47 ± 5.15 | 0.604 ± 0.064 |
| CH3 | 35.59 ± 3.89 | 27.07 ± 6.30 | 45.46 ± 5.17 | 0.582 ± 0.070 |
| RA-CH-2 | 35.39 ± 3.62 | 26.64 ± 5.98 | 45.19 ± 5.19 | 0.616 ± 0.064 |
| RA-GD | 35.33 ± 3.64 | 26.67 ± 5.95 | 45.16 ± 5.20 | 0.602 ± 0.069 |
| RA-SG | 35.40 ± 3.60 | 26.52 ± 5.17 | 45.10 ± 5.07 | 0.609 ± 0.063 |
| RA-YM | 35.50 ± 3.58 | 26.69 ± 5.80 | 45.15 ± 5.09 | 0.610 ± 0.063 |
| Yb2 | 35.34 ± 3.64 | 26.50 ± 5.74 | 45.12 ± 5.14 | 0.613 ± 0.062 |
| RA-JLLY | 35.45 ± 3.89 | 26.91 ± 6.25 | 45.34 ± 5.21 | 0.605 ± 0.067 |
| RA153 | 35.44 ± 3.61 | 26.57 ± 5.86 | 45.18 ± 5.18 | 0.604 ± 0.064 |
| RA17 | 35.52 ± 3.59 | 26.55 ± 5.77 | 45.14 ± 5.22 | 0.690 ± 0.064 |
| RCAD0122 | 35.40 ± 3.65 | 26.69 ± 5.86 | 45.21 ± 5.14 | 0.607 ± 0.064 |
| Category | Gene | Proteins | Strains |
|---|---|---|---|
| Ribosome | rplA | Large subunit ribosomal protein L1 | RA-CH-1, RA-GD, RA17 |
| rplB | Large subunit ribosomal protein L2 | + | |
| rplD | Large subunit ribosomal protein L4 | + | |
| rplE | Large subunit ribosomal protein L5 | + | |
| rplF | Large subunit ribosomal protein L6 | + | |
| rplL | Large subunit ribosomal protein L7/L12 | + | |
| rplI | Large subunit ribosomal protein L9 | + | |
| rplJ | Large subunit ribosomal protein L10 | + | |
| rplK | Large subunit ribosomal protein L11 | RA-CH-1 | |
| rplN | Large subunit ribosomal protein L14 | + | |
| rplO | Large subunit ribosomal protein L15 | + | |
| rplP | Large subunit ribosomal protein L16 | RA-CH-2, CH3, ATCC11845 | |
| rplQ | Large subunit ribosomal protein L17 | + | |
| rplR | Large subunit ribosomal protein L18 | + | |
| rplS | Large subunit ribosomal protein L19 | + | |
| rplU | Large subunit ribosomal protein L21 | + | |
| rplV | Large subunit ribosomal protein L22 | + | |
| rplX | Large subunit ribosomal protein L24 | + | |
| rpsA | Small subunit ribosomal protein S1 | + | |
| rpsB | Small subunit ribosomal protein S2 | + | |
| Ribosome | rpsC | Small subunit ribosomal protein S3 | + |
| rpsD | Small subunit ribosomal protein S4 | + | |
| rpsE | Small subunit ribosomal protein S5 | RA-CH-1, CH3 | |
| rpsG | Small subunit ribosomal protein S7 | + | |
| rpsH | Small subunit ribosomal protein S8 | Except RA17, RA-GD | |
| rpsI | Small subunit ribosomal protein S9 | + | |
| rpsK | Small subunit ribosomal protein S11 | + | |
| rpsO | Small subunit ribosomal protein S15 | + | |
| rpsP | Small subunit ribosomal protein S16 | CH3 | |
| rpsR | Small subunit ribosomal protein S18 | + | |
| Elongation factor | tuf | Elongation factor Tu | + |
| fusA | Elongation factor G | + | |
| tsf | Elongation factor Ts | + | |
| Chaperone | dnaK | Molecular chaperone DnaK | Except CH3 |
| groEL | Chaperonin GroEL | + | |
| tig | Trigger factor | + | |
| Enzymes | acnA | Aconitate hydratase | + |
| sucC | Succinyl-CoA synthetase β subunit | + | |
| sucD | Succinyl-CoA synthetase α subunit | + | |
| mdh | Malate dehydrogenase | + | |
| gapA | Glyceraldehyde 3-phosphate dehydrogenase | + | |
| ccoP | Cytochrome c oxidase cbb3-type subunit III | + | |
| ccp | Cytochrome c peroxidase | + | |
| atpA | F-type H+-transporting ATPase subunit α | RA-CH-1, RA-CH-2, RA17, ATCC11845 | |
| atpF | F-type H+-transporting ATPase subunit b | Except CH3 | |
| pncA | Nicotinamidase/pyrazinamidase | + | |
| ndk | Nucleoside-diphosphate kinase | + | |
| tlpA | Alkyl hydroperoxide reductase/thiol specific antioxidant/mal allergen | + | |
| ppiA | Peptidyl-prolyl isomerase | + | |
| dsrO | Molybdopterin-containing oxidoreductase | RA-CH-1 | |
| katE | Catalase | RA153, ATCC11845, Yb2, RA-SG | |
| ahpC | Peroxiredoxin | Except RA153, RA17 | |
| sdhB | Succinate dehydrogenase/fumarate reductase | RA153, RA17, RA-SG, RA-YM | |
| eno | Enolase | + | |
| Enzymes | nrfA | Nitrite reductase | RA-CH-2, RA153, RA17, RA-GD |
| dam | DNA adenine methylase | RA17, ATCC11845, RA-GD | |
| tatD | TatD DNase family protein | RA-CH-1 | |
| pabC | 4-Amino-4-deoxychorismate lyase | RA-JLLY | |
| ald | Alanine dehydrogenase | RA-JLLY | |
| ribBA | 3,4-Dihydroxy 2-butanone 4-phosphate synthase | RA-JLLY | |
| - | Peptidase s8 and s53 subtilisin kexin sedolisin | + | |
| - | Peptidase s46 | RA17 | |
| - | Putative FAD dependent oxidoreductase | RA17 | |
| - | Septum formation initiator | RA17 | |
| - | Serine protease | ATCC11845 | |
| - | Nodulation protein X acyltransferase 3 | ATCC11845 | |
| Binding protein | - | Cyclic nucleotide-binding protein | Except RA17 |
| Transport protein | arac | Transcriptional regulator | RA17 |
| Apoptosis protein | cys | Cytochrome c | Except RA17, RA-JLLY |
| Structure protein | gldl | Gliding motility protein gldl | RA17, ATCC11845 |
| ompH | Outer membrane protein | + | |
| ompa/motb | ompa/motb domain-containing protein | + | |
| ftnA | Ferritin | RA153, ATCC11845, Yb2, RCAD0122 | |
| hinT | Histidine triad (HIT) family protein | CH3 | |
| - | Phosphate-selective porin o and p protein | RA-CH-1 |
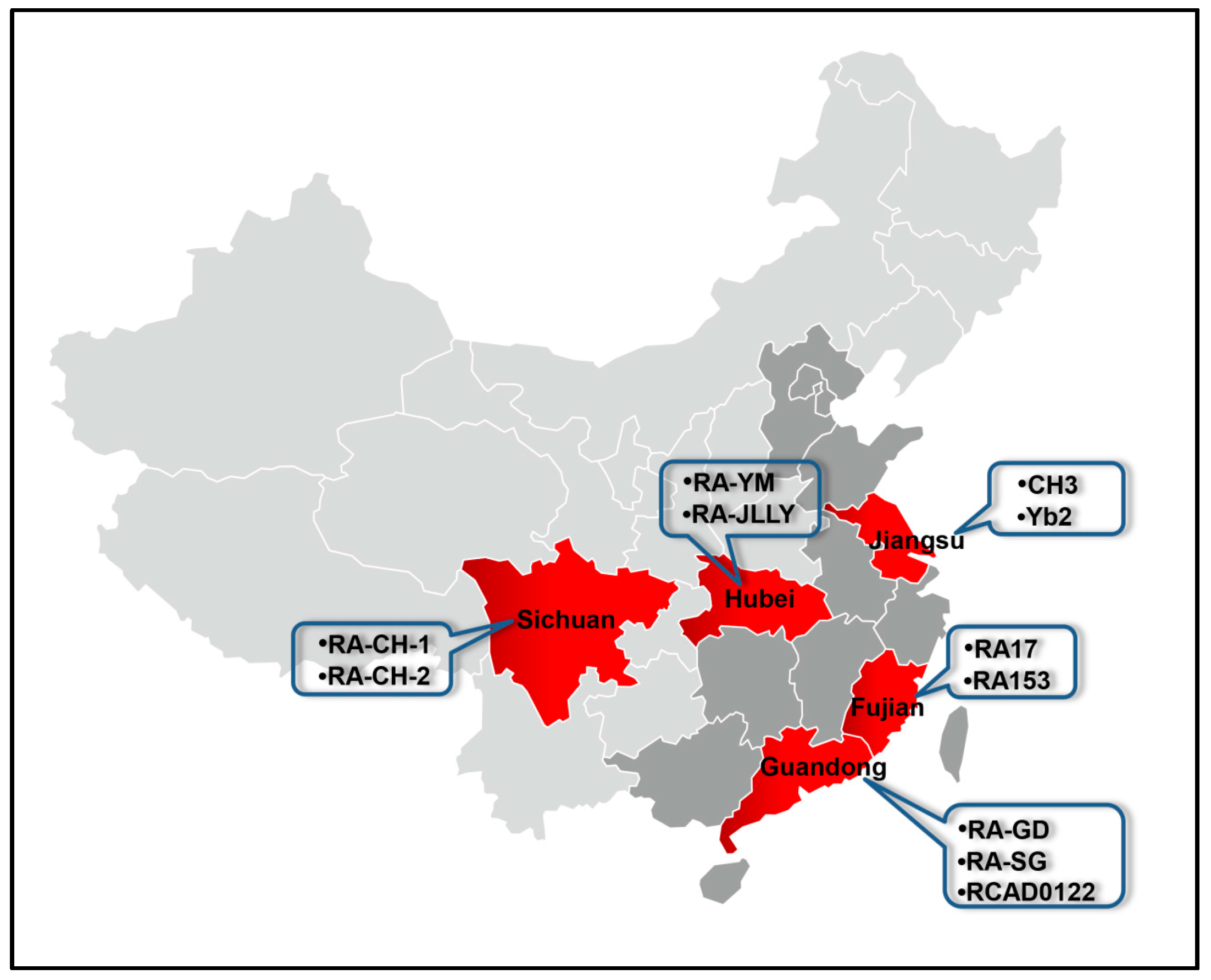
| Strain | Serotype | Geographic Location | Accession No. | CDS | CDS (>300 bp) | Reference |
|---|---|---|---|---|---|---|
| ATCC11845 | 6 | USA | CP003388. | 1941 | 1764 | [42,43] |
| RA-CH-1 | 1 | Sichuan | CP003787 | 2187 | 1953 | |
| CH3 | 1 | Jiangsu | CP006649 | 2181 | 1916 | [44] |
| RA-YM | 1 | Hubei | AENH00000000 | 2010 | 1796 | [45] |
| RA-CH-2 | 2 | Sichuan | CP004020 | 2044 | 1844 | [46] |
| RA-GD | 2 | Guangdong | CP002562 | 1985 | 1815 | [47] |
| Yb2 | 2 | Jiangsu | CP007204 | 2021 | 1877 | [48] |
| RA153 | 2 | Fujian | CP007504 | 1919 | 1730 | |
| RA17 | ND 1 | Fujian | CP007503 | 1656 | 1613 | |
| RA-SG | ND 1 | Guangdong | ANGF00000000 | 2066 | 1838 | [49] |
| RA-JLLY | ND 1 | Hubei | LAVB01000000 | 2089 | 1858 | [50] |
| RCAD0122 | ND 1 | Guangdong | LUDU00000000 | 2149 | 1892 | [51] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zhu, D.; Ma, G.; Liu, M.; Wang, M.; Jia, R.; Chen, S.; Sun, K.; Yang, Q.; Wu, Y.; et al. Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Riemerella anatipestifer. Int. J. Mol. Sci. 2016, 17, 1304. https://doi.org/10.3390/ijms17081304
Liu J, Zhu D, Ma G, Liu M, Wang M, Jia R, Chen S, Sun K, Yang Q, Wu Y, et al. Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Riemerella anatipestifer. International Journal of Molecular Sciences. 2016; 17(8):1304. https://doi.org/10.3390/ijms17081304
Chicago/Turabian StyleLiu, Jibin, Dekang Zhu, Guangpeng Ma, Mafeng Liu, Mingshu Wang, Renyong Jia, Shun Chen, Kunfeng Sun, Qiao Yang, Ying Wu, and et al. 2016. "Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Riemerella anatipestifer" International Journal of Molecular Sciences 17, no. 8: 1304. https://doi.org/10.3390/ijms17081304