
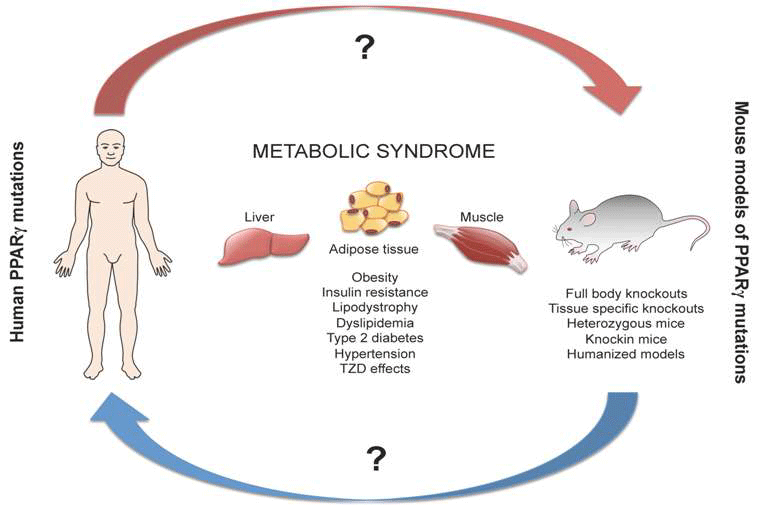
Is the Mouse a Good Model of Human PPARγ-Related Metabolic Diseases?

Abstract
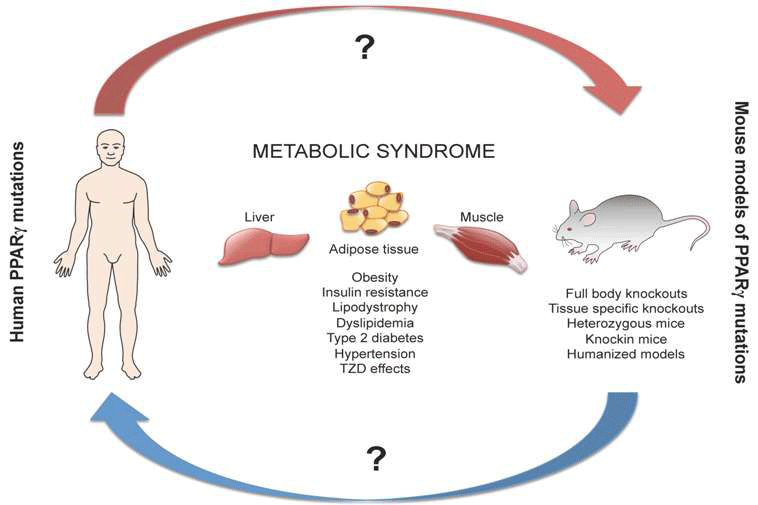
:
1. Introduction
2. Human Aspects of PPARγ in Metabolic Syndrome
2.1. PPARg Polymorphisms Related to Metabolic Traits without Lipodystrophy
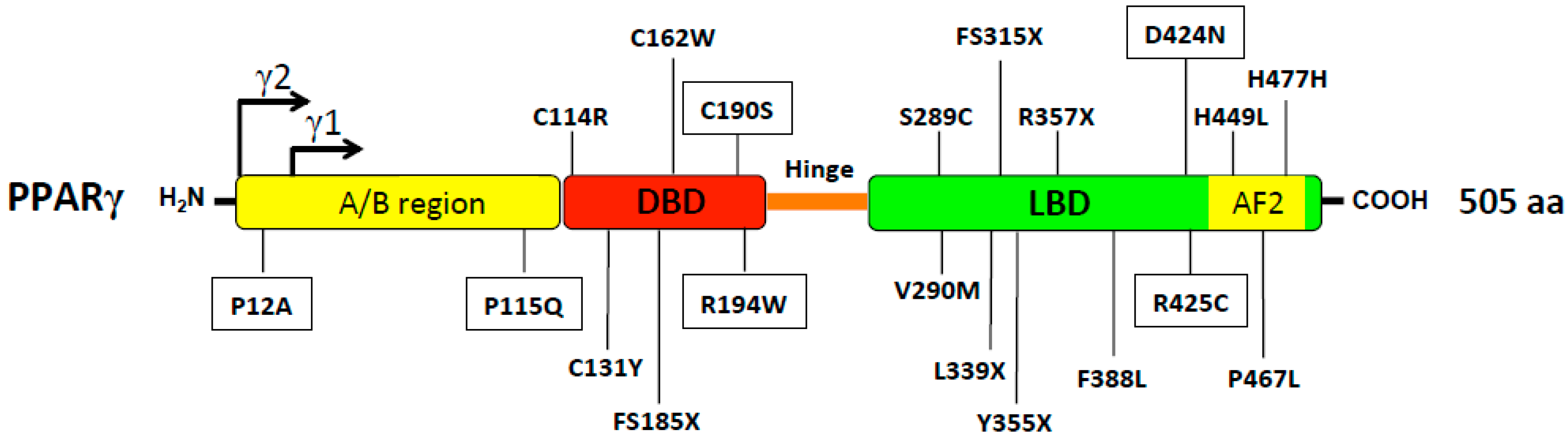
2.2. PPARg Mutations Associated with Lipodystrophy
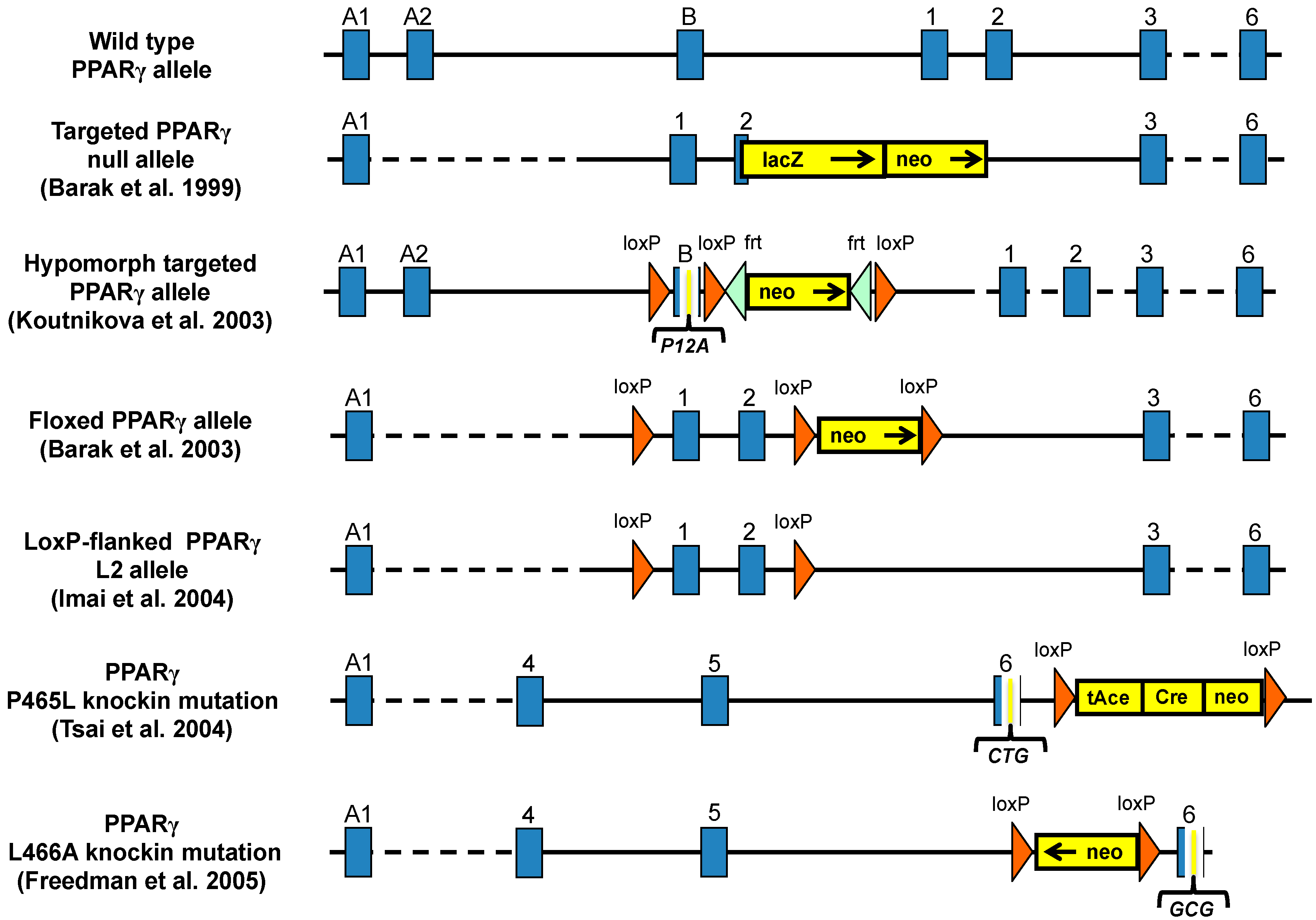
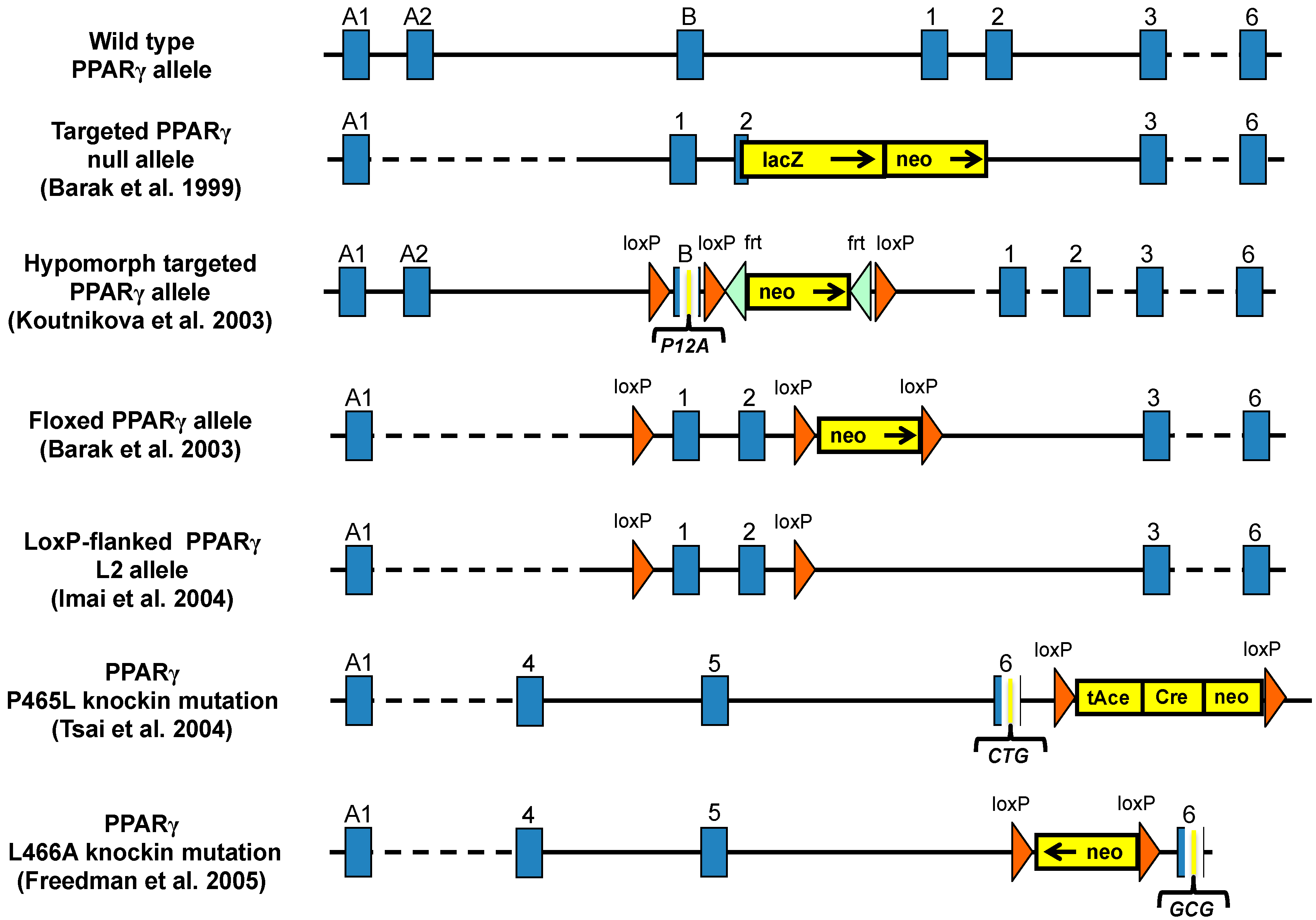
3. Mouse Models for Study the Role of PPARγ in Metabolic Diseases
3.1. PPARγ Full Body Knockout Mice
3.2. Heterozygous PPARγ Mice
3.3. Hypomorph Mouse Model
3.4. Ablation of PPARγ2 Isoform
3.5. PPARγ Mutant Mice
3.6. Tissue Specific Ablation of PPARγ
3.6.1. Adipose-Specific PPARγ Knockout
3.6.2. Muscle-Specific Ablation of PPARγ
3.6.3. Liver-Specific Disruption of PPARγ
3.6.4. PPARγ Ablation in Pancreatic Beta Cells
3.6.5. Disruption of PPARγ in Macrophages and Dendritic cells
4. Testing of Novel PPARγ Modulators in Mice
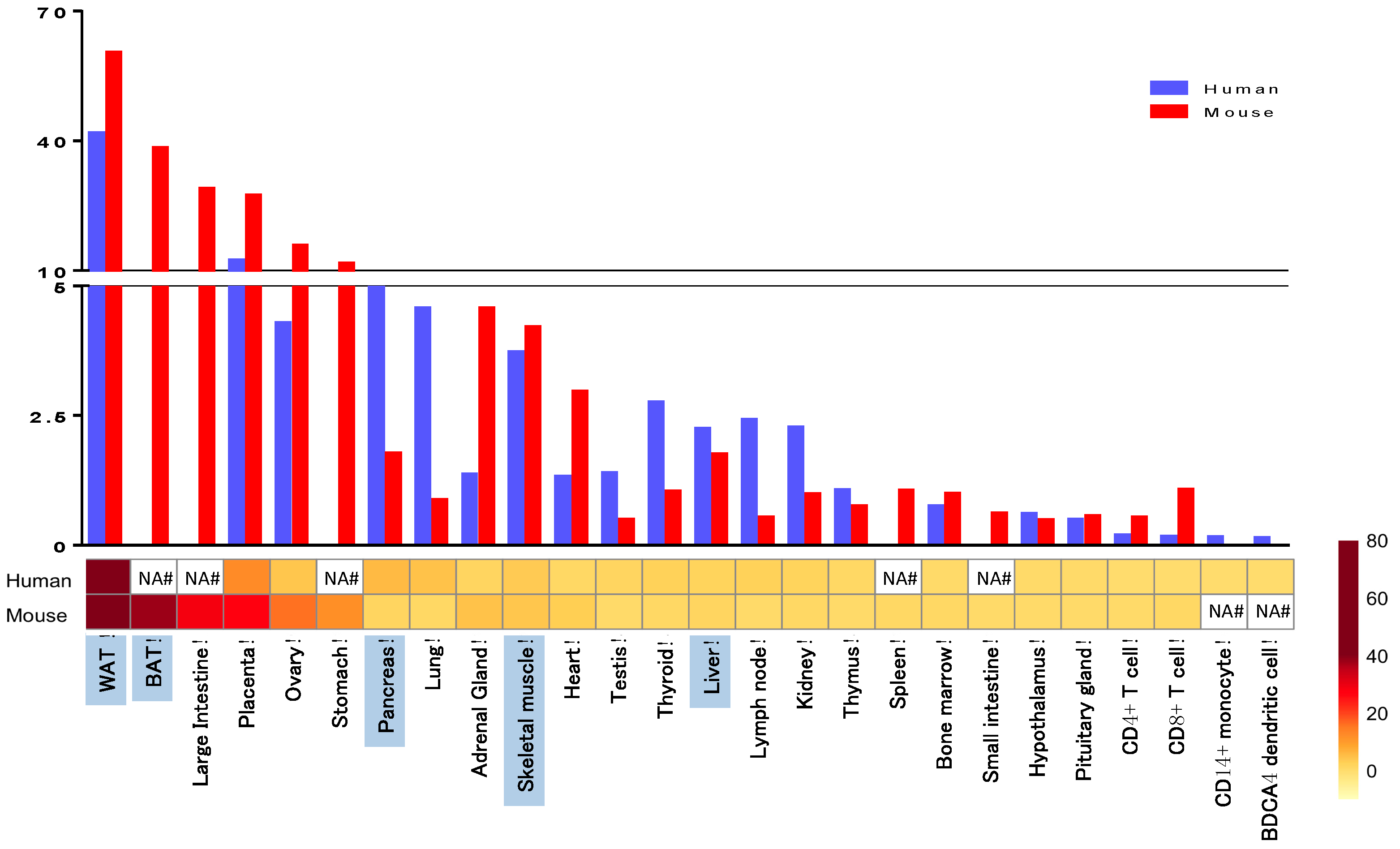
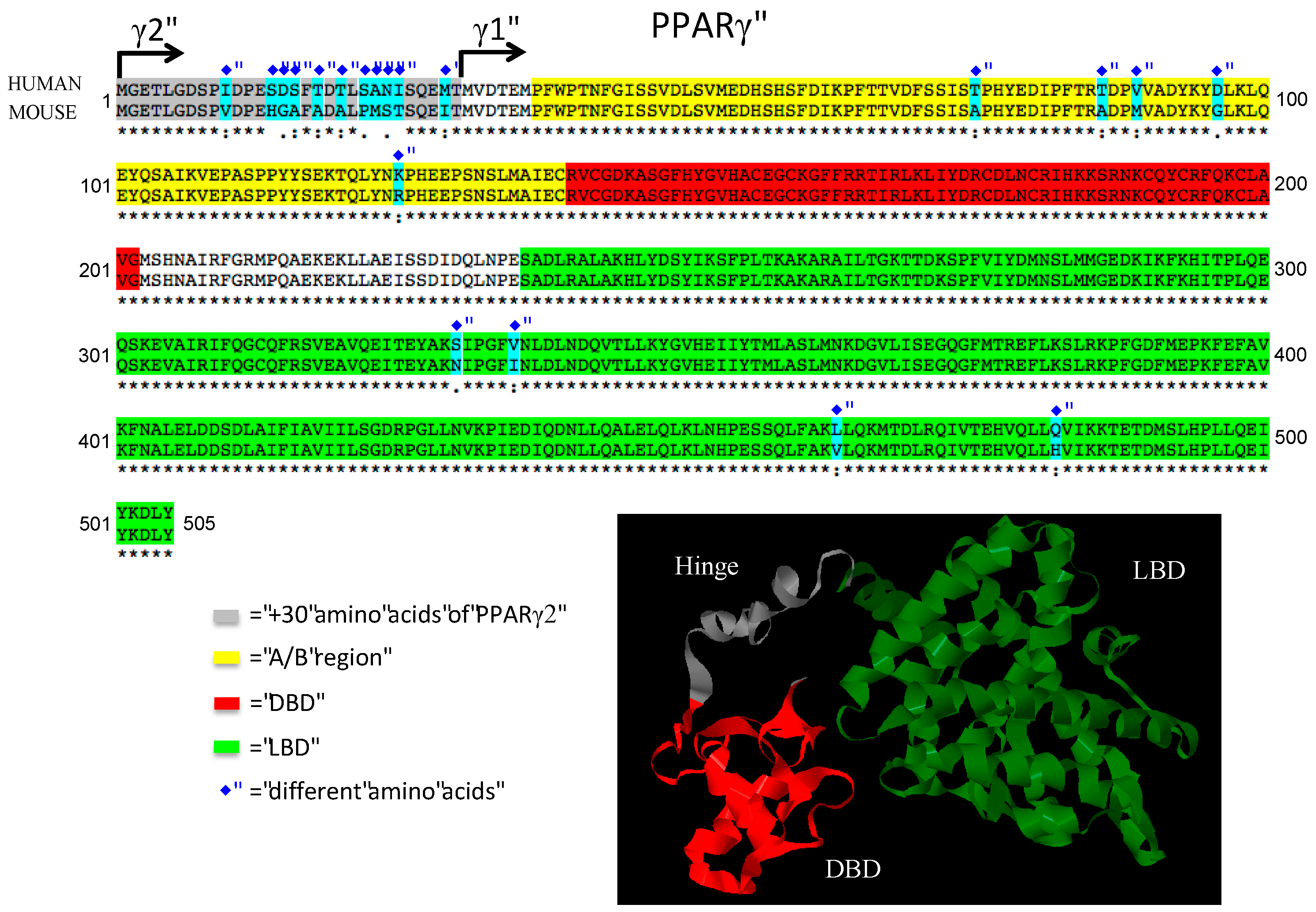
5. Comparison of Human and Mouse Findings
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the x-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of ppargamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.; Siersbaek, M.; Mandrup, S. Ppars: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Zhu, Y.; Alvares, K.; Huang, Q.; Rao, M.S.; Reddy, J.K. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J. Biol. Chem. 1993, 268, 26817–26820. [Google Scholar] [PubMed]
- Nadra, K.; Quignodon, L.; Sardella, C.; Joye, E.; Mucciolo, A.; Chrast, R.; Desvergne, B. Ppargamma in placental angiogenesis. Endocrinology 2010, 151, 4969–4981. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. Ppargamma promotes monocyte/macrophage differentiation and uptake of oxidized ldl. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized ldl regulates macrophage gene expression through ligand activation of ppargamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Szatmari, I.; Torocsik, D.; Agostini, M.; Nagy, T.; Gurnell, M.; Barta, E.; Chatterjee, K.; Nagy, L. Ppargamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood 2007, 110, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Temelkova-Kurktschiev, T.; Hanefeld, M.; Chinetti, G.; Zawadzki, C.; Haulon, S.; Kubaszek, A.; Koehler, C.; Leonhardt, W.; Staels, B.; Laakso, M. Ala12ala genotype of the peroxisome proliferator-activated receptor gamma2 protects against atherosclerosis. J. Clin. Endocrinol. Metab. 2004, 89, 4238–4242. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact ppar-gamma-rxr- nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Van Beekum, O.; Fleskens, V.; Kalkhoven, E. Posttranslational modifications of ppar-gamma: Fine-tuning the metabolic master regulator. Obesity 2009, 17, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. Ppargamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Degirolamo, C.; Mariani-Costantini, R.; Palasciano, G.; Moschetta, A. Lipid-sensing nuclear receptors in the pathophysiology and treatment of the metabolic syndrome. WIREs Syst. Biol. Med. 2011, 3, 562–587. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.; Henry, R.R. Thiazolidinedione safety. Expert. Opin. Drug Saf. 2012, 11, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, S.; Auwerx, J.; Argmann, C.A. Ppargamma in human and mouse physiology. Biochim. Biophys. Acta 2007, 1771, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamaki, J.; Mykkanen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A pro12ala substitution in ppargamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Gouda, H.N.; Sagoo, G.S.; Harding, A.H.; Yates, J.; Sandhu, M.S.; Higgins, J.P. The association between the peroxisome proliferator-activated receptor-gamma2 (pparg2) pro12ala gene variant and type 2 diabetes mellitus: A huge review and meta-analysis. Am. J. Epidemiol. 2010, 171, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Tonjes, A.; Stumvoll, M. The role of the pro12ala polymorphism in peroxisome proliferator-activated receptor gamma in diabetes risk. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Kreuzer, J.; Hamann, A.; Nawroth, P.P.; Dugi, K.A. The proline 12 alanine substitution in the peroxisome proliferator-activated receptor-gamma2 gene is associated with lower lipoprotein lipase activity in vivo. Diabetes 2002, 51, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Galbete, C.; Toledo, E.; Martinez-Gonzalez, M.A.; Martinez, J.A.; Guillen-Grima, F.; Marti, A. Pro12ala variant of the pparg2 gene increases body mass index: An updated meta-analysis encompassing 49,092 subjects. Obesity 2013, 21, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cook, N.R.; Cheng, S.; Erlich, H.A.; Lindpaintner, K.; Plutzky, J.; Zee, R.Y. Alanine for proline substitution in the peroxisome proliferator-activated receptor gamma-2 (pparg2) gene and the risk of incident myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.D.; Braund, P.S.; Burton, P.R.; Thompson, J.R.; Steeds, R.; Channer, K.; Cheng, S.; Lindpaintner, K.; Samani, N.J. Genotypes and haplotypes predisposing to myocardial infarction: A multilocus case-control study. Eur. Heart J. 2004, 25, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lou, Y.; Jin, W.; Liu, Y.; Lu, L.; Lu, G. The pro12ala polymorphism in the peroxisome proliferator-activated receptor gamma-2 gene (ppargamma2) is associated with increased risk of coronary artery disease: A meta-analysis. PLoS ONE 2012, 7, e53105. [Google Scholar] [CrossRef]
- Yang, Y.; Chan, L. Monogenic diabetes: What it teaches us on the common forms of type 1 and type 2 diabetes. Endocr. Rev. 2016, 37, 190–222. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.K.; LaValley, M.; Liu, C.T.; Rice, K.; An, P.; Liu, Y.; Miljkovic, I.; Rasmussen-Torvik, L.; Harris, T.B.; Province, M.A.; et al. Meta-analysis of gene-environment interaction: Joint estimation of snp and snp x environment regression coefficients. Genet. Epidemiol. 2011, 35, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, E.M.; Candido, A.P.; Castro, I.M.; Bastos, A.Q.; Machado-Coelho, G.L.; Freitas, R.N. Igf2, lepr, pomc, pparg, and ppargc1 gene variants are associated with obesity-related risk phenotypes in brazilian children and adolescents. Braz. J. Med. Biol. Res. 2015, 48, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, T.J.; Lin, E. The pro12ala polymorphism in the peroxisome proliferator-activated receptor gamma (pparg) gene in relation to obesity and metabolic phenotypes in a taiwanese population. Endocrine 2015, 48, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Dubinina, I.A.; Chistiakov, D.A.; Eremina, I.A.; Brovkin, A.N.; Zilberman, L.I.; Nikitin, A.G.; Kuraeva, T.L.; Nosikov, V.V.; Peterkova, V.A.; Dedov, II. Studying progression from glucose intolerance to type 2 diabetes in obese children. Diabetes Metab. Syndr. 2014, 8, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Kitamoto, A.; Kitamoto, T.; Mizusawa, S.; Teranishi, H.; So, R.; Matsuo, T.; Nakata, Y.; Hyogo, H.; Ochi, H.; et al. Association between type 2 diabetes genetic susceptibility loci and visceral and subcutaneous fat area as determined by computed tomography. J. Hum. Genet. 2012, 57, 305–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghera, D.K.; Demirci, F.Y.; Been, L.; Ortega, L.; Ralhan, S.; Wander, G.S.; Mehra, N.K.; Singh, J.; Aston, C.E.; Mulvihill, J.J.; et al. Pparg and adipoq gene polymorphisms increase type 2 diabetes mellitus risk in asian indian sikhs: Pro12ala still remains as the strongest predictor. Metabolism 2010, 59, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhang, R.; Wang, C.; Wang, J.; Ma, X.; Lu, J.; Qin, W.; Hou, X.; Wang, C.; Bao, Y.; et al. Pparg, kcnj11, cdkal1, cdkn2a-cdkn2b, ide-kif11-hhex, igf2bp2 and slc30a8 are associated with type 2 diabetes in a chinese population. PLoS ONE 2009, 4, e7643. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Potapov, V.A.; Khodirev, D.S.; Shamkhalova, M.S.; Shestakova, M.V.; Nosikov, V.V. The ppargamma pro12ala variant is associated with insulin sensitivity in russian normoglycaemic and type 2 diabetic subjects. Diab. Vasc. Dis. Res. 2010, 7, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Nakata, Y.; Katayama, Y.; Iemitsu, M.; Maeda, S.; Okura, T.; Kim, M.K.; Ohkubo, H.; Hotta, K.; Tanaka, K. Pparg genotype accounts for part of individual variation in body weight reduction in response to calorie restriction. Obesity 2009, 17, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.L.; Fingerlin, T.E.; Moss, L.K.; Barmada, M.M.; Ferrell, R.E.; Norris, J.M. Association of the peroxisome proliferator-activated receptor gamma gene with type 2 diabetes mellitus varies by physical activity among non-hispanic whites from colorado. Metabolism 2007, 56, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Ruchat, S.M.; Rankinen, T.; Weisnagel, S.J.; Rice, T.; Rao, D.C.; Bergman, R.N.; Bouchard, C.; Perusse, L. Improvements in glucose homeostasis in response to regular exercise are influenced by the pparg pro12ala variant: Results from the heritage family study. Diabetologia 2010, 53, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, A.; O'Dell, S.D.; Frost, G.S.; Griffin, B.A.; Lovegrove, J.A.; Jebb, S.A.; Sanders, T.A.; investigators, R.S. Interaction of pparg pro12ala with dietary fat influences plasma lipids in subjects at cardiometabolic risk. J. Lipid. Res. 2011, 52, 2298–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glamoclija, U.; Jevric-Causevic, A. Genetic polymorphisms in diabetes: Influence on therapy with oral antidiabetics. Acta Pharm. 2010, 60, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Chen, E.R.; Rajapurkar, S.R.; Safabakhsh, P.; Marinis, J.M.; Dispirito, J.R.; Emmett, M.J.; Briggs, E.R.; Fang, B.; Everett, L.J.; et al. Genetic variation determines ppargamma function and anti-diabetic drug response in vivo. Cell 2015, 162, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human ppargamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [PubMed]
- Black, M.H.; Wu, J.; Takayanagi, M.; Wang, N.; Taylor, K.D.; Haritunians, T.; Trigo, E.; Lawrence, J.M.; Watanabe, R.M.; Buchanan, T.A.; et al. Variation in pparg is associated with longitudinal change in insulin resistance in mexican americans at risk for type 2 diabetes. J. Clin. Endocrinol. Metab. 2015, 100, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz-Aydogan, H.; Kurnaz, O.; Kucukhuseyin, O.; Akadam-Teker, B.; Kurt, O.; Eronat, A.P.; Tekeli, A.; Bugra, Z.; Ozturk, O. Different effects of ppara, pparg and apoe snps on serum lipids in patients with coronary heart disease based on the presence of diabetes. Gene 2013, 523, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz-Aydogan, H.; Kurnaz, O.; Kurt, O.; Akadam-Teker, B.; Kucukhuseyin, O.; Tekeli, A.; Isbir, T. Effects of the pparg p12a and c161t gene variants on serum lipids in coronary heart disease patients with and without type 2 diabetes. Mol. Cell. Biochem. 2011, 358, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Oladi, M.; Nohtani, M.; Avan, A.; Mirhafez, S.R.; Tajbakhsh, A.; Ghasemi, F.; Asadi, A.; Elahdadi Salmani, M.; Mohammadi, A.; Hoseinzadeh, L.; et al. Impact of the c1431t polymorphism of the peroxisome proliferator activated receptor-gamma (ppar-gamma) gene on fasted serum lipid levels in patients with coronary artery disease. Ann. Nutr. Metab. 2015, 66, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Capaccio, D.; Ciccodicola, A.; Sabatino, L.; Casamassimi, A.; Pancione, M.; Fucci, A.; Febbraro, A.; Merlino, A.; Graziano, G.; Colantuoni, V. A novel germline mutation in peroxisome proliferator-activated receptor gamma gene associated with large intestine polyp formation and dyslipidemia. Biochim. Biophys. Acta 2010, 1802, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Demir, T.; Onay, H.; Savage, D.B.; Temeloglu, E.; Uzum, A.K.; Kadioglu, P.; Altay, C.; Ozen, S.; Demir, L.; Cavdar, U.; et al. Familial partial lipodystrophy linked to a novel peroxisome proliferator activator receptor -gamma (pparg) mutation, h449l: A comparison of people with this mutation and those with classic codon 482 lamin a/c (lmna) mutations. Diabet. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Auclair, M.; Vigouroux, C.; Boccara, F.; Capel, E.; Vigeral, C.; Guerci, B.; Lascols, O.; Capeau, J.; Caron-Debarle, M. Peroxisome proliferator-activated receptor-gamma mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Miehle, K.; Porrmann, J.; Mitter, D.; Stumvoll, M.; Glaser, C.; Fasshauer, M.; Hoffmann, K. Novel peroxisome proliferator-activated receptor gamma mutation in a family with familial partial lipodystrophy type 3. Clin. Endocrinol. 2016, 84, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Dyment, D.A.; Gibson, W.T.; Huang, L.; Bassyouni, H.; Hegele, R.A.; Innes, A.M. Biallelic mutations at pparg cause a congenital, generalized lipodystrophy similar to the berardinelli-seip syndrome. Eur. J. Med. Genet. 2014, 57, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Niu, T.; Ma, Y.; You, N.C.; Song, Y.; Sobel, E.M.; Hsu, Y.H.; Balasubramanian, R.; Qiao, Y.; Tinker, L.; et al. Common genetic variants in peroxisome proliferator-activated receptor-gamma (pparg) and type 2 diabetes risk among women's health initiative postmenopausal women. J. Clin. Endocrinol. Metab. 2013, 98, E600–E604. [Google Scholar] [CrossRef] [PubMed]
- Majithia, A.R.; Flannick, J.; Shahinian, P.; Guo, M.; Bray, M.A.; Fontanillas, P.; Gabriel, S.B.; Go, T.D.C.; Project, N.J.F.A.S.; Consortium, S.T.D.; et al. Rare variants in pparg with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 13127–13132. [Google Scholar] [CrossRef] [PubMed]
- Asselbergs, F.W.; Guo, Y.; van Iperen, E.P.; Sivapalaratnam, S.; Tragante, V.; Lanktree, M.B.; Lange, L.A.; Almoguera, B.; Appelman, Y.E.; Barnard, J.; et al. Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am. J. Hum. Genet. 2012, 91, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, L.; Attanasio, M.; Lucarini, L.; Sofi, F.; Marcucci, R.; Giglioli, C.; Valente, S.; Gensini, G.; Abbate, R.; Pepe, G. Ppargamma promoter polymorphisms and acute coronary syndrome. Atherosclerosis 2009, 205, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, R.; Bie, L.; Zhao, D.; Huang, C.; Hong, J. Association of the variants in the pparg gene and serum lipid levels: A meta-analysis of 74 studies. J. Cell. Mol. Med. 2015, 19, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Kokosar, M.; Benrick, A.; Perfilyev, A.; Fornes, R.; Nilsson, E.; Maliqueo, M.; Behre, C.J.; Sazonova, A.; Ohlsson, C.; Ling, C.; et al. Epigenetic and transcriptional alterations in human adipose tissue of polycystic ovary syndrome. Sci. Rep. 2016, 6, 22883. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.; Jansson, P.A.; Perfilyev, A.; Volkov, P.; Pedersen, M.; Svensson, M.K.; Poulsen, P.; Ribel-Madsen, R.; Pedersen, N.L.; Almgren, P.; et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014, 63, 2962–2976. [Google Scholar] [CrossRef] [PubMed]
- Garg, A. Lipodystrophies: Genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 2011, 96, 3313–3325. [Google Scholar] [CrossRef] [PubMed]
- Garg, A. Acquired and inherited lipodystrophies. N. Engl. J. Med. 2004, 350, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Semple, R.K.; Chatterjee, V.K.; O’Rahilly, S. Ppar gamma and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Garg, A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J. Clin. Endocrinol. Metab. 2002, 87, 408–411. [Google Scholar] [PubMed]
- Hegele, R.A.; Cao, H.; Frankowski, C.; Mathews, S.T.; Leff, T. Pparg f388l, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes 2002, 51, 3586–3590. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Tan, G.D.; Acerini, C.L.; Jebb, S.A.; Agostini, M.; Gurnell, M.; Williams, R.L.; Umpleby, A.M.; Thomas, E.L.; Bell, J.D.; et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes 2003, 52, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Ludtke, A.; Buettner, J.; Schmidt, H.H.; Worman, H.J. New pparg mutation leads to lipodystrophy and loss of protein function that is partially restored by a synthetic ligand. J. Med. Genet. 2007, 44, e88. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Schoenmakers, E.; Mitchell, C.; Szatmari, I.; Savage, D.; Smith, A.; Rajanayagam, O.; Semple, R.; Luan, J.; Bath, L.; et al. Non-DNA binding, dominant-negative, human ppargamma mutations cause lipodystrophic insulin resistance. Cell Metab. 2006, 4, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. Ppar gamma is required for placental, cardiac and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Duan, S.Z.; Ivashchenko, C.Y.; Whitesall, S.E.; D’Alecy, L.G.; Duquaine, D.C.; Brosius, F.C., 3rd; Gonzalez, F.J.; Vinson, C.; Pierre, M.A.; Milstone, D.S.; et al. Hypotension, lipodystrophy, and insulin resistance in generalized ppargamma-deficient mice rescued from embryonic lethality. J. Clin. Investig. 2007, 117, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Jia, Z.; Aoyagi, T.; McClain, D.; Mortensen, R.M.; Yang, T. Systemic ppargamma deletion impairs circadian rhythms of behavior and metabolism. PLoS ONE 2012, 7, e38117. [Google Scholar] [CrossRef]
- Hayashi, S.; Lewis, P.; Pevny, L.; McMahon, A.P. Efficient gene modulation in mouse epiblast using a sox2cre transgenic mouse strain. Mech. Dev. 2002, 119, S97–S101. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Murakami, K.; Motojima, K.; Komeda, K.; Ide, T.; Kubota, N.; Terauchi, Y.; Tobe, K.; et al. The mechanisms by which both heterozygous peroxisome proliferator-activated receptor gamma (ppargamma) deficiency and ppargamma agonist improve insulin resistance. J. Biol. Chem. 2001, 276, 41245–41254. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. Ppar gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Miles, P.D.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for ppar-gamma deficiency. J. Clin. Investig. 2000, 105, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Anghel, S.I.; Bedu, E.; Vivier, C.D.; Descombes, P.; Desvergne, B.; Wahli, W. Adipose tissue integrity as a prerequisite for systemic energy balance: A critical role for peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 29946–29957. [Google Scholar] [CrossRef] [PubMed]
- Koutnikova, H.; Cock, T.A.; Watanabe, M.; Houten, S.M.; Champy, M.F.; Dierich, A.; Auwerx, J. Compensation by the muscle limits the metabolic consequences of lipodystrophy in ppar gamma hypomorphic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 14457–14462. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; Dalla Nora, E.; Vidal-Puig, A.J. Mouse models of ppar-gamma deficiency: Dissecting ppar-gamma's role in metabolic homoeostasis. Biochem. Soc. Trans. 2005, 33, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fu, M.; Cui, T.; Xiong, C.; Xu, K.; Zhong, W.; Xiao, Y.; Floyd, D.; Liang, J.; Li, E.; et al. Selective disruption of ppargamma 2 impairs the development of adipose tissue and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2004, 101, 10703–10708. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, G.; Virtue, S.; Lelliott, C.; Boiani, R.; Campbell, M.; Christodoulides, C.; Perrin, C.; Jimenez-Linan, M.; Blount, M.; Dixon, J.; et al. The link between nutritional status and insulin sensitivity is dependent on the adipocyte-specific peroxisome proliferator-activated receptor-gamma2 isoform. Diabetes 2005, 54, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, G.; Gray, S.L.; Yetukuri, L.; Shimomura, K.; Virtue, S.; Campbell, M.; Curtis, R.K.; Jimenez-Linan, M.; Blount, M.; Yeo, G.S.; et al. Ppar gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 2007, 3, e64. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.S.; Kim, H.J.; Takahashi, N.; Kim, H.S.; Hagaman, J.R.; Kim, J.K.; Maeda, N. Hypertension and abnormal fat distribution but not insulin resistance in mice with p465l ppargamma. J. Clin. Investig. 2004, 114, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; Nora, E.D.; Grosse, J.; Manieri, M.; Stoeger, T.; Medina-Gomez, G.; Burling, K.; Wattler, S.; Russ, A.; Yeo, G.S.; et al. Leptin deficiency unmasks the deleterious effects of impaired peroxisome proliferator-activated receptor gamma function (p465l ppargamma) in mice. Diabetes 2006, 55, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.D.; Lee, E.J.; Park, Y.; Jameson, J.L. A dominant negative peroxisome proliferator-activated receptor-gamma knock-in mouse exhibits features of the metabolic syndrome. J. Biol. Chem. 2005, 280, 17118–17125. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.M.; Rhoades, B.; Shapiro, J.S.; Rich, A.S.; Kim, J.K.; Shulman, G.I.; Kaestner, K.H.; Lazar, M.A. Genetic modulation of ppargamma phosphorylation regulates insulin sensitivity. Dev. Cell 2003, 5, 657–663. [Google Scholar] [CrossRef]
- Heikkinen, S.; Argmann, C.; Feige, J.N.; Koutnikova, H.; Champy, M.F.; Dali-Youcef, N.; Schadt, E.E.; Laakso, M.; Auwerx, J. The pro12ala ppargamma2 variant determines metabolism at the gene-environment interface. Cell Metab. 2009, 9, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Huang, L.W.; Snow, K.J.; Ablamunits, V.; Hasham, M.G.; Young, T.H.; Paulk, A.C.; Richardson, J.E.; Affourtit, J.P.; Shalom-Barak, T.; et al. A mouse model of conditional lipodystrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 16627–16632. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takakuwa, R.; Marchand, S.; Dentz, E.; Bornert, J.M.; Messaddeq, N.; Wendling, O.; Mark, M.; Desvergne, B.; Wahli, W.; et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 4543–4547. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R.; Barrick, C.; Kim, K.A.; Lindner, J.; Blondeau, B.; Fujimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of ppargamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef] [PubMed]
- Sugii, S.; Olson, P.; Sears, D.D.; Saberi, M.; Atkins, A.R.; Barish, G.D.; Hong, S.H.; Castro, G.L.; Yin, Y.Q.; Nelson, M.C.; et al. Ppargamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc. Natl. Acad. Sci. USA 2009, 106, 22504–22509. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Suh, J.M.; Atkins, A.R.; Ahmadian, M.; Li, P.; Whyte, J.; He, M.; Juguilon, H.; Yin, Y.Q.; Phillips, C.T.; et al. A ppargamma-fgf1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature 2012, 485, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Berry, D.C.; Tang, W.; Graff, J.M. Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis. Cell Rep. 2014, 9, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Jiang, Y.; Graff, J.M. Emerging roles of adipose progenitor cells in tissue development, homeostasis, expansion and thermogenesis. Trend. Endocrinol. Metab. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; He, W.; Barak, Y.; Le, J.; Bandyopadhyay, G.; Olson, P.; Wilkes, J.; Evans, R.M.; Olefsky, J. Muscle-specific pparg deletion causes insulin resistance. Nat. Med. 2003, 9, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.W.; Chen, L.; Fisher, S.J.; Szanto, I.; Ristow, M.; Jozsi, A.C.; Hirshman, M.F.; Rosen, E.D.; Goodyear, L.J.; Gonzalez, F.J.; et al. Muscle-specific ppargamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J. Clin. Investig. 2003, 112, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Cha, B.S.; Ciaraldi, T.P.; Park, K.S.; Carter, L.; Mudaliar, S.R.; Henry, R.R. Impaired fatty acid metabolism in type 2 diabetic skeletal muscle cells is reversed by ppargamma agonists. Am. J. Physiol. Endocrinol. Metabol. 2005, 289, E151–E159. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Puig, A.; Jimenez-Linan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of ppar gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of ppargamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Pattou, F.; Kerr-Conte, J.; Gmyr, V.; Vandewalle, B.; Desreumaux, P.; Auwerx, J.; Schoonjans, K.; Lefebvre, J. Expression of peroxisome proliferator-activated receptor gamma (ppargamma) in normal human pancreatic islet cells. Diabetologia 2000, 43, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Welters, H.J.; McBain, S.C.; Tadayyon, M.; Scarpello, J.H.; Smith, S.A.; Morgan, N.G. Expression and functional activity of ppargamma in pancreatic beta cells. Brit. J. Pharmacol. 2004, 142, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Kim, J.W.; Kim, S.H.; Cha, J.Y.; Kim, K.S.; Ahn, Y.H. Identification and functional characterization of the peroxisomal proliferator response element in rat glut2 promoter. Diabetes 2000, 49, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Kulkarni, R.N.; Sarraf, P.; Ozcan, U.; Okada, T.; Hsu, C.H.; Eisenman, D.; Magnuson, M.A.; Gonzalez, F.J.; Kahn, C.R.; et al. Targeted elimination of peroxisome proliferator-activated receptor gamma in beta cells leads to abnormalities in islet mass without compromising glucose homeostasis. Mol. Cell. Biol. 2003, 23, 7222–7229. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Jetton, T.L.; Mortensen, R.M.; Duan, S.Z.; Peshavaria, M.; Leahy, J.L. In vivo and in vitro studies of a functional peroxisome proliferator-activated receptor gamma response element in the mouse pdx-1 promoter. J. Biol. Chem. 2008, 283, 32462–32470. [Google Scholar] [CrossRef] [PubMed]
- Vivas, Y.; Martinez-Garcia, C.; Izquierdo, A.; Garcia-Garcia, F.; Callejas, S.; Velasco, I.; Campbell, M.; Ros, M.; Dopazo, A.; Dopazo, J.; et al. Early peroxisome proliferator-activated receptor gamma regulated genes involved in expansion of pancreatic beta cell mass. BMC Med. Genom. 2011, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Welters, H.J.; El Ouaamari, A.; Kawamori, D.; Meyer, J.; Hu, J.; Smith, D.M.; Kulkarni, R.N. Rosiglitazone promotes ppargamma-dependent and -independent alterations in gene expression in mouse islets. Endocrinology 2012, 153, 4593–4599. [Google Scholar] [CrossRef] [PubMed]
- Szeles, L.; Torocsik, D.; Nagy, L. Ppargamma in immunity and inflammation: Cell types and diseases. Biochim. Biophys. Acta 2007, 1771, 1014–1030. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, I.; Rajnavolgyi, E.; Nagy, L. Ppargamma, a lipid-activated transcription factor as a regulator of dendritic cell function. Ann. N. Y. Acad. Sci. 2006, 1088, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Szanto, A.; Szatmari, I.; Szeles, L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol. Rev. 2012, 92, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.E.; Sakai, S.; Lambert, G.; Nicol, C.J.; Matsusue, K.; Pimprale, S.; Lee, Y.H.; Ricote, M.; Glass, C.K.; Brewer, H.B., Jr.; et al. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of abca1, abcg1, and apoe in macrophages and reduced cholesterol efflux. Mol. Cell. Biol. 2002, 22, 2607–2619. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific ppargamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage ppar gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. Stat6 transcription factor is a facilitator of the nuclear receptor ppargamma-regulated gene expression in macrophages and dendritic cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; You, R.; Yuan, X.; Frazier, M.V.; Porter, P.; Seryshev, A.; Hong, J.S.; Song, L.Z.; Zhang, Y.; Hilsenbeck, S.; et al. Agonistic induction of ppargamma reverses cigarette smoke-induced emphysema. J. Clin. Investig. 2014, 124, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.Z.; Usher, M.G.; Foley, E.L.t.; Milstone, D.S.; Brosius, F.C.; Mortensen, R.M. Sex dimorphic actions of rosiglitazone in generalised peroxisome proliferator-activated receptor-gamma (ppar-gamma)-deficient mice. Diabetologia 2010, 53, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, Y.; Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Nakayama, R.; Orikawa, Y.; Yoshioka, Y.; Tanaka, K.; Ueki, K.; Kadowaki, T. A novel peroxisome proliferator-activated receptor (ppar)alpha agonist and ppargamma antagonist, z-551, ameliorates high-fat diet-induced obesity and metabolic disorders in mice. J. Biol. Chem. 2015, 290, 14567–14581. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; Cesar, F.A.; de Oliveira, E.M.; Turato, W.M.; Tripodi, G.L.; Castilho, G.; Machado-Lima, A.; de Las Heras, B.; Bosca, L.; Rabello, M.M.; et al. New ppargamma partial agonist improves obesity-induced metabolic alterations and atherosclerosis in LDLr(-/-) mice. Pharmacol. Res. 2016, 104, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Li, B.; Grayson, B.E.; Matter, E.K.; Woods, S.C.; Seeley, R.J. A role for central nervous system ppar-gamma in the regulation of energy balance. Nat. Med. 2011, 17, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative epigenomic analysis of murine and human adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.F.; Jorgensen, M.; Chen, Y.; Nielsen, R.; Sandelin, A.; Mandrup, S. Cross species comparison of c/ebpalpha and ppargamma profiles in mouse and human adipocytes reveals interdependent retention of binding sites. BMC Genom. 2011, 12, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruchat, S.M.; Weisnagel, S.J.; Vohl, M.C.; Rankinen, T.; Bouchard, C.; Perusse, L. Evidence for interaction between pparg pro12ala and ppargc1a gly482ser polymorphisms in determining type 2 diabetes intermediate phenotypes in overweight subjects. Exp. Clin. Endocrinol. Diabetes 2009, 117, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of ppargamma by cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.; Zushin, P.J.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.P.; Spiegelman, B.M. An erk/cdk5 axis controls the diabetogenic actions of ppargamma. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Asterholm, I.W.; Halberg, N.; Scherer, P.E. Mouse models of lipodystrophy key reagents for the understanding of the metabolic syndrome. Drug Discov. Today Dis. Model. 2007, 4, 17–24. [Google Scholar] [CrossRef] [PubMed]
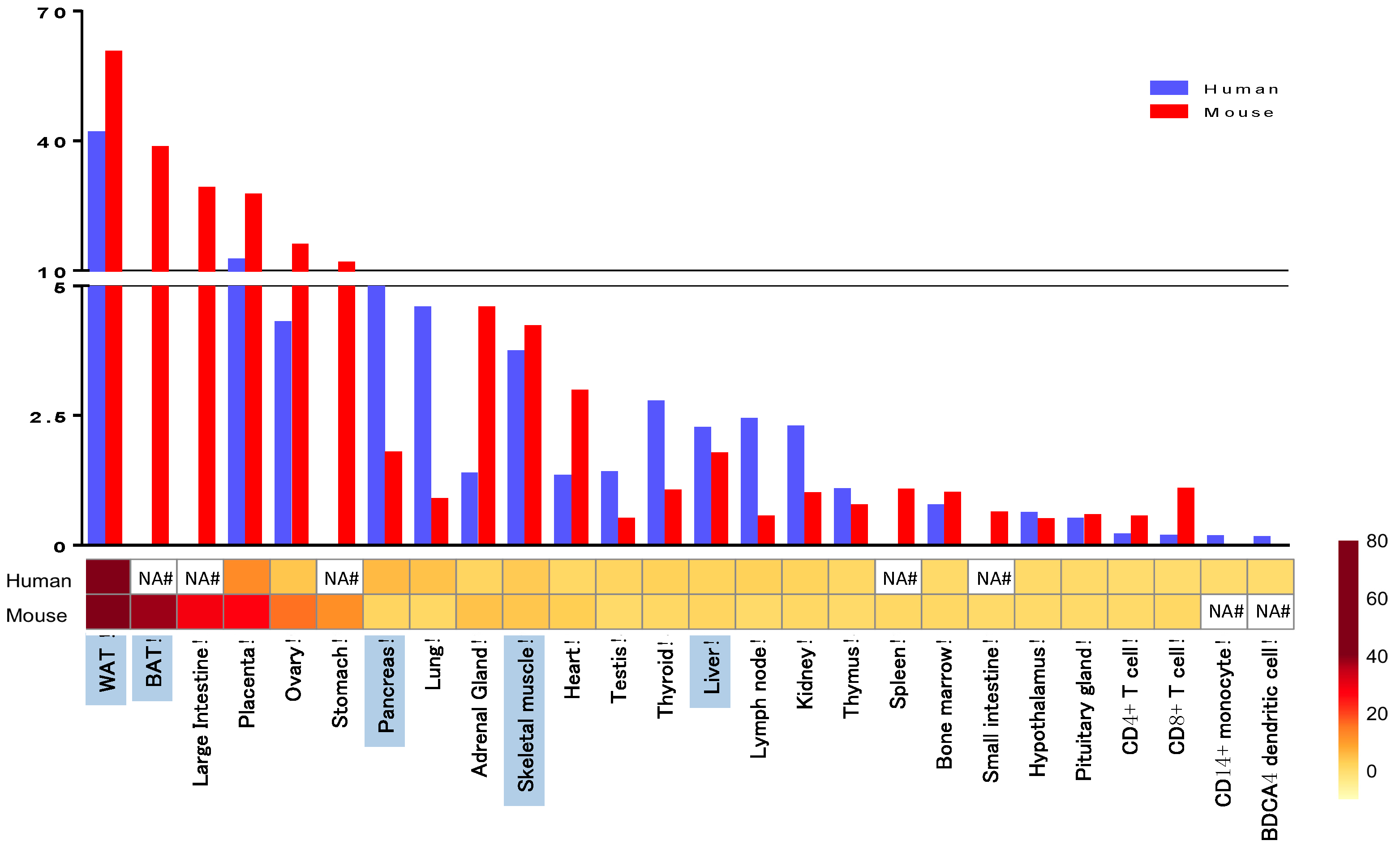
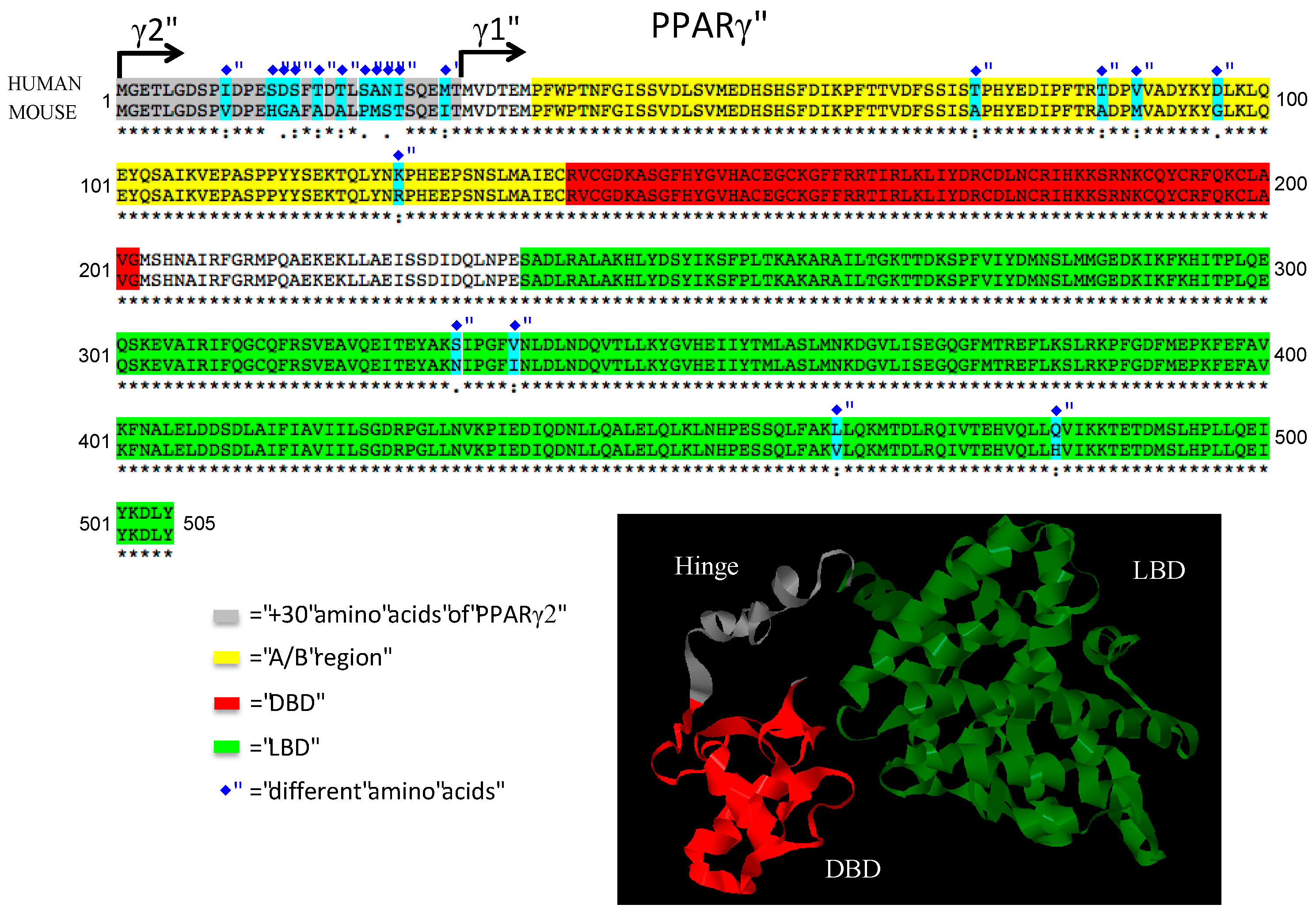
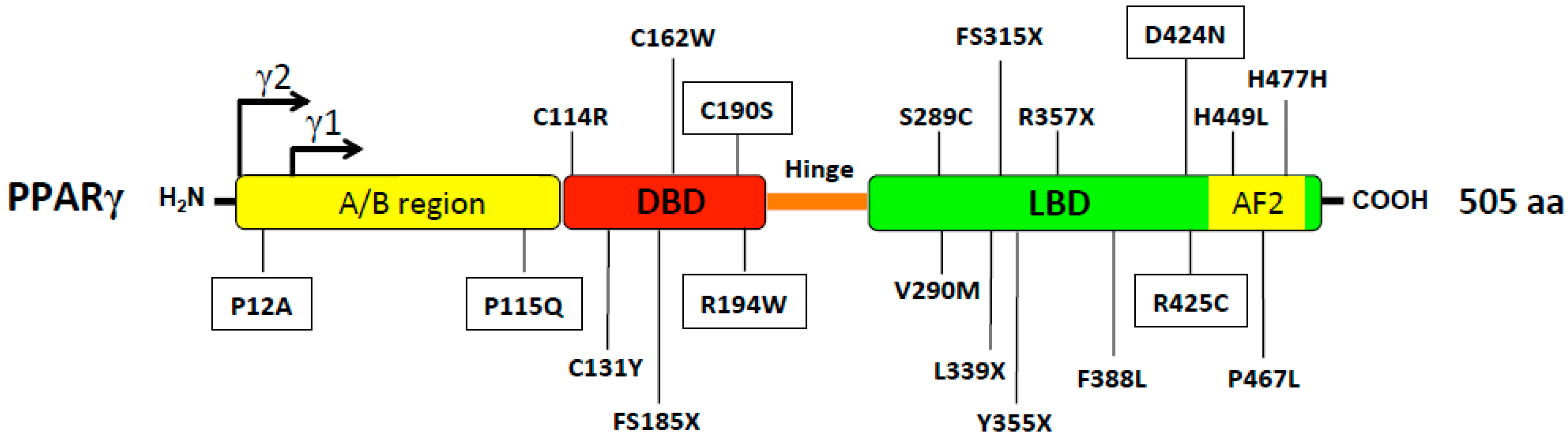

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorphism | Metabolic Traits Involved | References |
|---|---|---|
| Pro12Ala | T2D | [20,32] |
| Monogenic diabetes | [21] | |
| Higher BMI | [24,32] | |
| Altered insulin levels | [30] | |
| Insulin sensitivity | [36] | |
| BMI and insulin sensitivity in PCOS | [37] | |
| P467L V290M | Insulin resistance, liver steatosis, T2D and hypertension | [43] |
| Promoter variants polymorphism rs29722164 rs11128598 rs17793951 rs1151996 rs1175541 rs3856806 | Deterioration of B-cell function | [44] |
| V162 | Increase total cholesterol and LDL-cholesterol levels | [45] |
| C161T | CHD in patients with T2D | [46] |
| C1431T | Altered fasting serum lipids and risk factor for CHD | [47] |
| S289C | Dyslipidemia, obesity and hypertension | [48] |
| H449L | Hypertriglyceridemia, insulin resistance and hepatic steatosis, FPLD3 | [49] |
| R165T L339X | FPLD3 and severe hypertension | [50] |
| c.1040A > C | FPLD3, Diabetes Mellitus, hypertension and dyslipidemia | [51] |
| Biallelic mutation E138V and R164W | CGL, hypertriglyceridemia, diabetes mellitus, pancreatitis and renal failure | [52] |
| Features | Mouse Models | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| MORE- PG KO [69] | HET-PPARγ [72,75] | HYPO- PPARγ [76] | PPARγ2 KO [78] | Adipo PPARγ KO [88,89] | Sc.M. PPARγ KO [94,95] | Liver PPARγ KO [98] | β-cell PPARγ KO [103] | MΦ PPARγ KO [112] | |
| Obesity | No | ↓ | ↓ | No | ↓ (HFD) | ↑ (HFD) | No | No | ↑ (HFD) |
| Insuline resistance | Yes | IS | Yes | Yes (male) | unclear | Yes | Yes | No | Yes |
| Glucose tolerance | ↓ (male) | ND | ↓ | ↓ | ND | ↓ | ND | NC | ↓ (HFD) |
| Type 2 diabetes | Yes (male) | No | ND | ND | Yes | Yes | ND | No | ND |
| Lipodystrophy | Yes | No | Yes | Yes | Yes | No | No | ND | ND |
| Liver steatosis | No | No | No | No | Yes | ND | No | ND | No |
| Hypertension | hypoten. | ND | ND | ND | ND | ND | ND | ND | ND |
| Organomegaly | Yes | No | No | No | ND | Yes | No | ND | No |
| Food intake | NC | ↓ | NC | NC | ↑ (HFD) | ↓ | NC | ND | ND |
| Triglicerides | ↑ | ↓ | ↓ | NC | ↑ | ↑ | ↑ * | ND | NC |
| Free fatty acids | ↑ | ↓ | ↑ (fed) | ND | ↑ | ↑ | NC | ND | ND |
| Cholesterol | ND | ND | ND | ND | ND | ND | NC | ND | LDL ↓ |
| Glucose | ↑ | ND | ↑ (fed) | NC | NC | ↑ | ↑ * | NC | ↑ (HFD) |
| Insulin | ↑ | ↓ | ↑ | ND | ↑ | ↑ | ↑ * | NC | ↑ (HFD) |
| Leptin | ↓ | ↑ | ↓ | ↓ | ↓ | ↑ | ↑ * | ND | ↑ |
| Adiponectin | ↓ | ↑ | ↓ | ↓ | ↓ | ND | ↓ * | ND | ↓ |
| TZD effectiveness | ND | Yes | ND | Yes | partial | partial | Yes | Yes | Yes |
| Features | Human Mutants | Mouse Mutants | ||||
|---|---|---|---|---|---|---|
| P12A Mutant [20] | P467L Mutant [43,65] | F388L Mutant [64] | Biallelic E138V R164W [52] | P12A Mutant [85] | P465L Mutant [81] | |
| Obesity | Yes | No | No | No | No | No |
| Insuline resistance | Yes | Yes | Yes | Yes | IS | No |
| Glucose tolerance | ND | ↓ | ND | ND | ↑ | ↑ |
| Type 2 diabetes | Yes | Yes | Yes | Yes | No | No |
| Lipodystrophy | No | No | FPLD3 | CGL | No | redistr. |
| Liver steatosis | ND | ND | No | ND | ND | ND |
| Hypertension | ND | Yes | Yes | No | ND | Yes |
| Organomegaly | No | ND | No | Yes | No | ND |
| Food intake | ND | ND | ND | ND | NC | NC |
| Triglicerides | ↑ | ↑ | ↑ | ↑ | ↓ | NC |
| Free fatty acids | ND | ND | ND | ND | NC | NC |
| Cholesterol | ↑ | HDL ↓ | HDL ↓ | NC | ↓ | NC |
| Glucose | ↑ | ND | ↑ | ↑ | NC | NC |
| Insulin | ND | ↑ | ↑ | ↑ | NC | ↑ (HFD) |
| Leptin | ND | ND | ND | ↓ | NC | ND |
| Adiponectin | ND | ND | ND | ↓ | NC | ND |
| TZD effectiveness | ND | ND | partial | ND | partial | ND |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pap, A.; Cuaranta-Monroy, I.; Peloquin, M.; Nagy, L. Is the Mouse a Good Model of Human PPARγ-Related Metabolic Diseases? Int. J. Mol. Sci. 2016, 17, 1236. https://doi.org/10.3390/ijms17081236
Pap A, Cuaranta-Monroy I, Peloquin M, Nagy L. Is the Mouse a Good Model of Human PPARγ-Related Metabolic Diseases? International Journal of Molecular Sciences. 2016; 17(8):1236. https://doi.org/10.3390/ijms17081236
Chicago/Turabian StylePap, Attila, Ixchelt Cuaranta-Monroy, Matthew Peloquin, and Laszlo Nagy. 2016. "Is the Mouse a Good Model of Human PPARγ-Related Metabolic Diseases?" International Journal of Molecular Sciences 17, no. 8: 1236. https://doi.org/10.3390/ijms17081236