
Argon Induces Protective Effects in Cardiomyocytes during the Second Window of Preconditioning

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
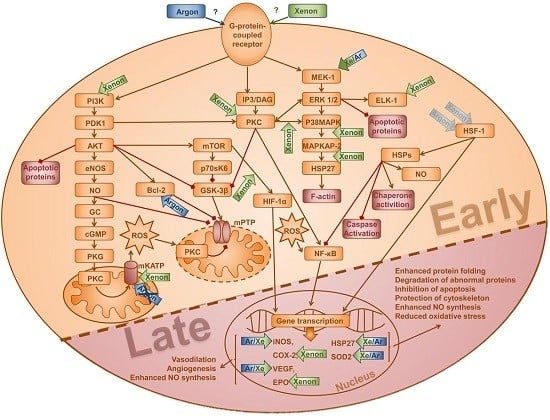
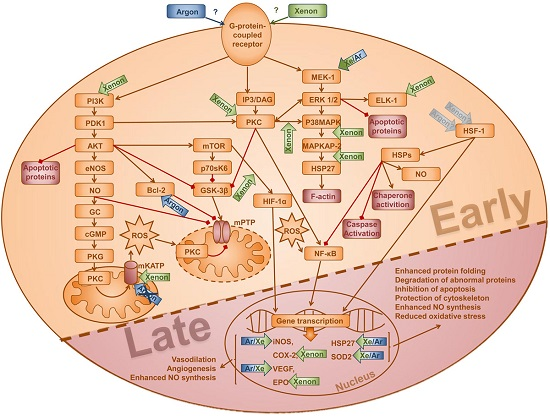
1. Introduction
2. Results
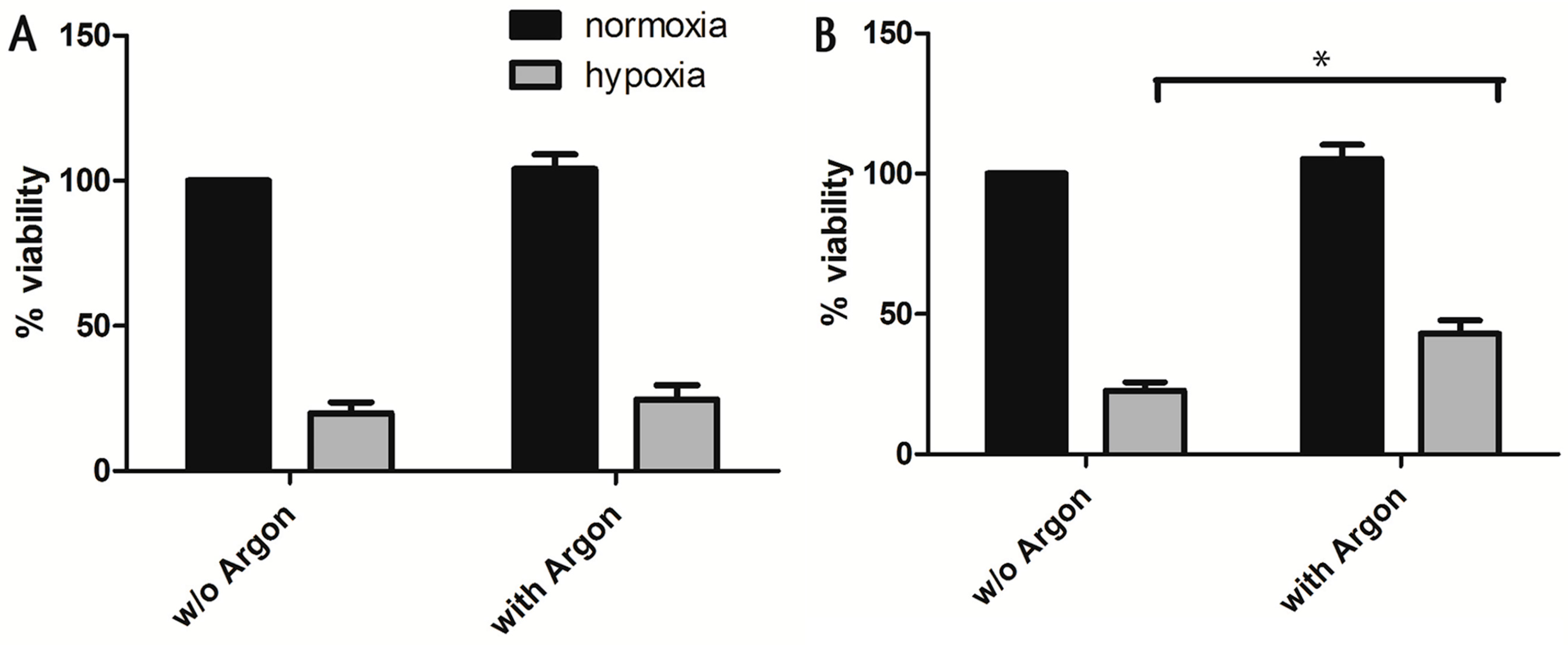
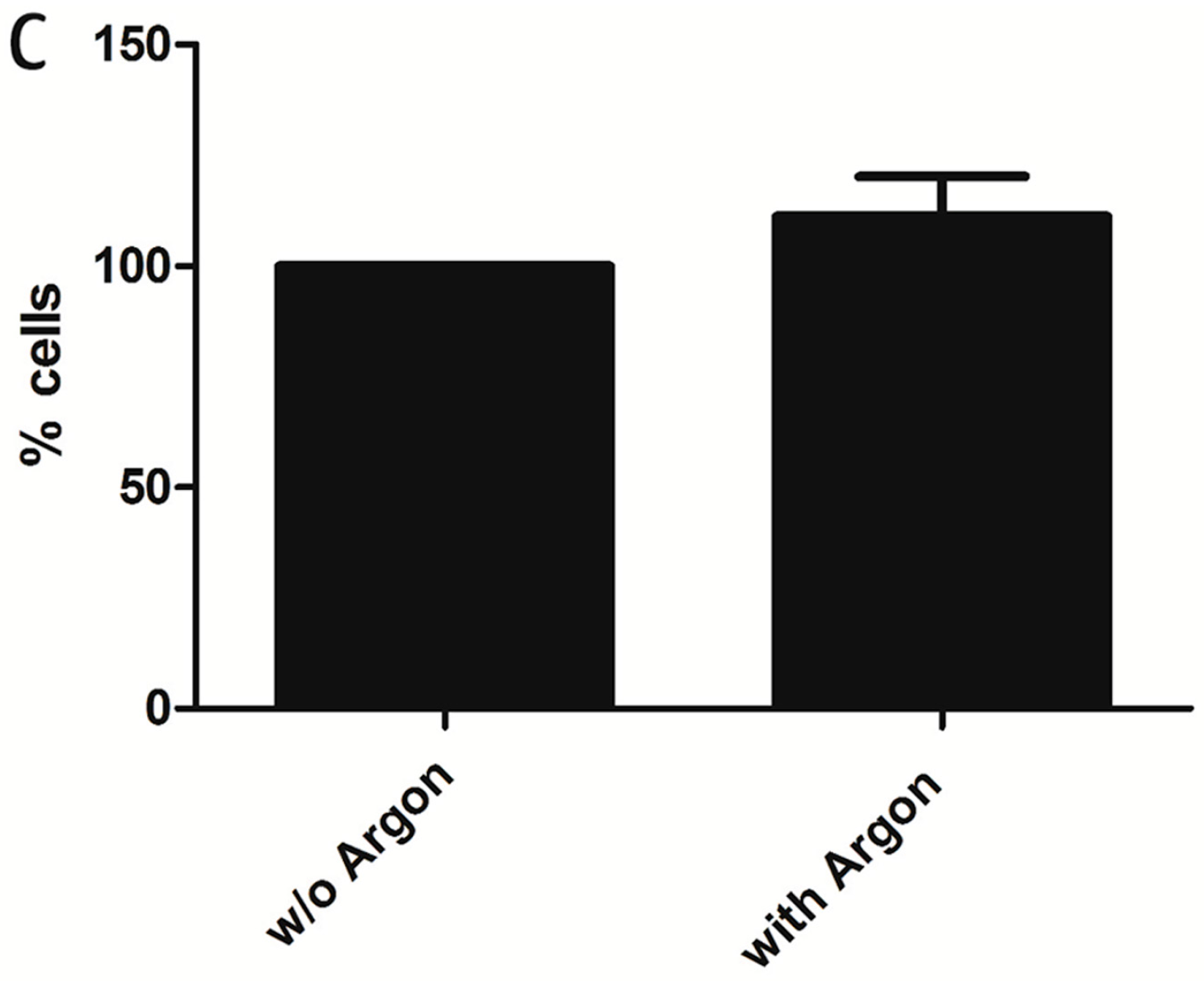
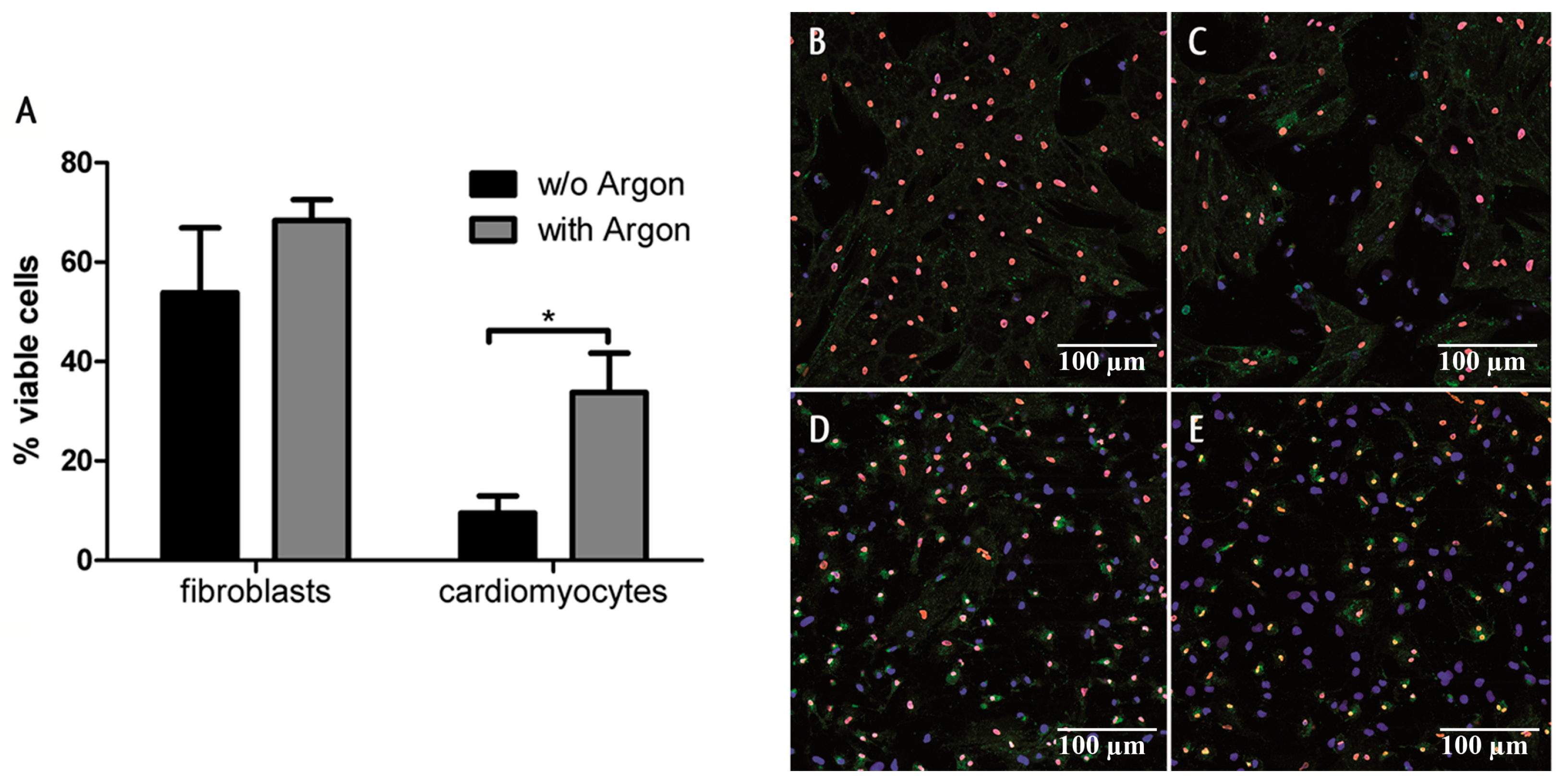
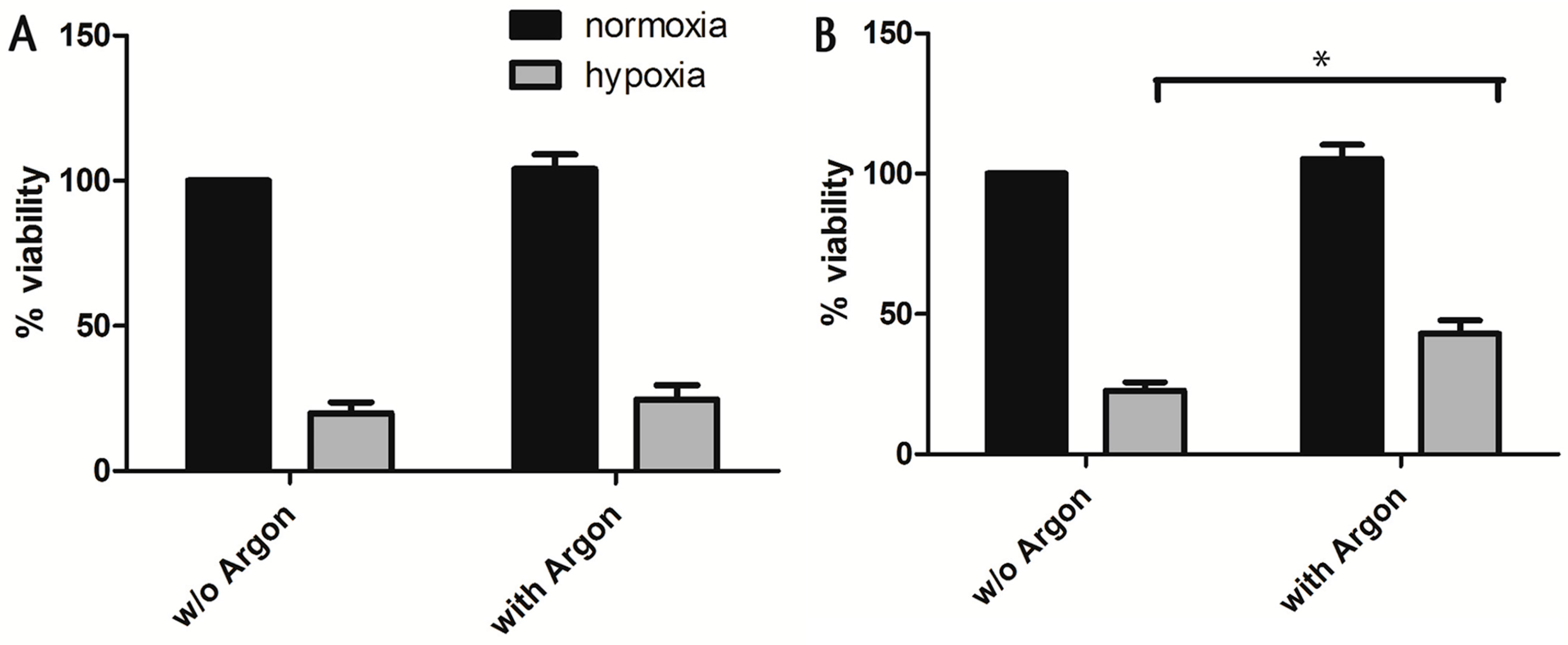
2.1. Cell Survival and Preconditioning Effects
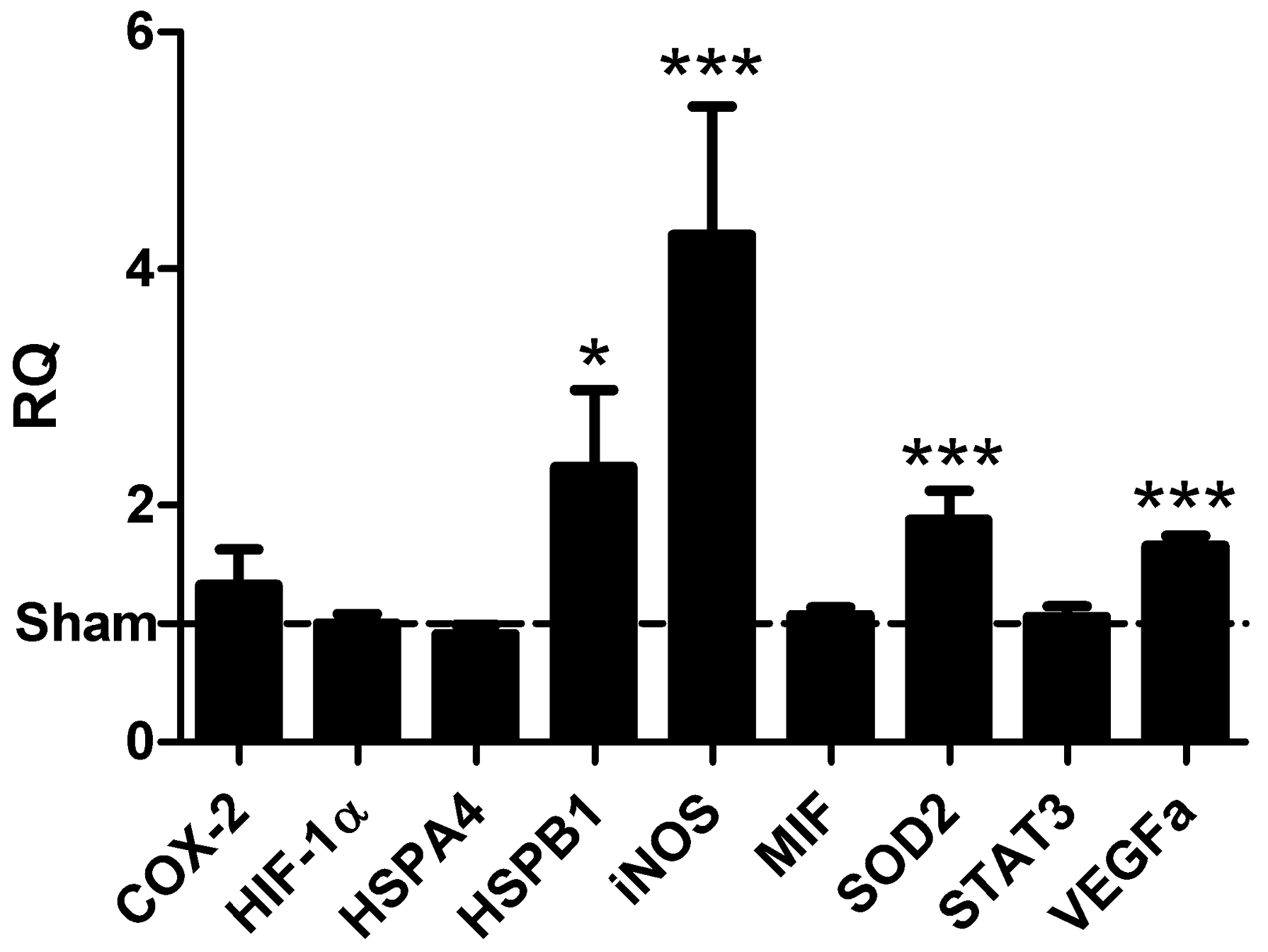
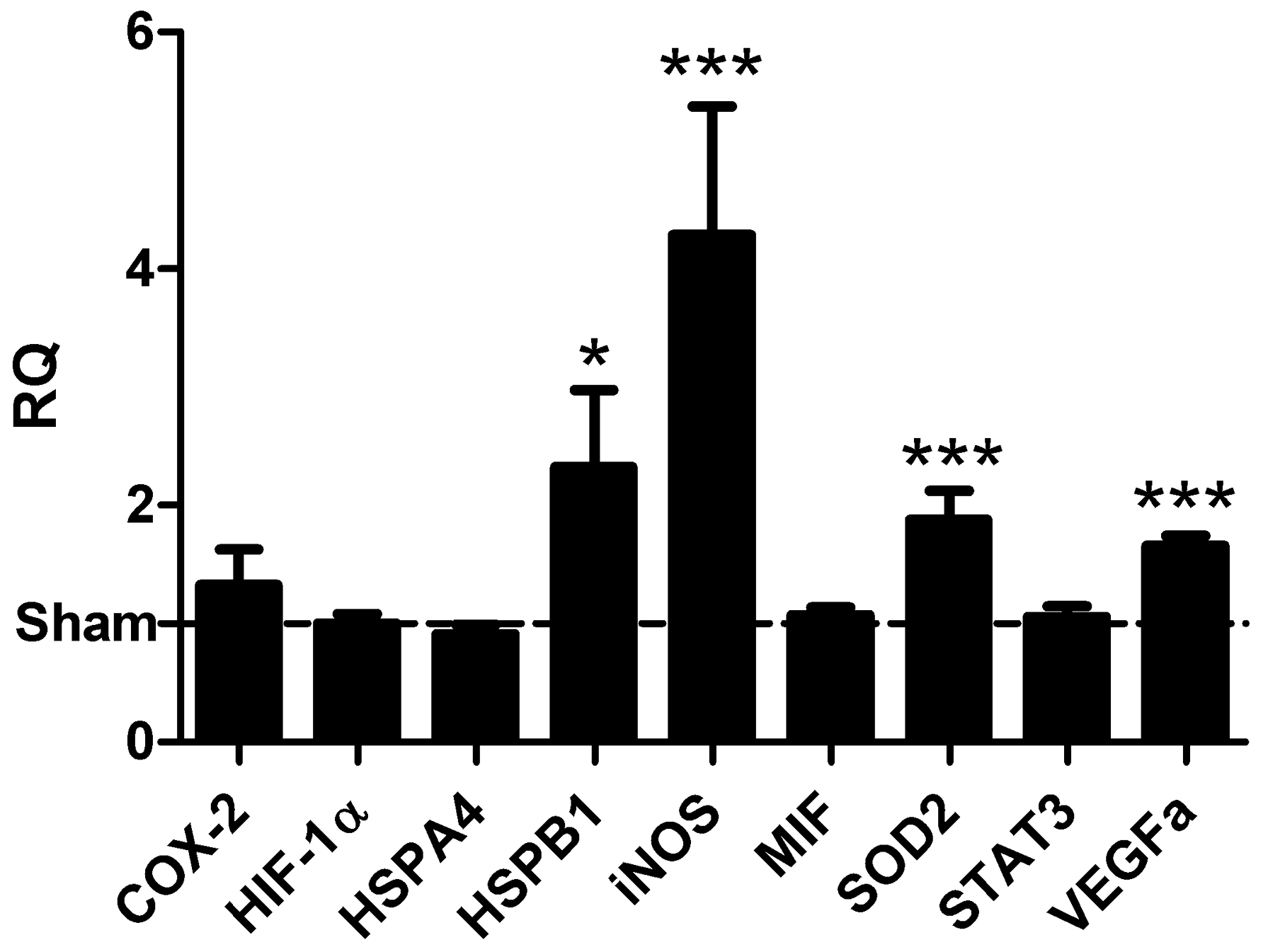
2.2. Induction of Gene Transcription
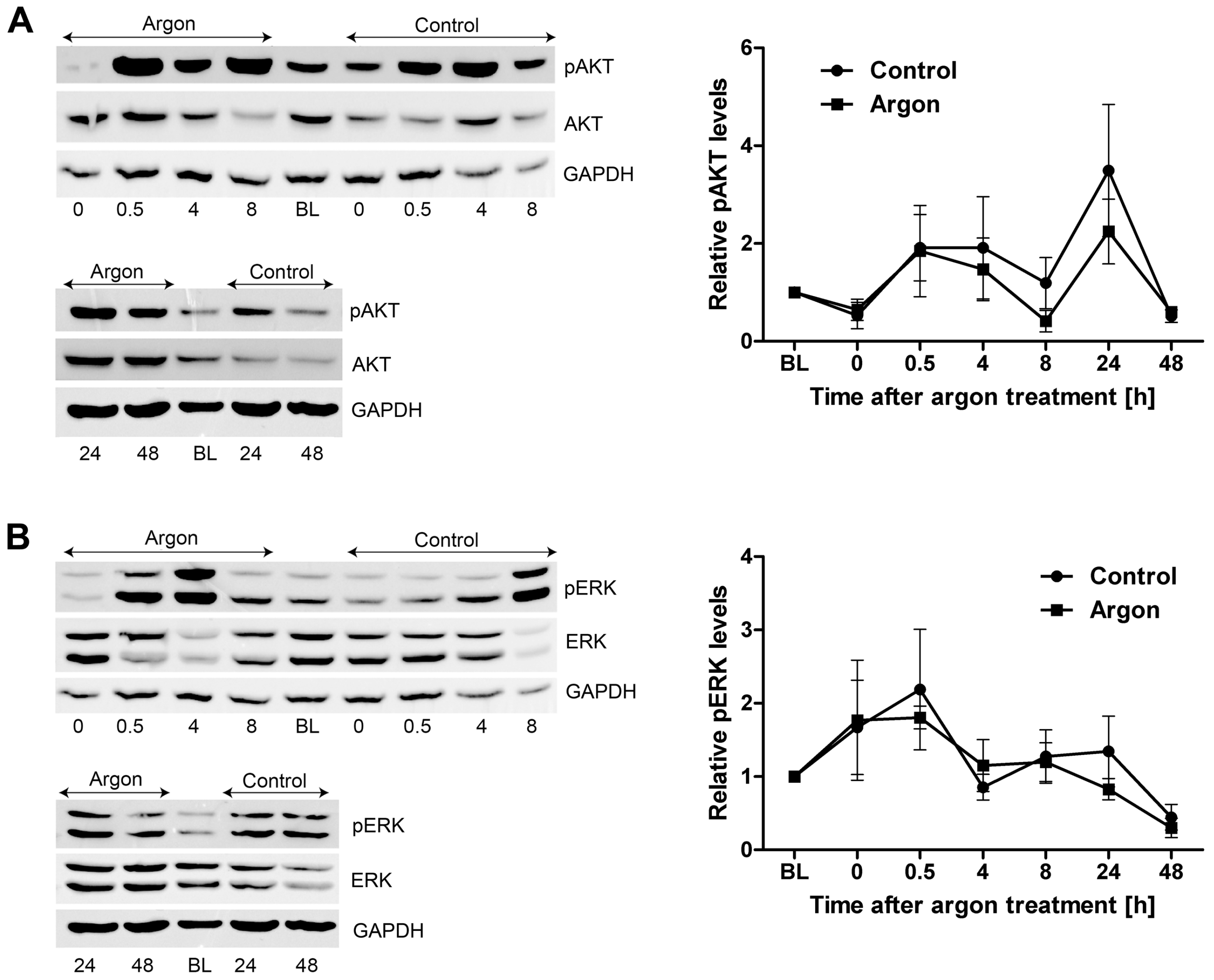
2.3. Protein Expression and Phosphorylation of Kinases
3. Discussion
Limitations
4. Materials and Methods
4.1. Ethics Approval
4.2. Reagents
4.3. Cell Culture
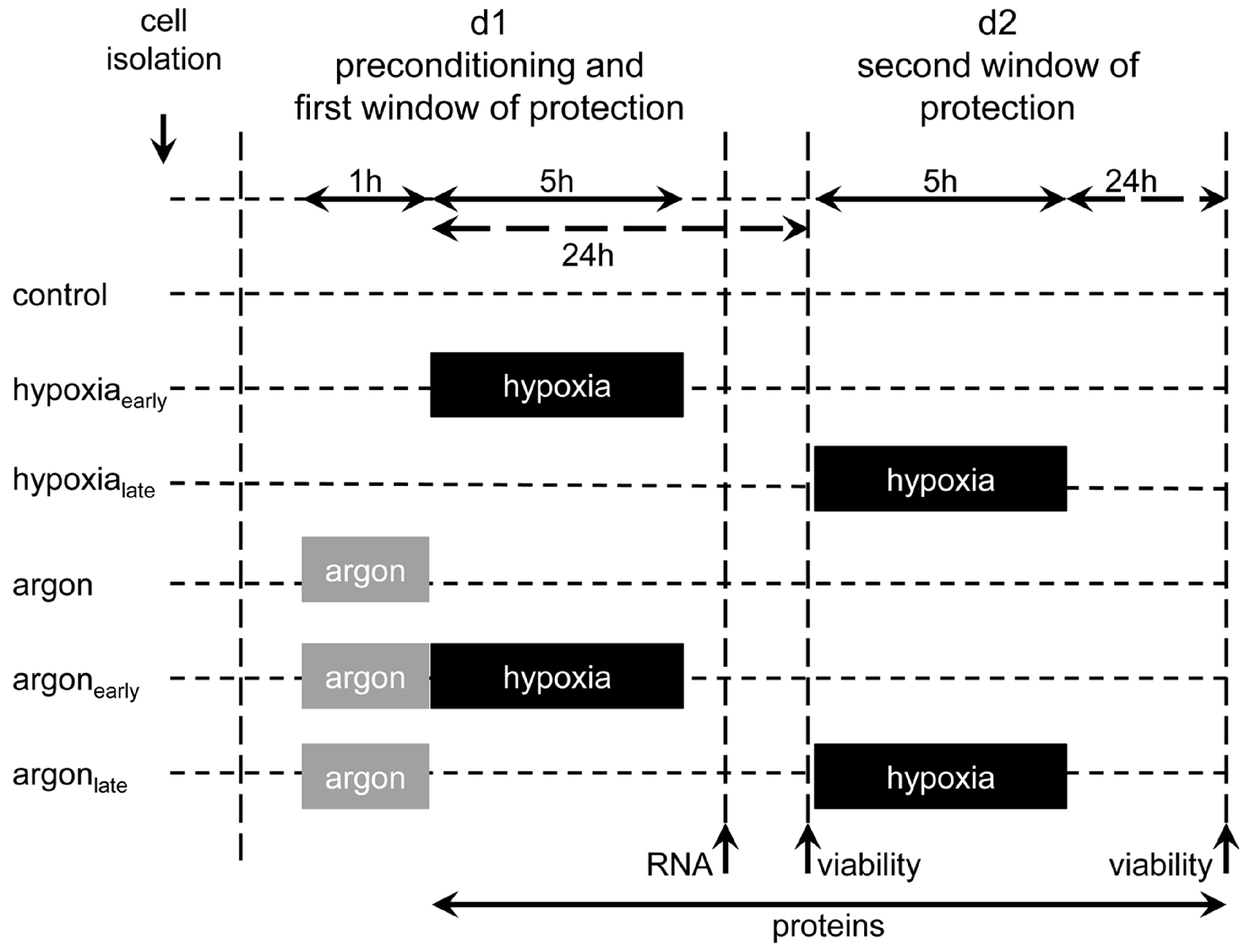
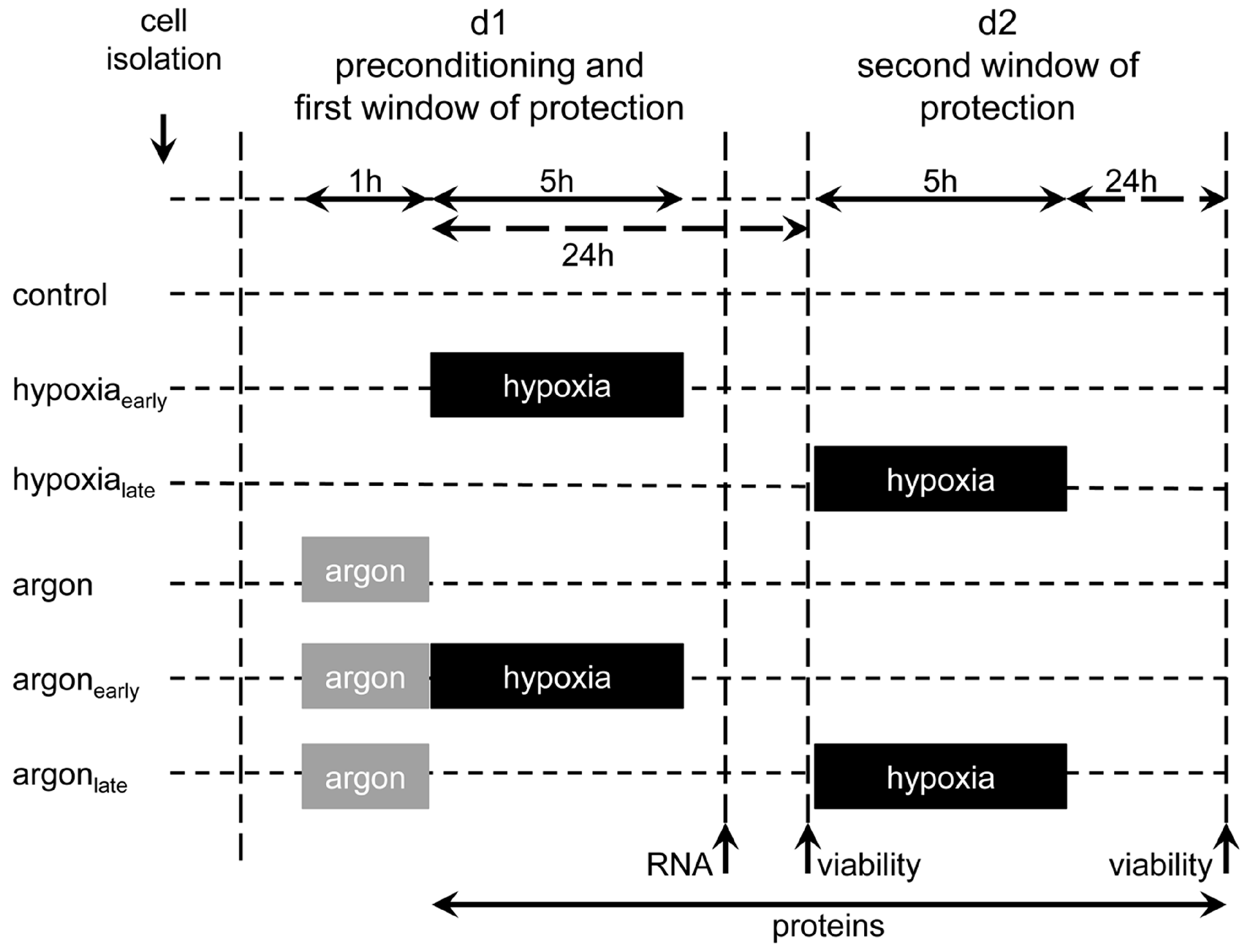
4.4. Experimental Set-up and Preconditioning
4.5. Cell Viability after Terminal Hypoxia
4.6. Proliferation Assay
4.7. Western Blotting
4.8. Quantitative Real Time Polymerase Chain Reaction
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bethesda, M.D. Nhlbi Morbidity and Mortality Chartbook; National Heart, Lung and Blood Institute: Bethesda, MD, USA, 2002. [Google Scholar]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart disease and stroke statistics—2014 update: A report from the american heart association. Circulation 2014, 129, e28–e292. [Google Scholar] [CrossRef] [PubMed]
- De Hert, S.G. Volatile anesthetics and cardiac function. Semin. Cardiothorac. Vasc. Anesth. 2006, 10, 33–42. [Google Scholar] [CrossRef] [PubMed]
- De Hert, S.G. Cardioprotection by volatile anesthetics: What about noncardiac surgery? J. Cardiothorac. Vasc. Anesth. 2011, 25, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. The second window of preconditioning (swop) where are we now? Cardiovasc. Drugs Ther. 2010, 24, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Gattullo, D.; Rastaldo, R.; Losano, G. Ischemic preconditioning: From the first to the second window of protection. Life Sci. 2001, 69, 1–15. [Google Scholar] [CrossRef]
- Yang, X.M.; Baxter, G.F.; Heads, R.J.; Yellon, D.M.; Downey, J.M.; Cohen, M.V. Infarct limitation of the second window of protection in a conscious rabbit model. Cardiovasc. Res. 1996, 31, 777–783. [Google Scholar] [CrossRef]
- Yellon, D.M.; Baxter, G.F. A "second window of protection" or delayed preconditioning phenomenon: Future horizons for myocardial protection? J. Mol. Cell. Cardiol. 1995, 27, 1023–1034. [Google Scholar] [CrossRef]
- Harris, K.; Armstrong, S.P.; Campos-Pires, R.; Kiru, L.; Franks, N.P.; Dickinson, R. Neuroprotection against traumatic brain injury by xenon, but not argon, is mediated by inhibition at the n-methyl-d-aspartate receptor glycine site. Anesthesiology 2013, 119, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Mio, Y.; Shim, Y.H.; Richards, E.; Bosnjak, Z.J.; Pagel, P.S.; Bienengraeber, M. Xenon preconditioning: The role of prosurvival signaling, mitochondrial permeability transition and bioenergetics in rats. Anesth. Analg. 2009, 108, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Soldatov, P.E.; D'Iachenko, A.I.; Pavlov, B.N.; Fedotov, A.P.; Chuguev, A.P. Survival of laboratory animals in argon-containing hypoxic gaseous environments. Aviakosm. Ekolog. Med. 1998, 32, 33–37. [Google Scholar] [PubMed]
- Weber, N.C.; Toma, O.; Wolter, J.I.; Obal, D.; Mullenheim, J.; Preckel, B.; Schlack, W. The noble gas xenon induces pharmacological preconditioning in the rat heart in vivo via induction of pkc-epsilon and p38 mapk. Br. J. Pharmacol. 2005, 144, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.C.; Stursberg, J.; Wirthle, N.M.; Toma, O.; Schlack, W.; Preckel, B. Xenon preconditioning differently regulates p44/42 mapk (erk 1/2) and p46/54 mapk (jnk 1/2 and 3) in vivo. Br. J. Anaesth. 2006, 97, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Limatola, V.; Ward, P.; Cattano, D.; Gu, J.; Giunta, F.; Maze, M.; Ma, D. Xenon preconditioning confers neuroprotection regardless of gender in a mouse model of transient middle cerebral artery occlusion. Neuroscience 2010, 165, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Lim, T.; Xu, J.; Tang, H.; Wan, Y.; Zhao, H.; Hossain, M.; Maxwell, P.H.; Maze, M. Xenon preconditioning protects against renal ischemic-reperfusion injury via hif-1alpha activation. J. Am. Soc. Nephrol. 2009, 20, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, M.; Jawad, N.; Li, Y.; Vizcaychipi, M.P.; Maze, M.; Ma, D. Effect of noble gases on oxygen and glucose deprived injury in human tubular kidney cells. Exp. Biol. Med. 2010, 235, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.C.; Frassdorf, J.; Ratajczak, C.; Grueber, Y.; Schlack, W.; Hollmann, M.W.; Preckel, B. Xenon induces late cardiac preconditioning in vivo: A role for cyclooxygenase 2? Anesth. Analg. 2008, 107, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Fahlenkamp, A.V.; Rossaint, R.; Haase, H.; Al Kassam, H.; Ryang, Y.M.; Beyer, C.; Coburn, M. The noble gas argon modifies extracellular signal-regulated kinase 1/2 signaling in neurons and glial cells. Eur. J. Pharmacol. 2012, 674, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Jawad, N.; Rizvi, M.; Gu, J.; Adeyi, O.; Tao, G.; Maze, M.; Ma, D. Neuroprotection (and lack of neuroprotection) afforded by a series of noble gases in an in vitro model of neuronal injury. Neurosci. Lett. 2009, 460, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, P.D.; Rossaint, J.; Rossaint, R.; Weis, J.; Fries, M.; Fahlenkamp, A.; Ryang, Y.M.; Grottke, O.; Coburn, M. Argon: Neuroprotection in in vitro models of cerebral ischemia and traumatic brain injury. Crit. Care 2009, 13, R206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagel, P.S.; Krolikowski, J.G.; Shim, Y.H.; Venkatapuram, S.; Kersten, J.R.; Weihrauch, D.; Warltier, D.C.; Pratt, P.F., Jr. Noble gases without anesthetic properties protect myocardium against infarction by activating prosurvival signaling kinases and inhibiting mitochondrial permeability transition in vivo. Anesth. Analg. 2007, 105, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Brucken, A.; Cizen, A.; Fera, C.; Meinhardt, A.; Weis, J.; Nolte, K.; Rossaint, R.; Pufe, T.; Marx, G.; Fries, M. Argon reduces neurohistopathological damage and preserves functional recovery after cardiac arrest in rats. Br. J. Anaesth. 2013, 110 (Suppl. 1), i106–i112. [Google Scholar] [CrossRef] [PubMed]
- Brucken, A.; Kurnaz, P.; Bleilevens, C.; Derwall, M.; Weis, J.; Nolte, K.; Rossaint, R.; Fries, M. Dose dependent neuroprotection of the noble gas argon after cardiac arrest in rats is not mediated by k(atp)-channel opening. Resuscitation 2014, 85, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Goebel, U. Argon: A novel therapeutic option to treat neuronal ischemia and reperfusion injuries? Neural Regen. Res. 2015, 10, 1043–1044. [Google Scholar] [PubMed]
- Ulbrich, F.; Kaufmann, K.; Roesslein, M.; Wellner, F.; Auwarter, V.; Kempf, J.; Loop, T.; Buerkle, H.; Goebel, U. Argon mediates anti-apoptotic signaling and neuroprotection via inhibition of toll-like receptor 2 and 4. PLoS ONE 2015, 10, e0143887. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Kaufmann, K.B.; Coburn, M.; Lagreze, W.A.; Roesslein, M.; Biermann, J.; Buerkle, H.; Loop, T.; Goebel, U. Neuroprotective effects of argon are mediated via an erk-1/2 dependent regulation of heme-oxygenase-1 in retinal ganglion cells. J. Neurochem. 2015, 134, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Schallner, N.; Coburn, M.; Loop, T.; Lagreze, W.A.; Biermann, J.; Goebel, U. Argon inhalation attenuates retinal apoptosis after ischemia/reperfusion injury in a time- and dose-dependent manner in rats. PLoS ONE 2014, 9, e115984. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.R.; Stokes, J.W.; Downing, P. Anaesthesia and the 'inert' gases with special reference to xenon. Anaesth. Intensive Care 1992, 20, 66–70. [Google Scholar] [PubMed]
- Das, D.K.; Maulik, N. Cardiac genomic response following preconditioning stimulus. Cardiovasc. Res. 2006, 70, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Reperfusion injury salvage kinase signalling: Taking a risk for cardioprotection. Heart Fail. Rev. 2007, 12, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.; Franks, N.P. Bench-to-bedside review: Molecular pharmacology and clinical use of inert gases in anesthesia and neuroprotection. Crit. Care 2010, 14, 229. [Google Scholar] [CrossRef] [PubMed]
- Hollig, A.; Schug, A.; Fahlenkamp, A.V.; Rossaint, R.; Coburn, M. Argon: Systematic review on neuro- and organoprotective properties of an "inert" gas. Int. J. Mol. Sci. 2014, 15, 18175–18196. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jones, W.K.; Xuan, Y.T.; Tang, X.L.; Bao, W.; Wu, W.J.; Han, H.; Laubach, V.E.; Ping, P.; Yang, Z.; et al. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible no synthase gene. Proc. Natl. Acad. Sci. USA 1999, 96, 11507–11512. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.C.; Toma, O.; Wolter, J.I.; Wirthle, N.M.; Schlack, W.; Preckel, B. Mechanisms of xenon- and isoflurane-induced preconditioning—A potential link to the cytoskeleton via the mapkapk-2/hsp27 pathway. Br. J. Pharmacol. 2005, 146, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.; Jarrett, N.C.; Hess, M.L.; Kukreja, R.C. Essential role of inducible nitric oxide synthase in monophosphoryl lipid a-induced late cardioprotection: Evidence from pharmacological inhibition and gene knockout mice. Circulation 1999, 99, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, K.; Sukhatme, V.P.; Shubeita, H.E.; Chien, K.R. Alpha- and beta-adrenergic stimulation induces distinct patterns of immediate early gene expression in neonatal rat myocardial cells. J. Biol. Chem. 1990, 265, 13809–13817. [Google Scholar] [PubMed]
- Yellon, D.M.; Downey, J.M. Preconditioning the myocardium: From cellular physiology to clinical cardiology. Physiol. Rev. 2003, 83, 1113–1151. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Manchikalapudi, S.; Tang, X.L.; Takano, H.; Qiu, Y.; Guo, Y.; Zhang, Q.; Jadoon, A.K. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ. Res. 1997, 81, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.R.; Rodriguez, E.; Tse, J.; Scholz, P.M. Effect of increased myocardial cyclic gmp induced by cyclic gmp-phosphodiesterase inhibition on oxygen consumption and supply of rabbit hearts. Clin. Exp. Pharmacol. Physiol. 1994, 21, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Gath, I.; Schwarz, P.; Closs, E.I.; Kleinert, H. Isoforms of nitric oxide synthase: Properties, cellular distribution and expressional control. Biochem. Pharmacol. 1995, 50, 1321–1332. [Google Scholar] [CrossRef]
- Xuan, Y.T.; Tang, X.L.; Qiu, Y.; Banerjee, S.; Takano, H.; Han, H.; Bolli, R. Biphasic response of cardiac no synthase isoforms to ischemic preconditioning in conscious rabbits. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2360–H2371. [Google Scholar] [PubMed]
- Goetzenich, A.; Hatam, N.; Preuss, S.; Moza, A.; Bleilevens, C.; Roehl, A.B.; Autschbach, R.; Bernhagen, J.; Stoppe, C. The role of hypoxia-inducible factor-1alpha and vascular endothelial growth factor in late-phase preconditioning with xenon, isoflurane and levosimendan in rat cardiomyocytes. Interact. Cardiovasc. Thorac. Surg. 2014, 18, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Redel, A.; Stumpner, J.; Smul, T.M.; Lange, M.; Jazbutyte, V.; Ridyard, D.G.; Roewer, N.; Kehl, F. Endothelial nitric oxide synthase mediates the first and inducible nitric oxide synthase mediates the second window of desflurane-induced preconditioning. J. Cardiothorac. Vasc. Anesth. 2013, 27, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Wakeno-Takahashi, M.; Otani, H.; Nakao, S.; Imamura, H.; Shingu, K. Isoflurane induces second window of preconditioning through upregulation of inducible nitric oxide synthase in rat heart. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2585–H2591. [Google Scholar] [CrossRef] [PubMed]
- Barral, J.M.; Broadley, S.A.; Schaffar, G.; Hartl, F.U. Roles of molecular chaperones in protein misfolding diseases. Semin. Cell. Dev. Biol. 2004, 15, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Buzzard, K.A.; Giaccia, A.J.; Killender, M.; Anderson, R.L. Heat shock protein 72 modulates pathways of stress-induced apoptosis. J. Biol. Chem. 1998, 273, 17147–17153. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Meyrick, B. Hypoxia increases hsp90 binding to enos via pi3k-akt in porcine coronary artery endothelium. Lab. Investig. 2004, 84, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, T.J.; Sievers, R.E.; Vissern, F.L.; Welch, W.J.; Wolfe, C.L. Heat shock protein induction in rat hearts: A role for improved myocardial salvage after ischemia and reperfusion? Circulation 1992, 85, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Heads, R.J.; Latchman, D.S.; Yellon, D.M. The molecular basis of ad r aptation to ischemia in the heart: The role of stress proteins and anti-oxidants in the ischemic and eperfused heart. EXS 1996, 76, 383–407. [Google Scholar] [PubMed]
- Bluhm, W.F.; Martin, J.L.; Mestril, R.; Dillmann, W.H. Specific heat shock proteins protect microtubules during simulated ischemia in cardiac myocytes. Am. J. Physiol. 1998, 275, H2243–H2249. [Google Scholar] [PubMed]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [PubMed]
- Bitar, K.N. Hsp27 phosphorylation and interaction with actin-myosin in smooth muscle contraction. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G894–G903. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta 2011, 1813, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, S.; Kuzuya, T.; Fuji, H.; Yamashita, N.; Oe, H.; Hori, M.; Suzuki, K.; Taniguchi, N.; Tada, M. Sublethal ischemia alters myocardial antioxidant activity in canine heart. Am. J. Physiol. 1993, 264, H33–H39. [Google Scholar] [PubMed]
- Rui, T.; Cepinskas, G.; Feng, Q.; Kvietys, P.R. Delayed preconditioning in cardiac myocytes with respect to development of a proinflammatory phenotype: Role of sod and nos. Cardiovasc. Res. 2003, 59, 901–911. [Google Scholar] [CrossRef]
- Yamashita, N.; Nishida, M.; Hoshida, S.; Kuzuya, T.; Hori, M.; Taniguchi, N.; Kamada, T.; Tada, M. Induction of manganese superoxide dismutase in rat cardiac myocytes increases tolerance to hypoxia 24 hours after preconditioning. J. Clin. Investig. 1994, 94, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhai, X.; Ashraf, M. Direct evidence that initial oxidative stress triggered by preconditioning contributes to second window of protection by endogenous antioxidant enzyme in myocytes. Circulation 1996, 93, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Crystal, G.J.; Malik, G.; Yoon, S.H.; Kim, S.J. Isoflurane late preconditioning against myocardial stunning is associated with enhanced antioxidant defenses. Acta Anaesthesiol. Scand. 2012, 56, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lian, C.W.; Fang, L.Q.; Liu, B.; Yang, B. Effects of xenon preconditioning against ischemia/reperfusion injury and oxidative stress in immature heart. Sichuan Da Xue Xue Bao Yi Xue Ban 2014, 45, 780–784. (In Chinese) [Google Scholar] [PubMed]
- Date, T.; Mochizuki, S.; Belanger, A.J.; Yamakawa, M.; Luo, Z.; Vincent, K.A.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Expression of constitutively stable hybrid hypoxia-inducible factor-1alpha protects cultured rat cardiomyocytes against simulated ischemia-reperfusion injury. Am. J. Physiol. Cell Physiol. 2005, 288, C314–C320. [Google Scholar] [CrossRef] [PubMed]
- Andersen, F.; Watanabe, H.; Bjarkam, C.; Danielsen, E.H.; Cumming, P. Pig brain stereotaxic standard space: Mapping of cerebral blood flow normative values and effect of mptp-lesioning. Brain Res. Bull. 2005, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Barac, D.Y.; Reisner, Y.; Silberman, M.; Zeevi-Levin, N.; Danon, A.; Salomon, O.; Shoham, M.; Shilkrut, M.; Kostin, S.; Schaper, J.; et al. Mechanical load induced by glass microspheres releases angiogenic factors from neonatal rat ventricular myocytes cultures and causes arrhythmias. J. Cell. Mol. Med. 2008, 12, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Munk, M.; Memon, A.A.; Goetze, J.P.; Nielsen, L.B.; Nexo, E.; Sorensen, B.S. Hypoxia changes the expression of the epidermal growth factor (egf) system in human hearts and cultured cardiomyocytes. PLoS ONE 2012, 7, e40243. [Google Scholar] [CrossRef] [PubMed]
- Von Harsdorf, R.; Li, P.F.; Dietz, R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 1999, 99, 2934–2941. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, O.; Ogawa, Y.; Itoh, H.; Suga, S.; Komatsu, Y.; Kishimoto, I.; Nishino, K.; Yoshimasa, T.; Nakao, K. Rapid transcriptional activation and early mrna turnover of brain natriuretic peptide in cardiocyte hypertrophy: Evidence for brain natriuretic peptide as an "emergency" cardiac hormone against ventricular overload. J. Clin. Investig. 1995, 96, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Eder, A.; Bonstrup, M.; Flato, M.; Mewe, M.; Schaaf, S.; Aksehirlioglu, B.; Schwoerer, A.P.; Uebeler, J.; Eschenhagen, T. Development of a drug screening platform based on engineered heart tissue. Circ. Res. 2010, 107, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Gerlier, D.; Thomasset, N. Use of mtt colorimetric assay to measure cell activation. J. Immunol. Methods 1986, 94, 57–63. [Google Scholar] [CrossRef]
- Jones, L.J.; Gray, M.; Yue, S.T.; Haugland, R.P.; Singer, V.L. Sensitive determination of cell number using the cyquant cell proliferation assay. J. Immunol. Methods 2001, 254, 85–98. [Google Scholar] [CrossRef]
- Cumming, G.; Fidler, F.; Vaux, D.L. Error bars in experimental biology. J. Cell Biol. 2007, 177, 7–11. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayer, B.; Soppert, J.; Kraemer, S.; Schemmel, S.; Beckers, C.; Bleilevens, C.; Rossaint, R.; Coburn, M.; Goetzenich, A.; Stoppe, C. Argon Induces Protective Effects in Cardiomyocytes during the Second Window of Preconditioning. Int. J. Mol. Sci. 2016, 17, 1159. https://doi.org/10.3390/ijms17071159
Mayer B, Soppert J, Kraemer S, Schemmel S, Beckers C, Bleilevens C, Rossaint R, Coburn M, Goetzenich A, Stoppe C. Argon Induces Protective Effects in Cardiomyocytes during the Second Window of Preconditioning. International Journal of Molecular Sciences. 2016; 17(7):1159. https://doi.org/10.3390/ijms17071159
Chicago/Turabian StyleMayer, Britta, Josefin Soppert, Sandra Kraemer, Sabrina Schemmel, Christian Beckers, Christian Bleilevens, Rolf Rossaint, Mark Coburn, Andreas Goetzenich, and Christian Stoppe. 2016. "Argon Induces Protective Effects in Cardiomyocytes during the Second Window of Preconditioning" International Journal of Molecular Sciences 17, no. 7: 1159. https://doi.org/10.3390/ijms17071159