
Chemical Structure-Biological Activity Models for Pharmacophores’ 3D-Interactions

Abstract
:
1. Introduction
2. The Special Computing Trace of Algebraic Structure-Activity Relationship (SAR) Method
2.1. General Special Computing Trace of the Algebraic Structure-Activity Relationship (SPECTRAL-SAR) Algorithm
- The orthogonality constraint of the molecular descriptors’ states involved in Equation (2),also restrains the classes of molecular descriptors to be considered; for instance, the electronegativity and chemical hardness are ideal candidates because of their orthogonal and complementary conceptually nature [47,48];
- Introducing the variational principle selecting from the pool of various predicted endpoints of Equation (2), from those of linear, bilinear, up to those that are multi-linear in nature, e.g.,with the general form given by the SPECTRAL-SAR algorithm [16,17,42]:with the vectors , , …, orthogonally constructed, i.e.,assuring that and are orthogonal, along the general Gram–Schmidt expressions:so that the vector is orthogonal on all previous ones.
2.2. Results of SPECTRAL-SAR in Pharmacophores’ 3D-Interaction
3. The Minimal Topological Difference Method
3.1. General MTD Algorithm
- molecular superposition, which identifies similarities within a series of molecules or describes a “pharmacophoric constellation” of atoms,
- and identifying and describing the positions that are equivalent.
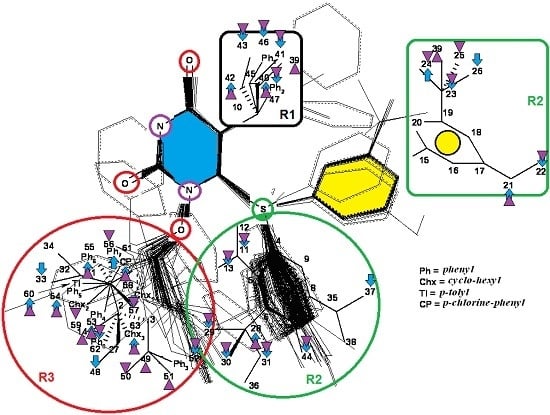
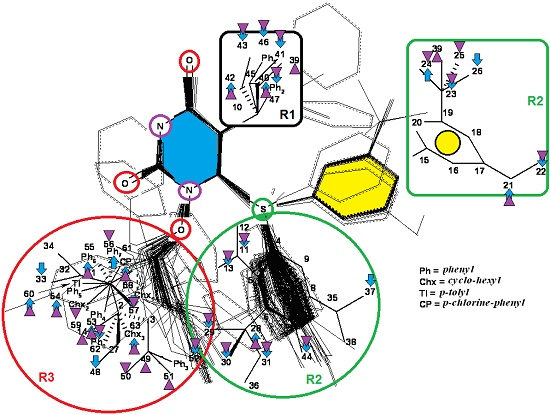
3.2. Results of MTD in Pharmacophores’ 3D-Interaction
- the mono-parametric model of hydrophobicity:
- the mono-parametric model when the steric parameter is:
- the combined correlation:
4. New Pharmacophores in Severe Genetic Disorders
4.1. 3D-QSAR Modeling for Severe Genetic Disorders
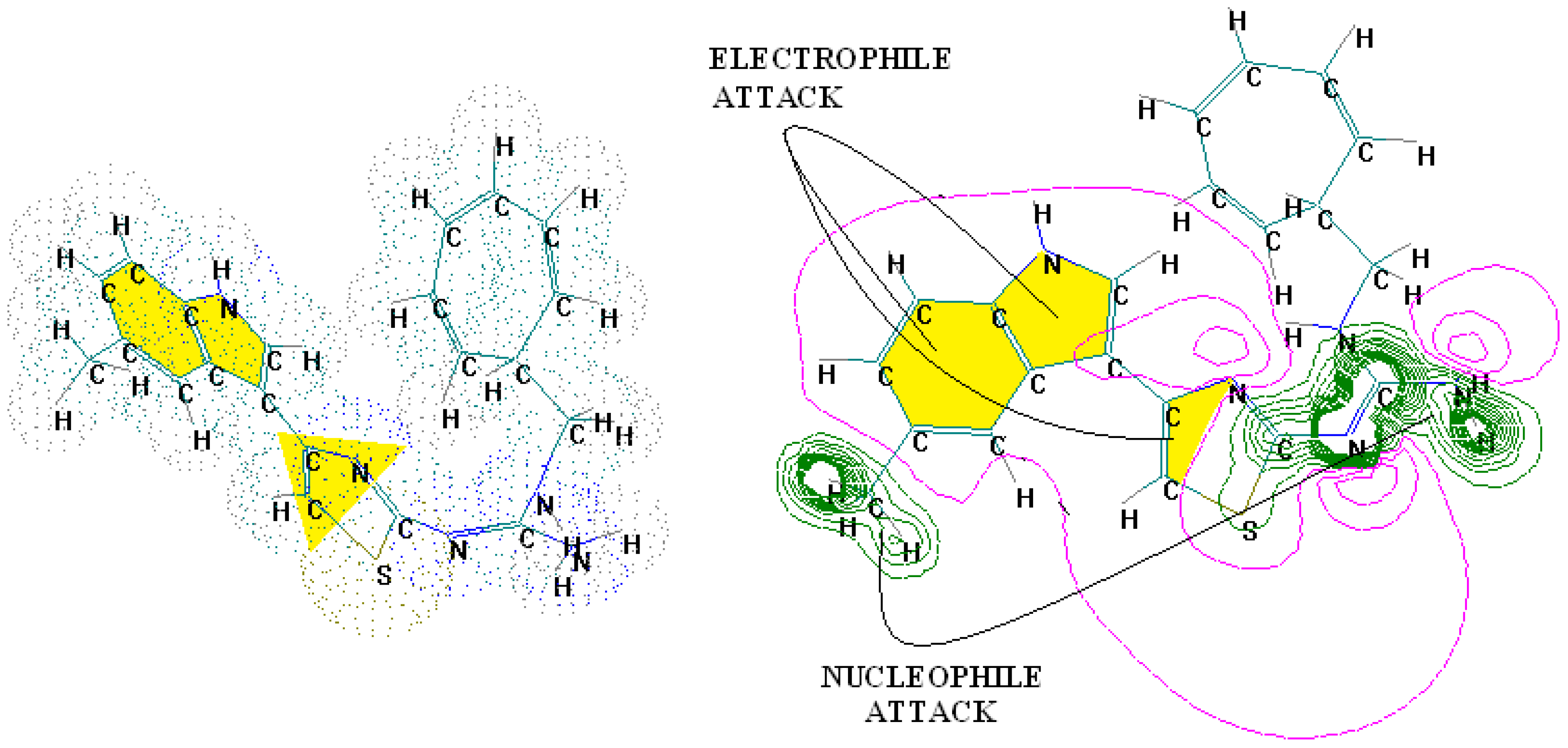
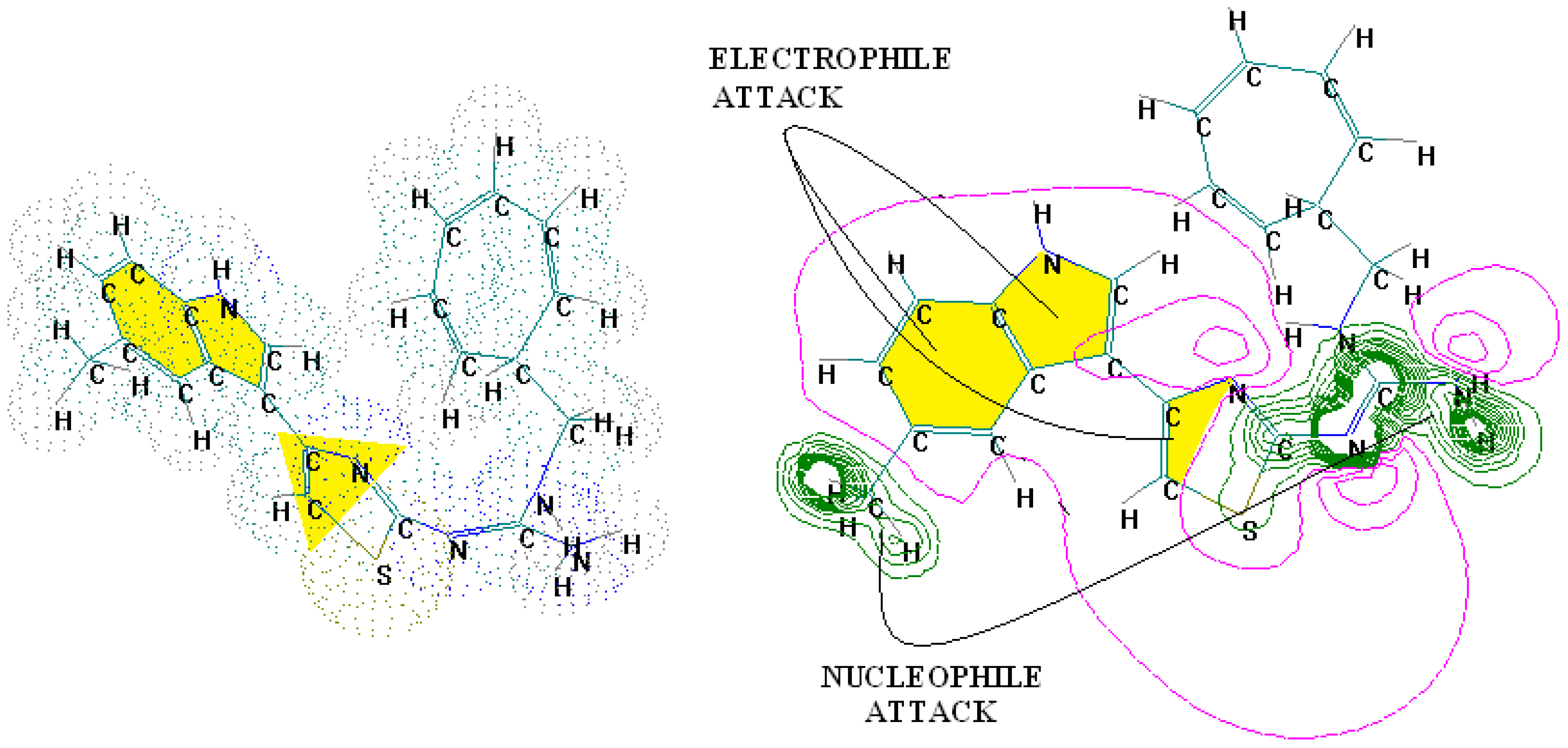
4.2. 3D-QSAR Predictions in Pharmacophores’ 3D-Interaction
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| (Q)SA(/P)R | quantitative structure-activity (/property) relationships |
| HIV | human immunodeficiency virus |
| HAART | highly-active antiretroviral therapy |
| AIDS | acquired immune deficiency syndrome |
| RT | reverse transcriptase |
| FDA | Federal Drug Agency |
| CB | chemical-biological |
| A | activity |
| L | ligand |
| R | receptor |
| LR | Ligand-eceptor complex |
References
- Pandey, A.V. Bioinformatics tools and databases for the study of human growth hormone. Endocr. Dev. 2012, 23, 71–85. [Google Scholar] [PubMed]
- Koedrith, P.; Kim, H.; Weon, J.; Seo, Y.R. Toxicogenomic approaches for understanding molecular mechanisms of heavy metal mutagenicity and carcinogenicity. Int. J. Hyg. Environ. Health 2013, 216, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Odejinmi, S.I.; Vankayalapati, H.; Wierenga, K.J.; Lai, K. Innovative therapy for Classic Galactosemia—Tale of two HTS. Mol. Genet. Metab. 2012, 105, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Hurko, O. Target-based drug discovery, genetic diseases, and biologics. Neurochem. Int. 2012, 61, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, B.; Gallo, A.; Le Strat, Y.; Lu, L.; Gorwood, P. Genetics of dopamine receptors and drug addiction: A comprehensive review. Behav. Pharmacol. 2009, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, I.; Boku, S.; Takahashi, Y. Psychopharmacology of atypical antipsychotic drugs: From the receptor binding profile to neuroprotection and neurogenesis. Psychiatry Clin. Neurosci. 2015, 69, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Lei, S. Cross interaction of dopaminergic and adrenergic systems in neural modulation. Int. J. Physiol. Pathophysiol. Pharmacol. 2014, 6, 137–142. [Google Scholar] [PubMed]
- McKinney, J.D.; Richard, A.; Waller, C.; Newman, M.C.; Gerberick, F. The practice of structure activity relationships (SAR) in toxicology. Toxicol. Sci. 2000, 56, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Das, R.N. A review on principles, theory and practices of 2D-QSAR. Curr. Drug Metab. 2014, 15, 346–379. [Google Scholar] [CrossRef] [PubMed]
- Organisation for Economic Co-Operation and Development (OECD). Guidance Document on the Validation of (Quantitative) Structure-Activity Relationships [(Q)SAR] Models; OECD: Paris, France, 2007. [Google Scholar]
- Avram, S.; Berner, H.; Milac, A.L.; Wolschann, P. Quantitative structure—Activity relationship studies on membrane receptors inhibition by antipsychotic drugs. Application to schizophrenia treatment. Monatshefte Chem. Chem. Mon. 2008, 139, 407–426. [Google Scholar] [CrossRef]
- Bostrom, J.; Bohm, M.; Gundertofte, K.; Klebe, G. A 3D QSAR study on a set of dopamine D4 receptor antagonists. J. Chem. Inf. Comput. Sci. 2003, 43, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Duda-Seiman, D.; Borcan, F.; Wolschann, P. QSAR-CoMSIA applied to antipsychotic drugs with their dopamine D2 and serotonine 5HT2A membrane receptors. J. Serbian Chem. Soc. 2011, 76, 263–281. [Google Scholar] [CrossRef]
- Oprea, T.I.; Waller, C.L. Theoretical and practical aspects of three-dimensional quantitative structure-activity relationships. In Reviews in Computational Chemistry; John Wiley & Sons, Inc.: New York, NY, USA, 2007; pp. 127–182. [Google Scholar]
- Putz, M.V. QSAR & SPECTRAL-SAR in Computational Ecotoxicology; Apple Academics & CRC Press: Oakville & Toronto, ON, Canada; Point Pleasant, NJ, USA, 2012. [Google Scholar]
- Putz, M.V.; Lacrămă, A.M. Introducing spectral structure activity relationship (S-SAR) analysis. Application to ecotoxicology. Int. J. Mol. Sci. 2007, 8, 363–391. [Google Scholar] [CrossRef]
- Chiriac, A.; Ciubotariu, D.; Funar-Timoftei, S.; Kurunczi, L.; Mracec, M.; Mracec, M.; Szabadai, Z.; Şeclăman, E.; Simon, Z. QSAR and 3D-QSAR in timişoara. 1972–2005. Rev. Roum. Chim. 2006, 51, 79–99. [Google Scholar]
- Duda-Seiman, C; Duda-Seiman, D.; Dragoş, D.; Medeleanu, M.; Careja, V.; Putz, M.V.; Lacrămă, A.M.; Chiriac, A.; Nuţiu, R.; Ciubotariu, D. Design of Anti-HIV ligands by means of minimal topological difference (MTD) method. Int. J. Mol. Sci. 2006, 7, 537–555. [Google Scholar]
- Avram, S.; Buiu, C.; Duda-Seiman, C.; Borcan, F.; Mihailescu, D. Evaluation of the pharmacological descriptors related to the induction of antidepressant activity and its prediction by QSAR/QRAR methods. Mini Rev. Med. Chem. 2012, 12, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Milac, A.L.; Mihailescu, D. 3D-QSAR study indicates an enhancing effect of membrane ions on psychiatric drugs targeting serotonin receptor 5-HT1A. Mol. BioSyst. 2012, 8, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring the QSAR Hydrophobic, Electronic and Steric Constants; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Dearden, J.C. In silico prediction of drug toxicity. J. Comput. Aided Mol. Des. 2003, 17, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Helma, C. (Ed.) Predictive Toxicology; Taylor & Francis: Abingdon, UK, 2005.
- Tong, W.; Hong, H.; Xie, Q.; Shi, L.; Fang, H.; Perkins, R. Assessing QSAR limitations—A regulatory perspective. Curr. Comput. Aided Drug Des. 2005, 1, 195–205. [Google Scholar] [CrossRef]
- Leonard, J.T.; Roy, K. On selection of training and test sets for the development of predictive QSAR models. QSAR Comb. Sci. 2006, 25, 235–251. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Roy, K. On some aspects of validation of predictive quantitative structure-activity relationship models. Expert Opin. Drug Discov. 2007, 2, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Put, R.; Vander Heyden, Y. Review on modelling aspects in reversed-phase liquid chromatographic quantitative structure-retention relationships. Anal. Chim. Acta 2007, 602, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.P.; Leonard, J.T.; Roy, K. Exploring the impact of size of training sets for the development of predictive QSAR models. Chemometr. Intell. Lab. Syst. 2008, 90, 31–42. [Google Scholar] [CrossRef]
- Ajmani, S.; Jadhav, K.; Kulkarni, S.A. Group-based QSAR (G-QSAR): Mitigating interpretation challenges in QSAR. QSAR Comb. Sci. 2008, 28, 36–51. [Google Scholar] [CrossRef]
- Roy, P.P.; Paul, S.; Mitra, I.; Roy, K. On two novel parameters for validation of predictive QSAR models. Molecules 2009, 14, 1660–1701. [Google Scholar]
- Manoharan, P.; Vijayan, R.S.K.; Ghoshal, N. Rationalizing fragment based drug discovery for BACE1: Insights from FB-QSAR, FB-QSSR, multi objective (MO-QSPR) and MIF studies. J. Comput. Aided Mol. Des. 2010, 24, 843–864. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef] [PubMed]
- Ballante, F.; Ragno, R. 3-D QSAutogrid/R: An alternative procedure to build 3-D QSAR models. Methodologies and applications. J. Chem. Inf. Model. 2012, 52, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Brown, N. Bioisosteres in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Dirac, P.A.M. The Principles of Quantum Mechanics, 2nd ed.; Clarendon Press: Oxford, UK, 1947. [Google Scholar]
- Zeilinger, A. Experiment and the foundations of quantum physics. Rev. Mod. Phys. 1999, 71, S288–S297. [Google Scholar] [CrossRef]
- Moyer, M. Quantum Entanglement, Photosynthesis and Better Solar Cells. Scientific American 2009. Available online: http://www.scientificamerican.com/article.cfm?id=quantum-entanglement-and-photo (accessed on 23 February 2013).
- Collini, E.; Wong, C.Y.; Wilk, K.E.; Curmi, P.M.; Brumer, P.; Scholes, G.D. Coherently wired light-harvesting in photosynthetic marine algae at ambient temperature. Nature 2010, 463, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V. Quantum Nanochemistry: Vol V. Quantum Structure-Activity Relationship (Qu-SAR); Apple Academic & CRC Press: Oakville & Toronto, ON, Canada; Waretown, NJ, USA, 2016. [Google Scholar]
- Lacrămă, A.M.; Putz, M.V.; Ostafe, V. A Spectral-SAR model for the anionic-cationic interaction in ionic liquids: Application to Vibrio fischeri ecotoxicity. Int. J. Mol. Sci. 2007, 8, 842–863. [Google Scholar] [CrossRef]
- Chicu, S.A.; Putz, M.V. Köln-Timişoara molecular activity combined models toward interspecies toxicity assessment. Int. J. Mol. Sci. 2009, 10, 4474–4497. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Putz, A.M.; Barou, R. Spectral-SAR realization of OECD-QSAR principles. Int. J. Chem. Model. 2011, 3, 173–190. [Google Scholar]
- Putz, A.M.; Putz, M.V. Spectral-structure activity relationship (Spectral-SAR) assessment of ionic liquids’ in silico ecotoxicity. In Ionic Liquids—New Aspects for the Future; Kadokawa, J., Ed.; InTech: Rijeka, Croatia; New York, NY, USA; Shanghai, China, 2013. [Google Scholar]
- Putz, M.V. Electronegativity and chemical hardness: Different patterns in quantum chemistry. Curr. Phys. Chem. 2011, 1, 111–139. [Google Scholar] [CrossRef]
- Putz, M.V. Chemical Orthogonal Spaces. In Mathematical Chemistry Monographs; Faculty of Science University of Kragujevac: Kragujevac, Serbia, 2012; Volume 14. [Google Scholar]
- OECD. Report from the Expert Group on (Quantitative) Structure-Activity Relationships [(Q)SARs] on the Principles for the Validation of (Q)SARs; Series on Testing and Assessment, No. 49; OECD: Paris, France, 2004; p. 206. Available online: http://www.oecd.org/env/ehs/risk-assessment/guidancedocumentsandreportsrelatedtoqsars.htm (accessed on 23 February 2013).
- OECD. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; Series on Testing and Assessment, No. 69; OECD: Paris, France; Volume 207, p. 154. Available online: http://www.oecd.org/env/ehs/risk-assessment/guidancedocumentsandreportsrelatedtoqsars.htm (accessed on 23 February 2013).
- Putz, M.V.; Putz, A.M. DFT chemical reactivity driven by biological activity: Applications for the toxicological fate of chlorinated PAHs. Struct. Bond. 2013, 150, 181–232. [Google Scholar]
- Hammett, L.P. The effect of structure upon the reactions of organic compounds. Benzene derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry; Plenum: New York, NY, USA, 1978. [Google Scholar]
- Hansch, C.; Leo, A. Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley-Interscience: New York, NY, USA, 1979. [Google Scholar]
- Prabhakar, Y.S.; Solomon, V.R.; Gupta, M.K.; Katti, S.B. QSAR studies on thiazolidines: A biologically privileged scaffold. Top. Heterocycl. Chem. 2006, 4, 161–249. [Google Scholar]
- Putz, M.V.; Duda-Seiman, C.; Duda-Seiman, D.M.; Putz, A.M. Turning SPECTRAL-SAR into 3D-QSAR analysis. Application on H+K+-ATPase inhibitory activity. Int. J. Chem. Model. 2008, 1, 45–62. [Google Scholar]
- Horn, J. The proton-pump inhibitors: Similarities and differences. Clin. Ther. 2000, 22, 266–280. [Google Scholar] [CrossRef]
- Vanderhoff, B.T.; Tahboub, R.M. Proton pump inhibitors: An update. Am. Fam. Physician 2002, 66, 273–280. [Google Scholar] [PubMed]
- Sachs, G.; Shin, J.M.; Howden, C.W. Review article: The clinical pharmacology of proton pump inhibitors. Aliment. Pharmacol. Ther. 2006, 23 (Suppl. S2), 2–8. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.S.; Shah, A.K.; Bariwal, J.; Shelke, S.M.; Kale, A.P.; Jagtap, J.R.; Bhosale, A.V. Recent advances in proton pump inhibitors and management of acid-peptic disorders. Bioorg. Med. Chem. 2007, 15, 1181–1205. [Google Scholar] [CrossRef] [PubMed]
- Thom, R. Stabilitè Structurelle et Morphogènése; Benjamin-Addison-Wesley: New York, NY, USA, 1973. [Google Scholar]
- Viret, J. Reaction of the organism to stress: The survival attractor concept. Acta Biotheor. 1994, 42, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Lacorre, P. Predation and generation processes through a new representation of the cusp catastrophe. Acta Biotheor. 1997, 45, 93–115. [Google Scholar] [CrossRef]
- Viret, J. Topological approach of Jungian psychology. Acta Biotheor. 2010, 58, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Cerf, R. Catastrophe theory enables moves to be detected towards and away from self-organization: The example of epileptic seizure onset. Biol. Cybern. 2006, 94, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Putz, M.V.; Lazea, M.; Putz, A.M.; Seiman-Duda, C. Introducing catastrophe-QSAR. Application on modeling molecular mechanisms of pyridinone derivative-type HIV non-nucleoside reverse transcriptase inhibitors. Int. J. Mol. Sci. 2011, 12, 9533–9569. [Google Scholar] [CrossRef] [PubMed]
- Aerts, D.; Czachor, M.; Gabora, L.; Kuna, M.; Posiewnik, A.; Pykacz, J.; Syty, M. Quantum morphogenesis: A variation on Thom’s catastrophe theory. Phys. Rev. 2003, 67. [Google Scholar] [CrossRef]
- Ciubotariu, D.; Deretey, E.; Oprea, T.I.; Sulea, T.; Simon, Z.; Kurunczi, L.; Chiriac, A. Multiconformational minimal steric difference. structure-acetylcholinesterase hydrolysis rates relations for acetic acid esters. Quant. Struct. Act. Relatsh. 1993, 12, 367–372. [Google Scholar] [CrossRef]
- Simon, Z.; Chiriac, A.; Ciubotariu, D. Metoda MTD (The MTD method, in Romanian language). In Relaţii Cantitative Structură Chimică—Activitate Biologică (QSAR). Metoda MTD (Quantitative Chemical Structure—Biological Activity Relationships Studies (QSAR). The MTD Method, in Romanian Language); Chiriac, A., Ciubotariu, D., Simon, Z., Eds.; Chapter 5; Mirton Publishing House: Timişoara, Romania, 1996. [Google Scholar]
- Ciubotariu, D.; Gogonea, V.; Medeleanu, M. QSAR Studies by Molecular Descriptions; Diudea, V.M., Ed.; Chapter 10; Nova Science Publishing Inc.: Huntington, NY, USA, 2001. [Google Scholar]
- Ciubotariu, D.; Medeleanu, M.; Vlaia, V.; Olariu, T.; Ciubotariu, C.; Dragos, D.; Corina, S. Molecular van der waals space and topological indices from the distance matrix. Molecules 2004, 9, 1053–1078. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, T.; Tanaka, H.; Baba, M. A novel lead for specific anti-HIV-1 agents: 1-[(2-Hydroxyethoxy)methyl]-6-(phenylthio)thymine. J. Med. Chem. 1989, 32, 2507–2509. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Tanaka, H.; de Clercq, E. Highly specific inhibition of human immunodeficiency virus type 1 by a novel 6-substituted acyclouridine derivative. Biochem. Biophys. Res. Commun. 1989, 165, 1375–1381. [Google Scholar] [CrossRef]
- Tanaka, H.; Hirayama, M.; Suzuki, M.; Miyasaka, T.; Matsuda, A.; Ueda, T. A lithiation route to C-5 substitution of an imidazole nucleoside and its application to the synthesis of 3-deazaguanosine. Tetrahedron 1986, 42, 1971–1980. [Google Scholar] [CrossRef]
- Tanaka, H.; Takashima, H.; Ubasawa, M.; Sekiya, K.; Nitta, I.; Baba, M.; Shigeta, S.; Walker, R.T.; de Clercq, E.; Miyasaka, T. Structure-activity relationships of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine analogues: Effect of substitutions at the C-6 phenyl ring and at the C-5 position on anti-HIV-1 activity. J. Med. Chem. 1992, 35, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Duda-Seiman, C.; Duda-Seiman, D.; Hegheş, A.; Nuţiu, R.; Ciubotariu, D.; Suceveanu, N. Modelarea compusilor pirimidinici cu activitate anti-HIV (Molecular modeling of pyrimidinic compounds with anti-HIV activity). J. Med. Pharm. 2004, 50, 144–149. [Google Scholar]
- Tanaka, H.; Baba, M.; Ubasawa, M.; Takashima, H.; Sekiya, K.; Nitta, I.; Shigeta, S.; Walker, R.T.; de Clercq, E.; Miyasaka, T. Synthesis and anti-HIV activity of 2-, 3-, and 4-substituted analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT). J. Med. Chem. 1991, 34, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Baba, M.; Saito, S.; Miyasaka, T.; Takashima, H.; Sekiya, K.; Ubasawa, M.; Nitta, I.; Walker, R.T. Specific anti-HIV-1 “acyclonucleosides” which cannot be phosphorylated: Synthesis of some deoxy analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J. Med. Chem. 1991, 34, 1508–1511. [Google Scholar] [CrossRef] [PubMed]
- HyperChem 7.01. In Program Package; Hypercube Inc.: Gainesville, FL, USA, 2002.
- Dean, P.M. Molecular Foundations of Drug-Receptor Interactions, 1st ed.; Cambridge University Press: Cambridge, UK, 1987; Volume 1. [Google Scholar]
- Putz, M.V. Residual-QSAR. Implications for genotoxic carcinogenesis. Chem. Cent. J. 2011, 5, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Putz, A.M.; Putz, M.V. Spectral inverse quantum (Spectral-IQ) method for modeling mesoporous systems: Application on silica films by FTIR. Int. J. Mol. Sci. 2012, 13, 15925–15941. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.X.; Li, Z.G.; Xie, H.Y.; Gao, J.R.; Zou, J.W. Quantitative structure-activity relationship analysis of aryl alkanol piperazine derivatives with antidepressant activities. Eur. J. Med. Chem. 2009, 44, 4367–4375. [Google Scholar] [CrossRef] [PubMed]
- Shelke, S.M.; Bhosale, S.H. Synthesis, antidepressant evaluation and QSAR studies of novel 2-(5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)-N-(substituted phenyl)acetamides. Bioorg. Med. Chem. Lett. 2010, 20, 4661–4664. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.C.; Salum, L.B.; Honorio, K.M.; Andricopulo, A.D.; da Silva, A.B. Pharmacophore-based 3D QSAR studies on a series of high affinity 5-HT1A receptor ligands. Eur. J. Med. Chem. 2010, 45, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Chitta, A.; Sivan, S.K.; Manga, V. 3D QSAR based design of novel oxindole derivative as 5HT7 inhibitors. J. Recept. Signal Transduct. Res. 2014, 34, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, S.H.; Kanhed, A.M.; Dash, R.C.; Suryawanshi, M.R.; Mahadik, K.R. Design, synthesis, pharmacological evaluation and computational studies of 1-(biphenyl-4-yl)-2-[4-(substituted phenyl)-piperazin-1-yl]ethanones as potential antipsychotics. Eur. J. Med. Chem. 2014, 74, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Milac, A.; Mernea, M.; Mihailescu, D.; Putz, M.V.; Buiu, C. Structure-biological function relationship extended to mitotic arrest-deficient 2-like protein mad2 native and mutants-new opportunity for genetic disorder control. Int. J. Mol. Sci. 2014, 15, 21381–21400. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Purisima, E.O. Molecular surface generation using a variable-radius solvent probe. Proteins 2006, 62, 244–261. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I. Property distribution of drug-related chemical databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA 1984, 81, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Lee, K.J. A comparative study of the second-order hydrophobic moments for globular proteins: The consensus scale of hydrophobicity and the CHARMM partial atomic charges. Int. J. Mol. Sci. 2011, 12, 8449–8465. [Google Scholar] [CrossRef] [PubMed]
- Rawat, N.; Biswas, P. Hydrophobic moments, shape, and packing in disordered proteins. J. Phys. Chem. B 2012, 116, 6326–6335. [Google Scholar] [CrossRef] [PubMed]
- Thaipisuttikul, P.; Ittasakul, P.; Waleeprakhon, P.; Wisajun, P.; Jullagate, S. Psychiatric comorbidities in patients with major depressive disorder. Neuropsychiatr. Dis. Treat. 2014, 10, 2097–2103. [Google Scholar] [PubMed]
- American Psychiatric Association (APA). Diagnostic and Statistical Manual of Mental Disorders; DSM-IV-TR: Washington, DC, USA, 2000. [Google Scholar]
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. J. Neuropsychopharmacol. 2008, 11, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Savil, M.; Banks, C.; Khanom, H.; Priebe, S. Do negative symptoms of schizophrenia change over time? A meta-analysis of longitudinal data. Psychol. Med. 2014, 26, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Michael, K.D.; Liu, Y.; Del Giovane, C.; Qin, B.; Cohen, D.; Gentile, S.; Xie, P. Systematic review of management for treatment-resistant depression in adolescents. BMC Psychiatry 2014, 14, 340. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.D.; Reilly, K.; Isenberg, K.; Villa, K.F. Antipsychotic patterns of use in patients with schizophrenia: Polypharmacy versus monotherapy. BMC Psychiatry 2014, 14, 341. [Google Scholar] [CrossRef] [PubMed]
- Campiani, G.; Butini, S.; Fattorusso, C.; Trotta, F.; Gemma, S.; Catalanotti, B.; Nacci, V.; Fiorini, I.; Cagnotto, A.; Mereghetti, I.; et al. Novel atypical antipsychotic agents: Rational design, an efficient palladium-catalyzed route, and pharmacological studies. J. Med. Chem. 2005, 48, 1705–1708. [Google Scholar] [CrossRef] [PubMed]
- Zyta, J.; Jaszczyszyn, A.; Swiatek, P.; Gasiorowski, K.; Malinka, W. Synthesis, pro-apoptotic activity and 2D-QSAR studies of new analogues of fluphenazine. Acta Pol. Pharm. 2014, 71, 49–58. [Google Scholar] [PubMed]
- Koshland, D.E.; Guacci, V. Sister chromatid cohesion: The beginning of a long and beautiful relationship. Curr. Opin. Cell Biol. 2000, 12, 297–301. [Google Scholar] [CrossRef]
- Yu, H. Structural activation of Mad2 in the mitotic spindle checkpoint: The two-state Mad2 model versus the Mad2 template model. J. Cell Biol. 2006, 173, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Ricke, R.M.; van Deursen, J.M. Correction of microtubule-kinetochore attachment errors: Mechanisms and role in tumor suppression. Semin. Cell Dev. Biol. 2011, 22, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Bolanos-Garcia, V.M.; Wu, Q.; Ochi, T.; Chirgadze, D.Y.; Sibanda, B.L.; Blundell, T.L. Spatial and temporal organization of multi-protein assemblies: Achieving sensitive control in information-rich cell-regulatory systems. Math. Phys. Eng. Sci. 2012, 370, 3023–3039. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sun, H.; Tomchick, D.R.; Yu, H.; Luo, X. Structure of human Mad1 C-terminal domain reveals its involvement in kinetochore targeting. Proc. Natl. Acad. Sci. USA 2012, 109, 6549–6554. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yu, H. Protein metamorphosis: The two-state behavior of Mad2. Structure 2008, 16, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, B.; Liu, C.J.; Tomchick, D.R.; Machius, M.; Rizo, J.; Yu, H.; Luo, X. Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol. 2008, 6, e50. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yu, H. Mutual regulation between the spindle checkpoint and APC/C. Semin. Cell Dev. Biol. 2011, 22, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Holland, A.J.; Fachinetti, D.; Kulukian, A.; Cetin, B.; Cleveland, D.W. Catalytic assembly of the mitotic checkpoint inhibitor BubR1-Cdc20 by a Mad2-induced functional switch in Cdc20. Mol. Cell 2013, 51, 92–104. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
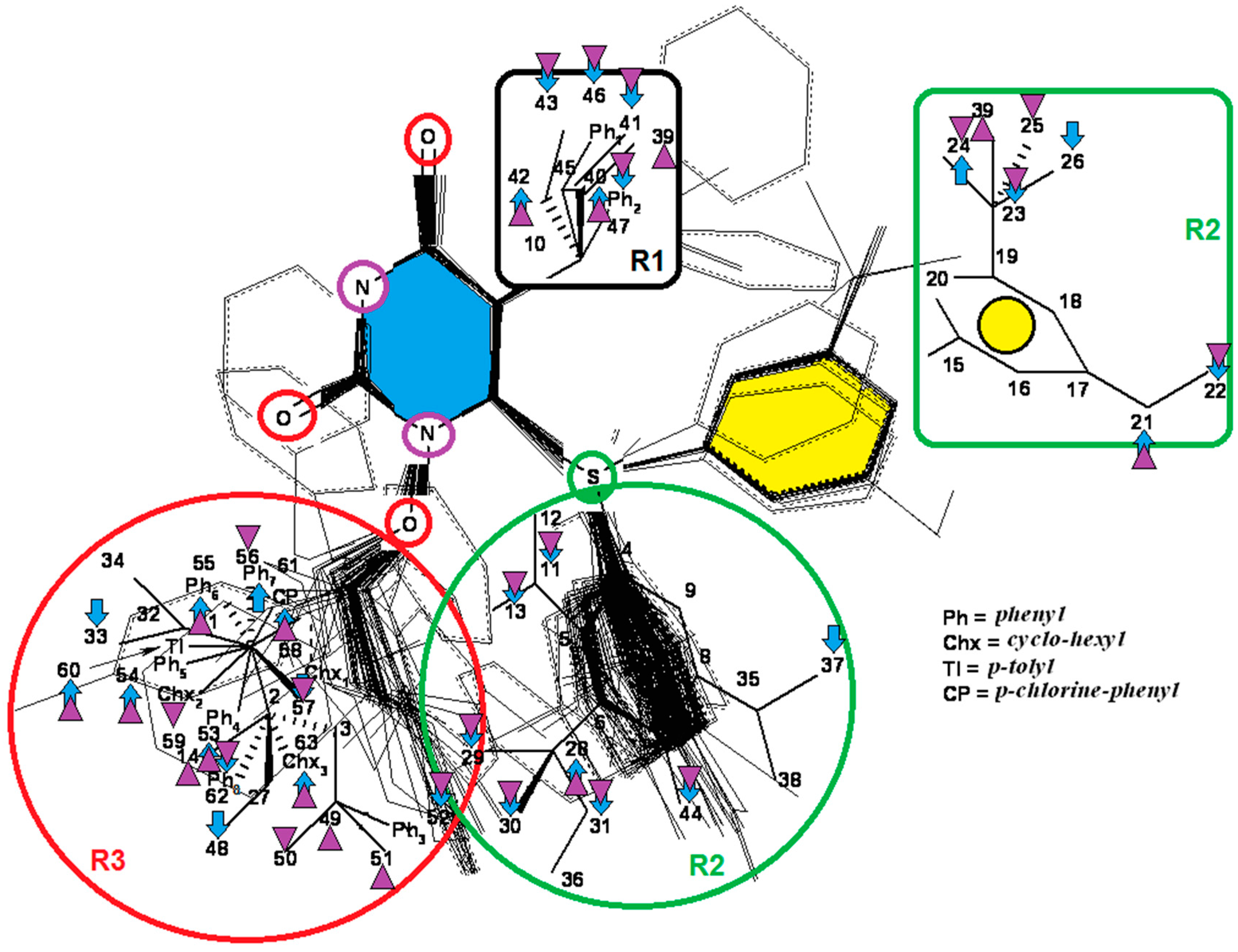{kind=link}
| Name of Descriptors | Chemical and Physical Considerations of Descriptors | Reference |
|---|---|---|
| Dipole-mag | electronic descriptor involved in ligand-receptor interactions | [84] |
| SASA | = total solvent accessible surface area | [85] |
| FOSA | = hydrophobic component of the total solvent accessible surface area (saturated carbon and attached hydrogen) | [85] |
| FISA | = hydrophilic component of the total solvent accessible surface area (SASA on N, O, H on heteroatoms, and carbonyl C) | [85] |
| Glob | globularity descriptor | [85] |
| CoMFA, electrostatic descriptor | the electrostatic interactions between a probe atom, usually an sp3-carbon atom with a +1 charge, and the ligands are calculated at uniform grid points following the Coulombic function | [86,87] |
| CoMFA, steric descriptor | the steric interactions between a probe atom, usually an sp3-carbon atom with a +1 charge, and the ligands are calculated at uniform grid points following the Lennard–Jones function | [86,87] |
| Descriptors | Chemical and Physical Considerations of Descriptors | Reference |
|---|---|---|
| Steric and hydrogen bonding interaction energies | the energies calculated with the water probe contain the steric and hydrogen bonding interaction energies, supplied by the presence of sodium, potassium, calcium and iron | [21] |
| EA | electron affinity | [88] |
| BBB | blood brain barrier | [88] |
| QPlogBB | brain/blood partition coefficient | [88] |
| CoMFA/CoMSIA, electrostatic descriptor | The electrostatic interactions between a probe atom, usually a sp3-carbon atom with a +1 charge, and the ligands are calculated at uniform grid points following the Coulombic function | [14] |
| CoMFA/CoMSIA, steric descriptor | The steric interactions between a probe atom, usually an sp3-carbon atom with a +1 charge, and the ligands are calculated at uniform grid points following the Lennard–Jones function | [14] |
| Classes of Antidepressants | Chemical Structure | Molecules Name | Chemical Classes |
|---|---|---|---|
| Selective serotonin reuptake inhibitors (SSRIs) |  | sertraline | tetrahydronaphthalenes |
 | paroxetine | piperidines | |
 | fluvoxamine | benzenes | |
 | escitalopram | benzofurans | |
| Serotonin norepinephrine reuptake inhibitors (SNRIs) |  | venlafaxine | phenols |
 | desvenlafaxine | phenols | |
 | duloxetine | naphthalenes | |
| Newer generation of drugs |  | clozapine | benzodiazepines |
 | ziprasidone | phenethylamines | |
 | paliperidone | benzoxazoles | |
 | risperidone | benzoxazoles | |
 | quetiapine | benzothiazepines | |
 | olanzapine | benzodiazepine |
| QSAR Models | q2 (Cross-Validated r2) | r2 (Fitted Correlation) | Root Mean Square Error (RMSE) | Cross-Validated RMSE | Fischer Test |
|---|---|---|---|---|---|
| QSAR Model 1 | 0.53 | 0.82 | 0.15 | 0.27 | 13.22 |
| QSAR Model 2 | 0.65 | 0.83 | 0.14 | 0.20 | 10.03 |
| QSAR Model 3 | 0.60 | 0.90 | 0.10 | 0.25 | 10.23 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putz, M.V.; Duda-Seiman, C.; Duda-Seiman, D.; Putz, A.-M.; Alexandrescu, I.; Mernea, M.; Avram, S. Chemical Structure-Biological Activity Models for Pharmacophores’ 3D-Interactions. Int. J. Mol. Sci. 2016, 17, 1087. https://doi.org/10.3390/ijms17071087
Putz MV, Duda-Seiman C, Duda-Seiman D, Putz A-M, Alexandrescu I, Mernea M, Avram S. Chemical Structure-Biological Activity Models for Pharmacophores’ 3D-Interactions. International Journal of Molecular Sciences. 2016; 17(7):1087. https://doi.org/10.3390/ijms17071087
Chicago/Turabian StylePutz, Mihai V., Corina Duda-Seiman, Daniel Duda-Seiman, Ana-Maria Putz, Iulia Alexandrescu, Maria Mernea, and Speranta Avram. 2016. "Chemical Structure-Biological Activity Models for Pharmacophores’ 3D-Interactions" International Journal of Molecular Sciences 17, no. 7: 1087. https://doi.org/10.3390/ijms17071087