
Comparative Analysis of the Chloroplast Genomic Information of Cunninghamia lanceolata (Lamb.) Hook with Sibling Species from the Genera Cryptomeria D. Don, Taiwania Hayata, and Calocedrus Kurz

Abstract
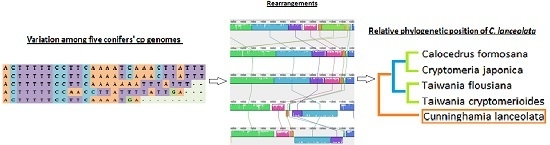
:
1. Introduction
2. Results and Discussion
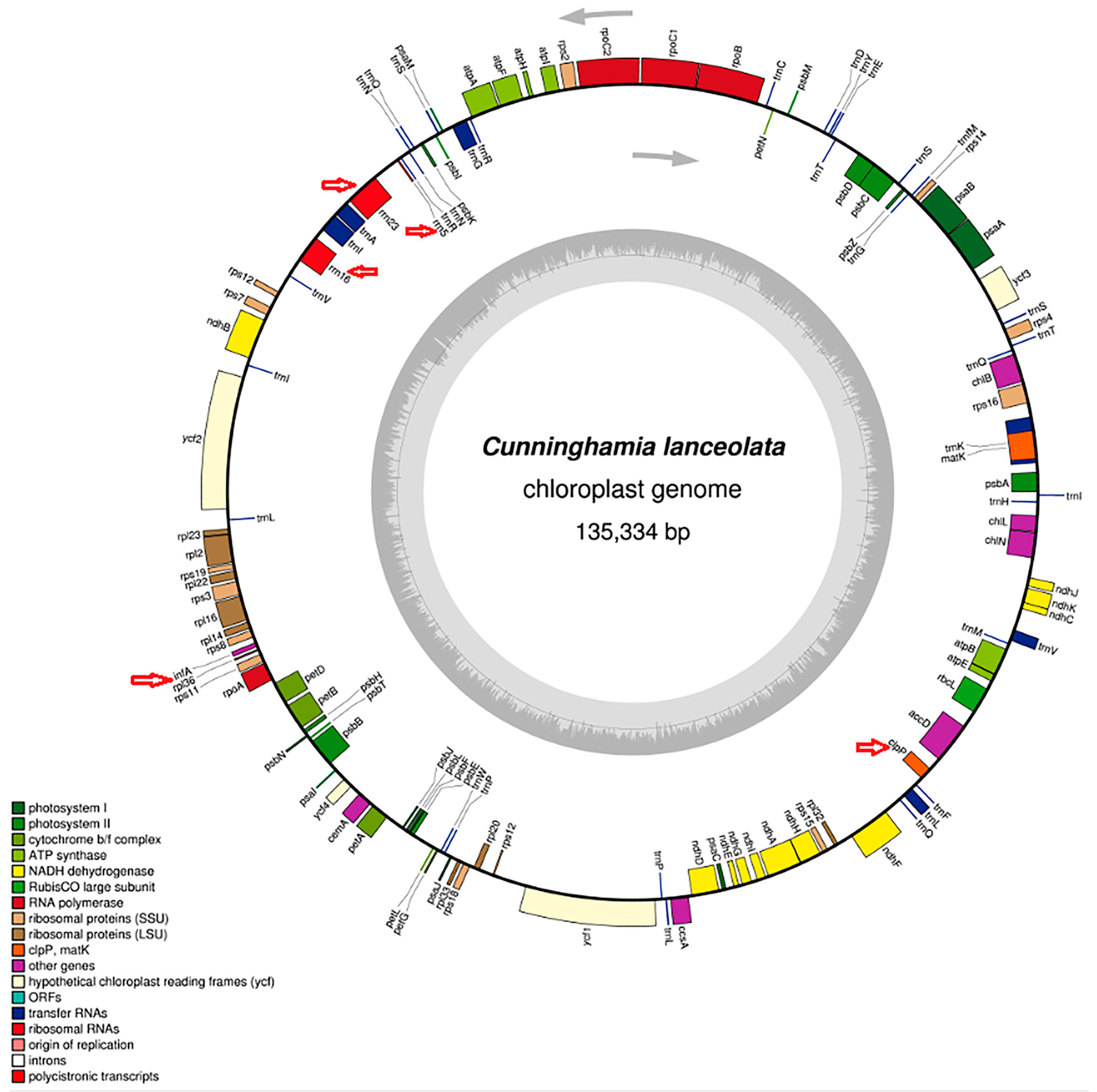
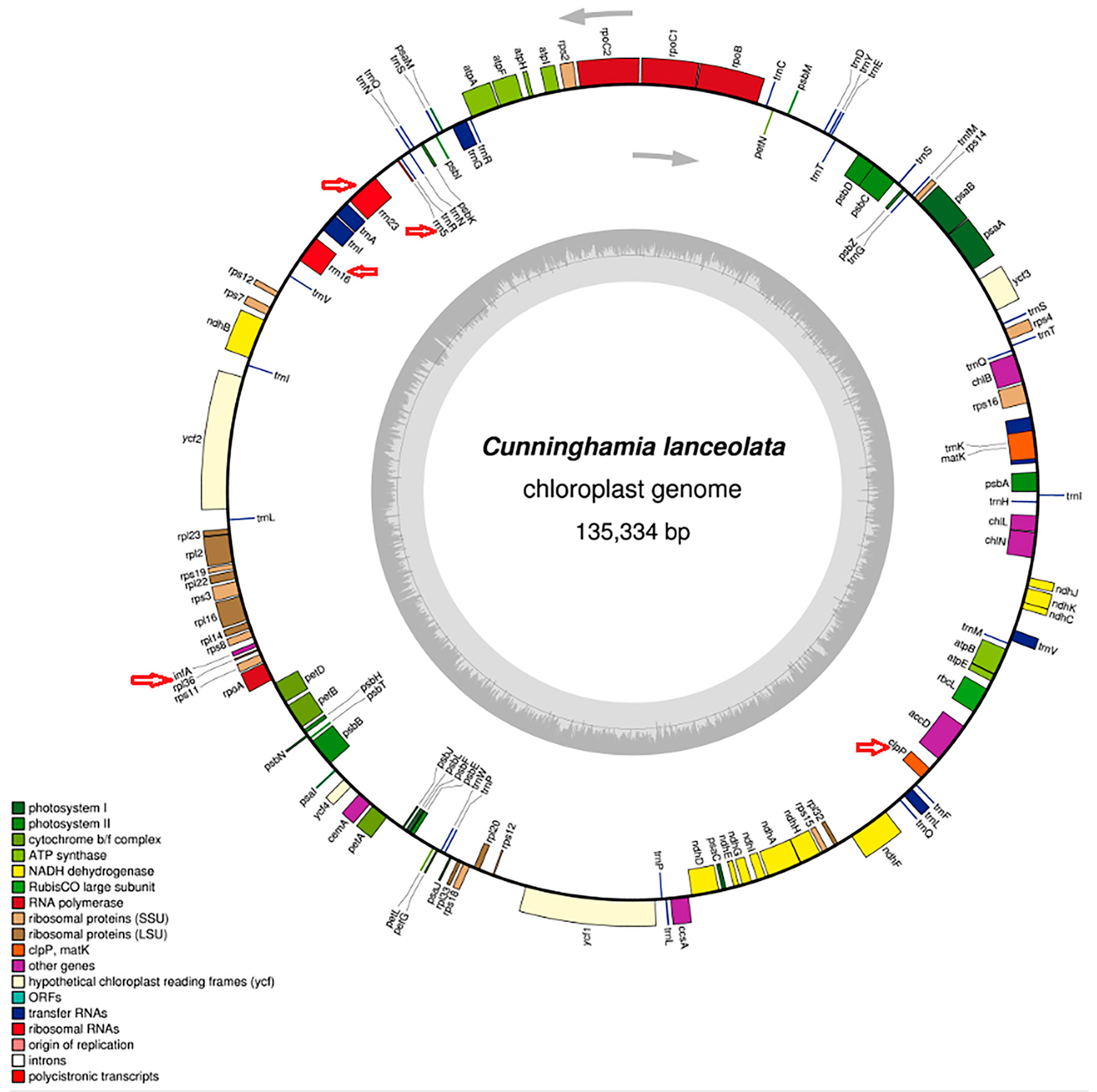
2.1. Re-Characterization of the Cunninghamia lanceolata Chloroplast Genome
2.2. Repeats Analysis
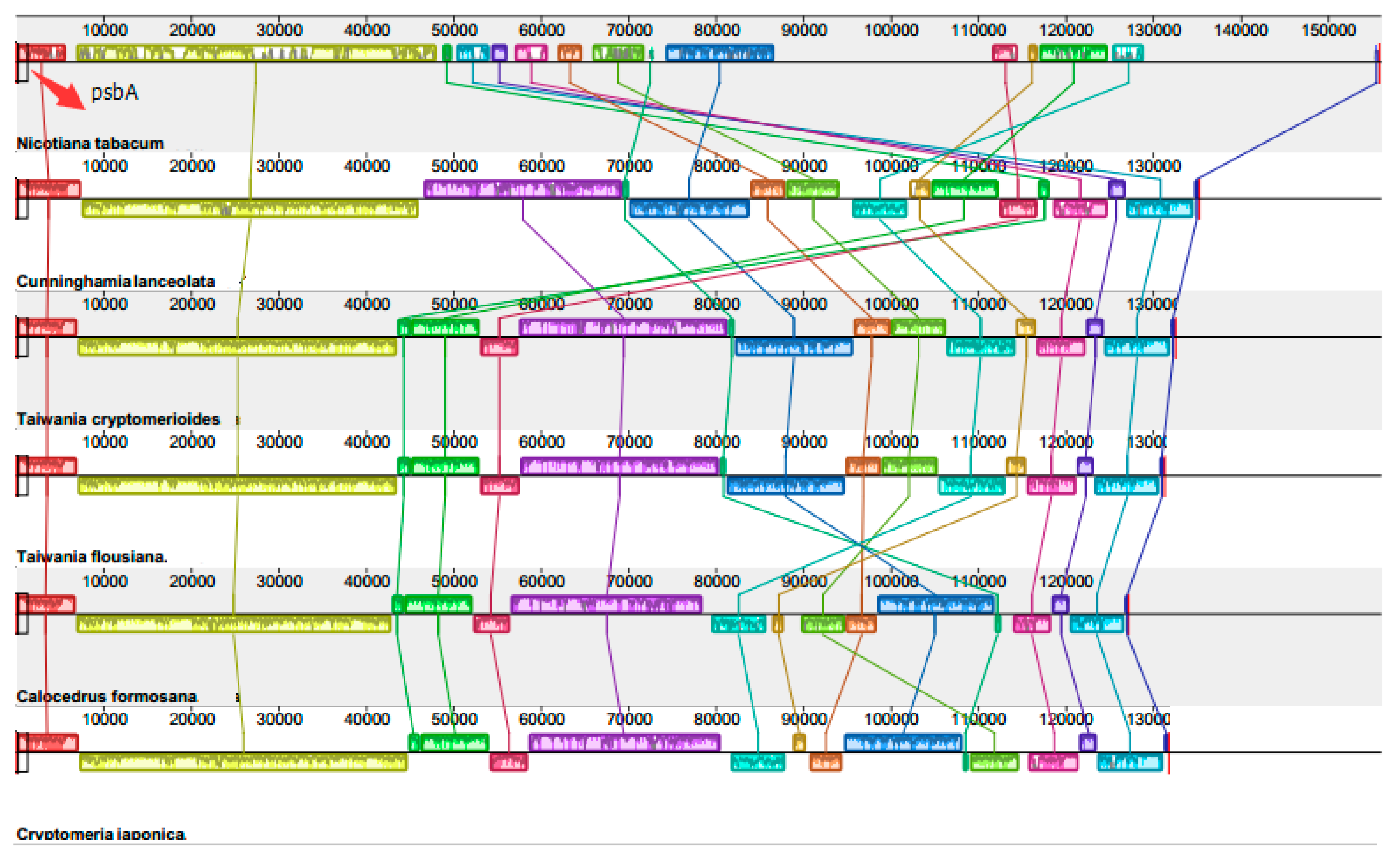
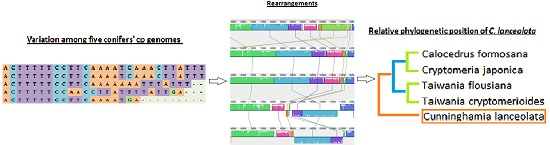
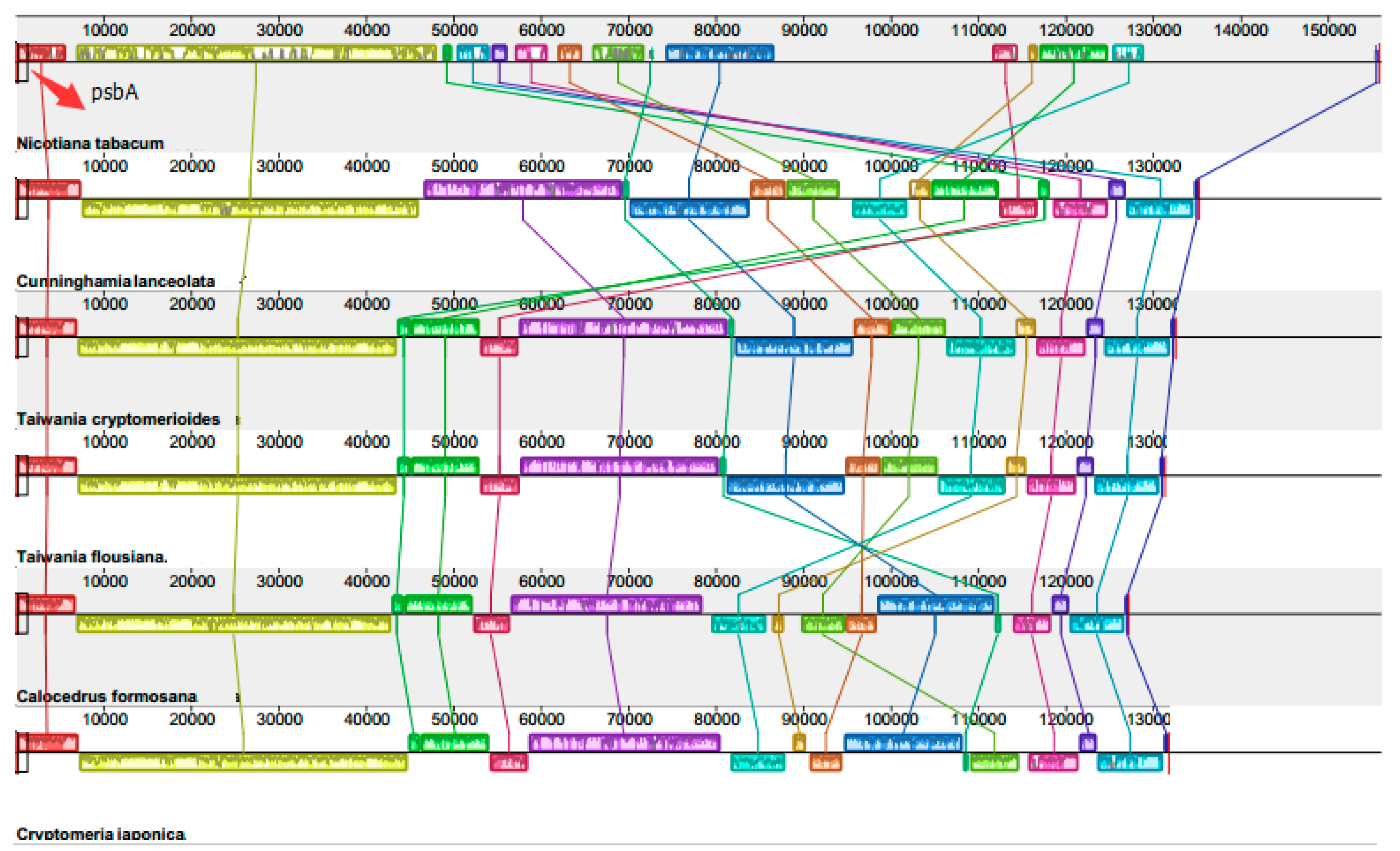
2.3. Chloroplast Genome Rearrangements
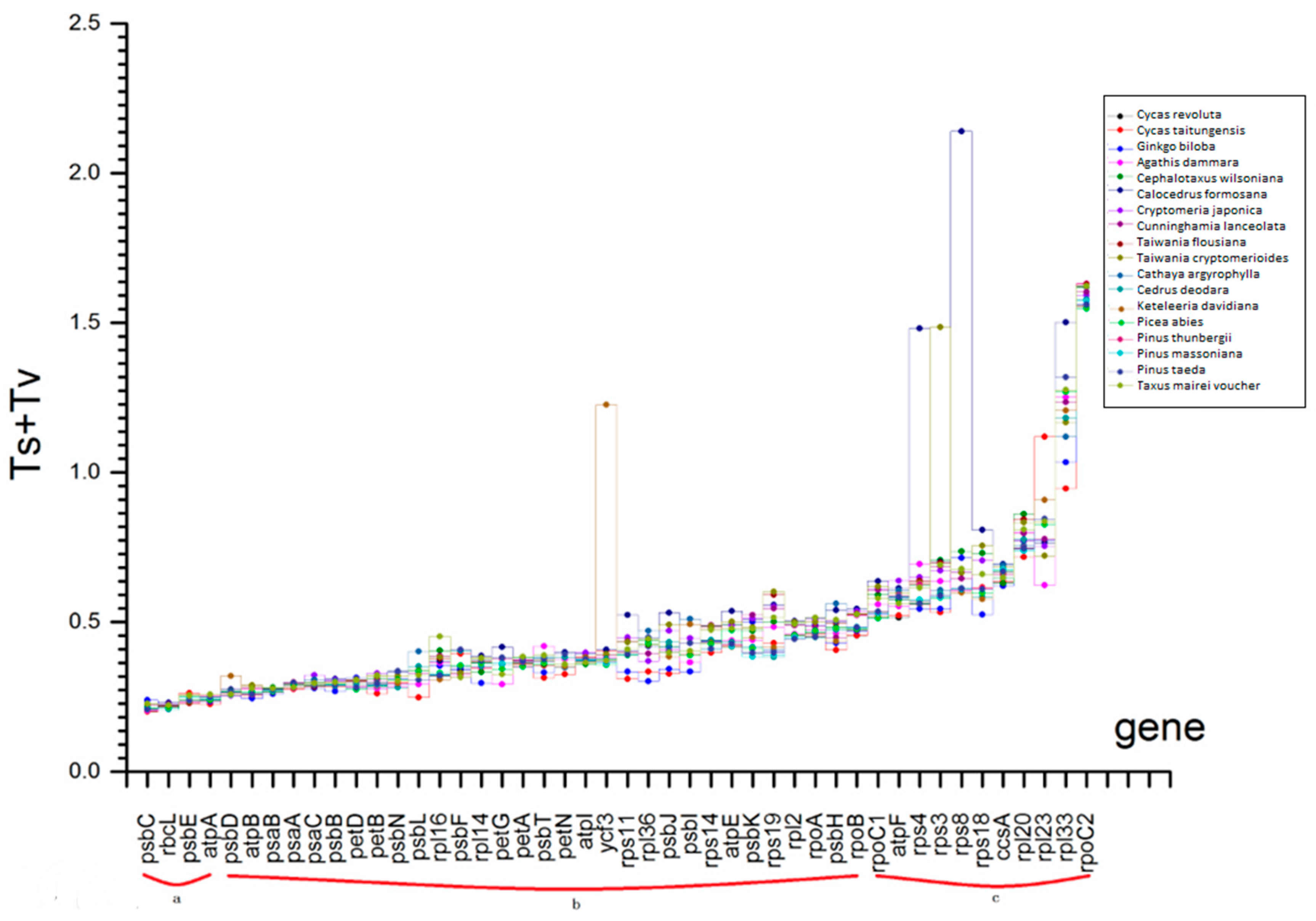
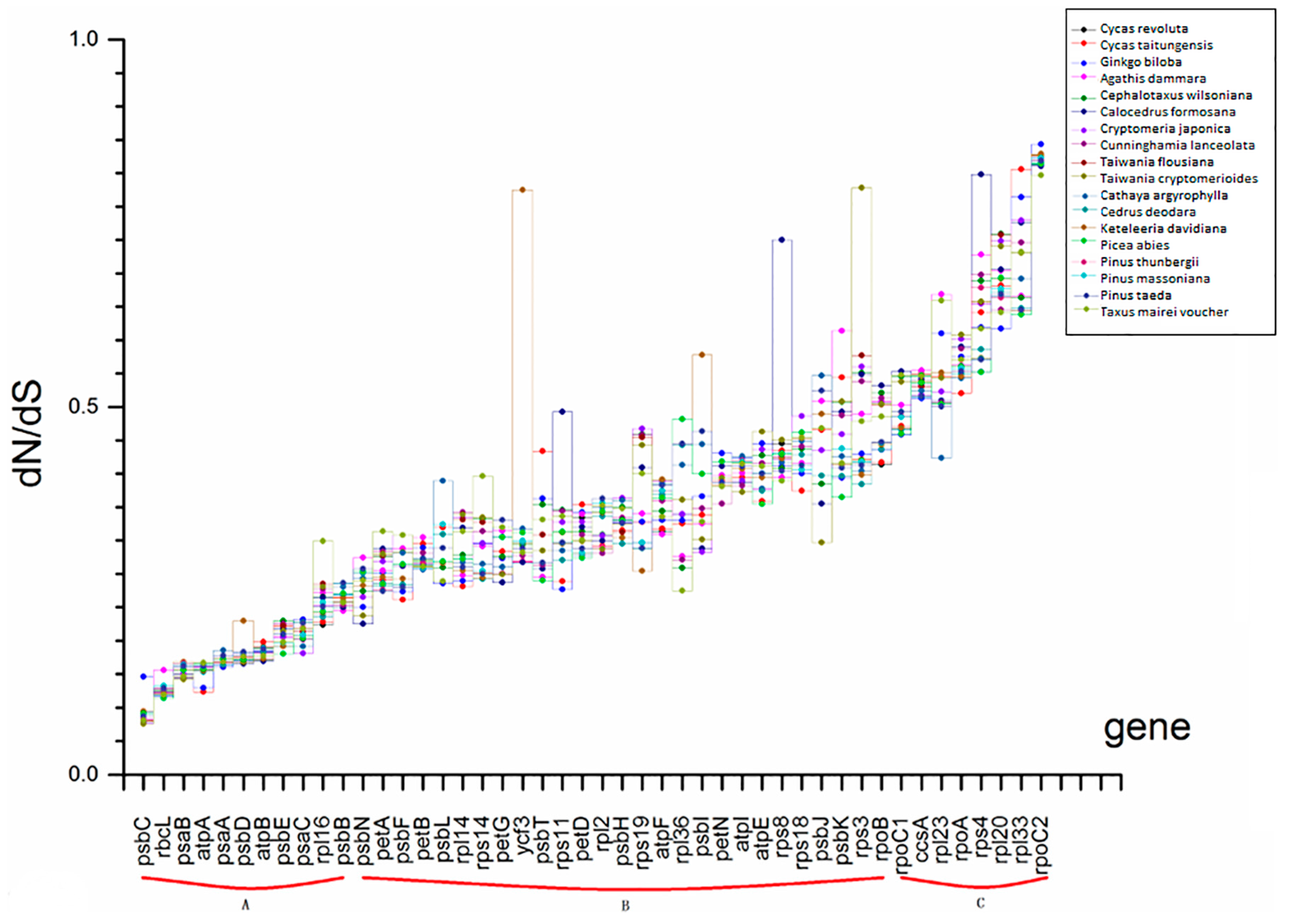
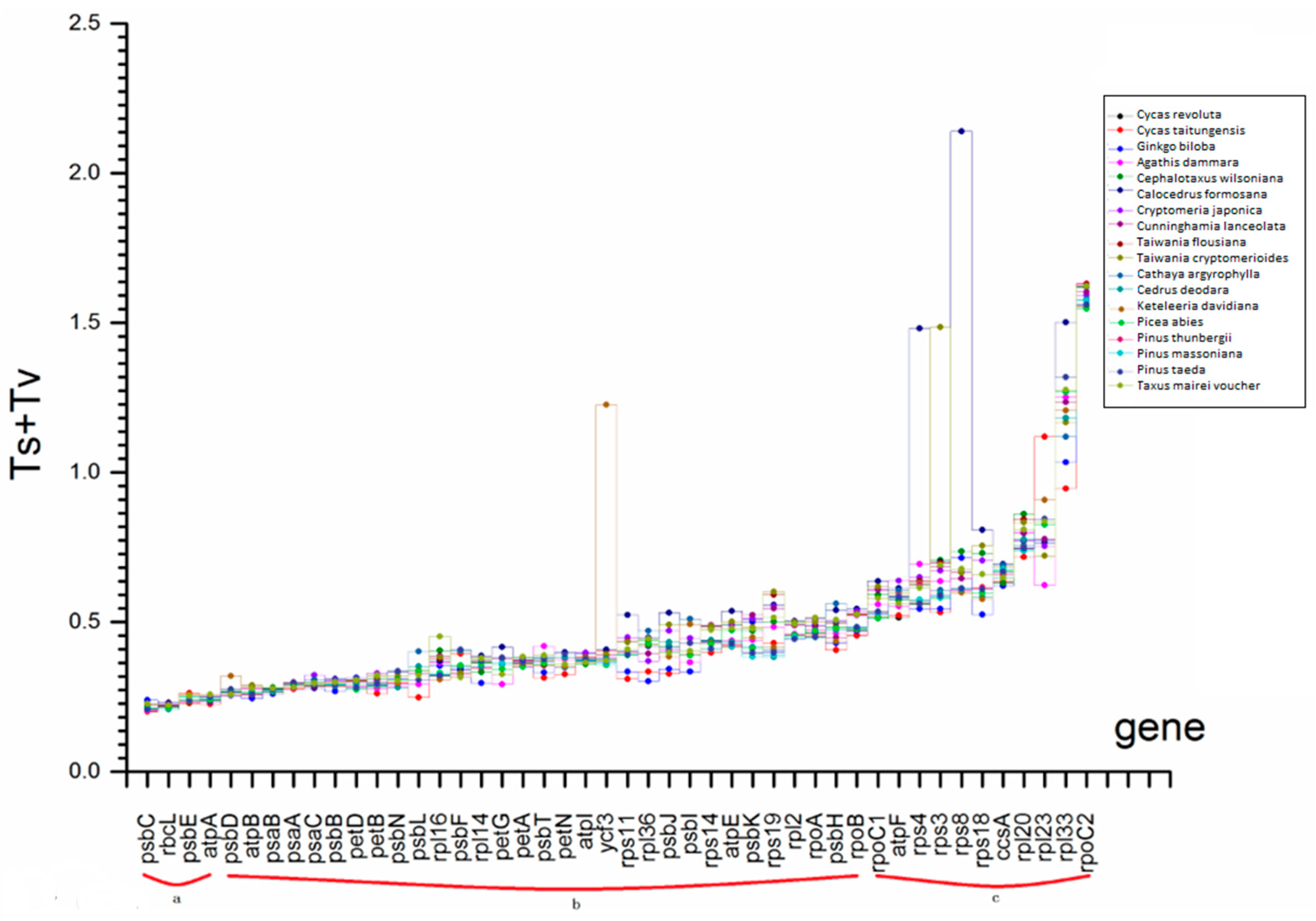
2.4. Selection Force and Substitution Rate Assessment
2.5. Phylogenetic Indication Based on Gene Function, Selection Force and Substitution Rate
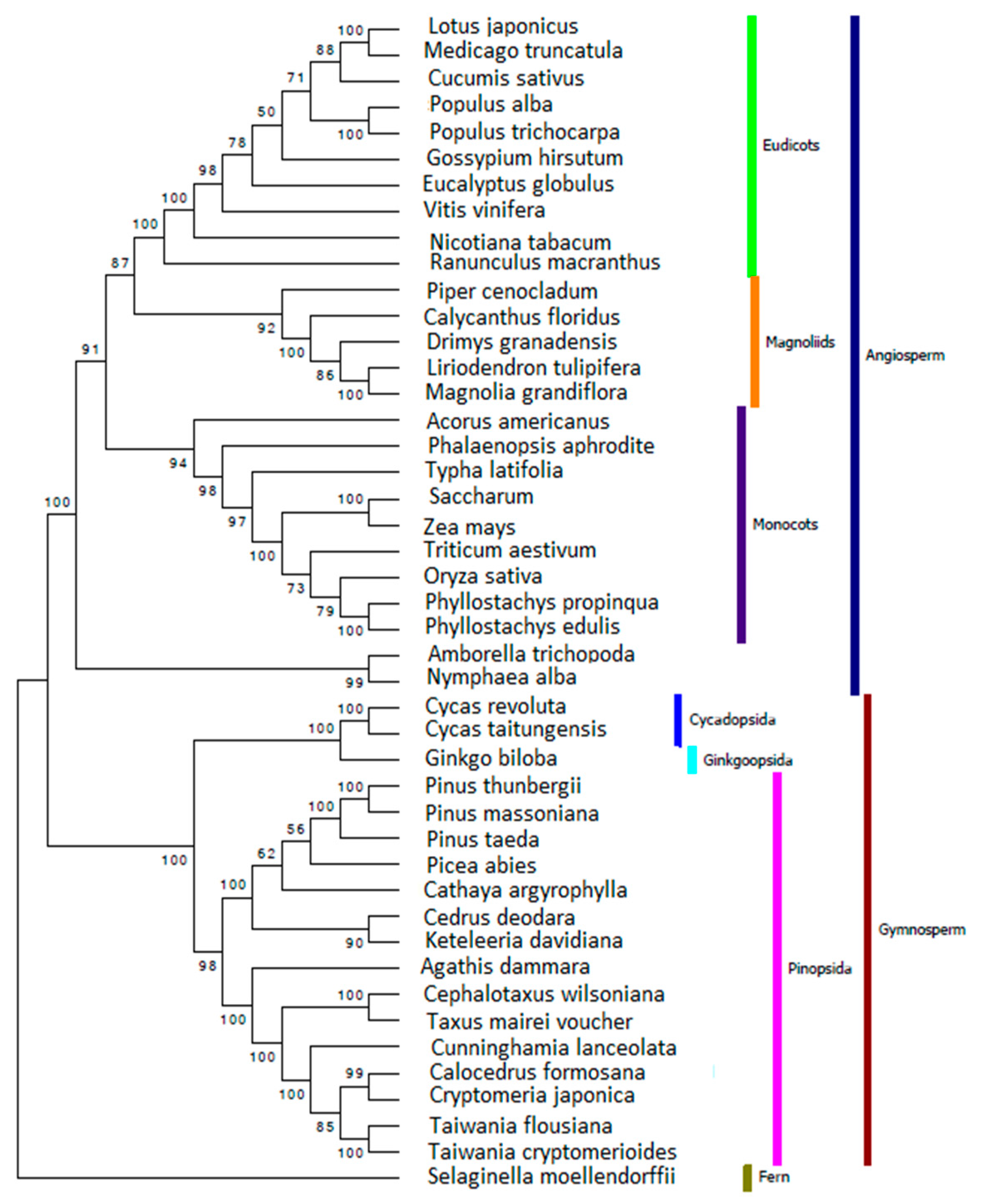
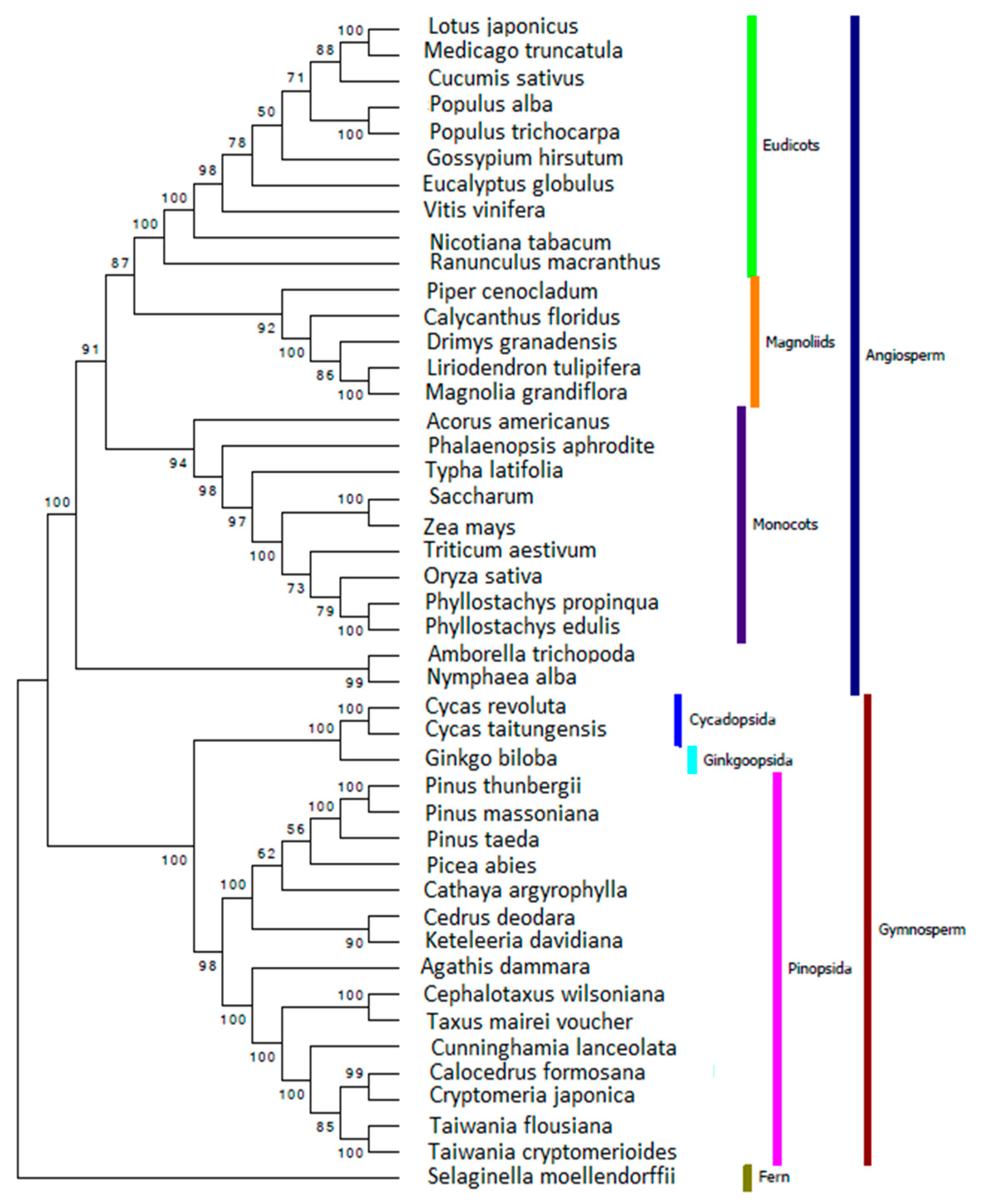
2.6. Reconstructing the Phylogenetic Relationships for Gymnosperm Based on Chloroplast Genome
3. Materials and Methods
3.1. Genome Sequence Collection
3.2. Re-Visiting the Chloroplast Genome
3.3. IR Identification and Sequence Repeat Analysis
3.4. Comparative Analysis of Chloroplast Genomes
3.5. Selection Force and Substitution Rate Assessment
3.6. Phylogenetic Indication Based on Gene Function, Selection Force and Substitution Rate
3.7. Reconstructing the Phylogenetic Relationships for Gymnosperms Based on Chloroplast Genome
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LSC | large single copy |
| SSC | small single copy |
| IR | inverted repeat |
| ML | maximum likelihood |
References
- Pilger, R. Gymnospermae: Coniferae. In Die Natureüchen Pflanzenfamilien, 2nd ed.; Fischer, E., Claussen, P., Harms, H., Prantl, K., Engler, A., Eds.; W. Engelmann: Leipzig, Germany, 1926; pp. 121–407. [Google Scholar]
- Stefanoviac, S.; Jager, M.; Deutsch, J.; Broutin, J.; Masselot, M. Phylogenetic relationships of conifers inferred from partial 28S rRNA gene sequences. Am. J. Bot. 1998, 85, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Horiuchi, J. Cunninghamia nodensis sp. nov., from the Palaeogene Noda Group, northeast Japan. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 1978, 54, 589–594. [Google Scholar] [CrossRef]
- Kilpper, K. Koniferen aus den Tertiären Deckschichten des Niederrheinischen Hauptflözes, 3.Taxodiaceae und Cupressaceae. Palaeontogr. Abt. B 1968, 124, 102–111. [Google Scholar]
- Ferguson, D.K. On the phytogeography of Coniferales in the European Cenozoic. Palaeogeogr. Palaeoclimatol. Palaeoecol. 1967, 3, 73–110. [Google Scholar] [CrossRef]
- Florin, R. The distribution of conifer and taxad genera in time and space. Acta Horti Bergiani 1963, 20, 121–312. [Google Scholar]
- Endo, R. A Collection of Plant Fossils; The Asakura Publishing Co., Ltd.: Tokyo, Japan, 1966. [Google Scholar]
- Meng, X.; Chen, F.; Deng, S. Fossil Plant Cunninghamia asiatica (Krassilov) Comb. Nov. Acta Bot. Sin. 1988, 30, 649–654. [Google Scholar]
- Zeng, W. Plate tectonics on the relationship between the flora of the southeastern China and the North America. J. Xiamen Univ. Nat. Sci. 1989, 28, 410–413. [Google Scholar]
- Chen, Y.; Shi, J. Some fundamental problems on the genetic improvement of Chinese fir. J. Nanjing For. Univ. Nat. Sci. Ed. 1983, 4, 6–19. [Google Scholar]
- Shi, J.; Zhen, Y.; Zheng, R. Proteome profiling of early seed development in Cunninghamia lanceolata (Lamb.) Hook. J. Exp. Bot. 2010, 61, 2367–2381. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, Z.; Boyd, S.; Williams, D. Chemical composition of decomposing stumps in successive rotation of Chinese fir (Cunninghamia lanceolata (Lamb.) Hook.) plantations. Chin. Sci. Bull. 2005, 50, 2581–2586. [Google Scholar] [CrossRef]
- Wang, G.; Gao, Y.; Yang, L.; Shi, J. Identification and analysis of differentially expressed genes in differentiating xylem of Chinese fir (Cunninghamia lanceolata) by suppression subtractive hybridization. Genome 2007, 50, 1141–1155. [Google Scholar] [PubMed]
- Wang, G.; Gao, Y.; Wang, J.; Yang, L.; Song, R.; Li, X.; Shi, J. Overexpression of two cambium-abundant Chinese fir (Cunninghamia lanceolata) α-expansin genes ClEXPA1 and ClEXPA2 affect growth and development in transgenic tobacco and increase the amount of cellulose in stem cell walls. Plant Biotechnol. J. 2011, 9, 486–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Liu, W.; Luo, Z.; Wang, P.; Zhang, Y.; Zheng, R.; Shi, J. Transcriptome characteristics and six alternative expressed genes positively correlated with the phase transition of annual cambial activities in Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook). PLoS ONE 2013, 8, 71562. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Su, Q.; Zheng, R.; Liu, G.; Lu, Y.; Bian, L.; Chen, J.; Shi, J. ClRTL1 encodes a Chinese Fir RNase III-like protein involved in regulating shoot branching. Int. J. Mol. Sci. 2015, 16, 25691–25710. [Google Scholar] [CrossRef] [PubMed]
- Trontin, J.-F.; Klimaszewska, K.; Morel, A.; Hargreaves, C.; Lelu-Walter, M.-A. Molecular aspects of conifer zygotic and somatic embryo development: a review of genome-wide approaches and recent insights. In In Vitro Embryogenesis in Higher Plants; Springer Science+Business Media LLC: New York, NY, USA, 2016; pp. 167–207. [Google Scholar]
- Chaw, S.-M.; Chang, C.-C.; Chen, H.-L.; Li, W.-H. Dating the monocot-dicot divergence and the origin of core eudicots using whole chloroplast genomes. J. Mol. Evol. 2004, 58, 424–441. [Google Scholar] [PubMed]
- Jakobsson, M.; Säll, T.; Lind-Halldén, C.; Halldén, C. The evolutionary history of the common chloroplast genome of Arabidopsis thaliana and A. suecica. J. Evol. Biol. 2007, 20, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Muse, S.V. Examining rates and patterns of nucleotide substitution in plants. Plant Mol. Biol. 2000, 42, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-B.; Kwon, Y.; Yu, H.-J.; Lim, K.-B.; Seo, J.-H.; Mun, J.-H. The complete chloroplast genome of Phalaenopsis “Tiny Star”. Mitochondrial DNA 2016, 27, 1300–1302. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Palmer, J.D. Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In Molecular Systematics of Plants; Springer: New York, NY, USA, 1992; pp. 14–35. [Google Scholar]
- Lavin, M.; Doyle, J.J.; Palmer, J.D. Evolutionary significance of the loss of the chloroplast-DNA inverted repeat in the Leguminosae subfamily Papilionoideae. Evolution 1990, 44, 390–402. [Google Scholar] [CrossRef]
- Wu, C.S.; Wang, Y.N.; Hsu, C.Y.; Lin, C.P.; Chaw, S.M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Chaw, S.M. Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): Evolution towards shorter intergenic spacers. Plant Biotechnol. J. 2014, 12, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Hallick, R.B.; Hong, L.; Drager, R.G.; Favreau, M.R.; Monfort, A.; Orsat, B.; Spielmann, A.; Stutz, E. Complete sequence of Euglena gracilis chloroplast DNA. Nucleic Acids Res. 1993, 21, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M. The Chloroplast Genome; Springer: New York, NY, USA, 1992. [Google Scholar]
- NCBI, Complete Genomes: Eukaryota, 2016. Available online: http://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?opt=plastid&taxid=2759&sort=Genome (accessed on 15 April 2016).
- Kluge, A.G. A concern for evidence and a phylogenetic hypothesis of relationships among Epicrates (Boidae, Serpentes). Syst. Zool. 1989, 38, 7–25. [Google Scholar] [CrossRef]
- Lu, Y.; Ran, J.H.; Guo, D.M.; Yang, Z.Y.; Wang, X.Q. Phylogeny and divergence times of gymnosperms inferred from single-copy nuclear genes. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liu, T.; Liu, C.; Zhou, F.; Lai, X.E.; Hu, D.; Chen, J.; Huang, S. The complete chloroplast genome sequence of Cunninghamia lanceolata. Mitochondrial DNA 2015. [Google Scholar] [CrossRef]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: Diversified genomic structure of coniferous species. BMC Plant Biol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-H.; Liu, Q.; Hu, W.; Wang, T.; Xue, Q.; Messing, J. Dynamics of chloroplast genomes in green plants. Genomics 2015, 106, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.M.; Duvall, M.R. The chloroplast genome of Anomochloa marantoidea (Anomochlooideae; Poaceae) comprises a mixture of grass-like and unique features. Am. J. Bot. 2010, 97, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Tsudzuki, T.; Sugiura, M. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc. Nat. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Stoebe, B.; Goremykin, V.; Hansmann, S.; Hasegawa, M.; Kowallik, K.V. Gene transfer to the nucleus and the evolution of chloroplasts. Nature 1998, 393, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qi, Z.C.; Zhao, Y.P.; Fu, C.X.; Xiang, Q.Y.J. Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales—Influences of gene partitions and taxon sampling. Mol. Phylogenet. Evol. 2012, 64, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.V. Natural selection and phylogenetic analysis. Proc. Natl. Acad. Sci. USA 2009, 106, 8799–8800. [Google Scholar] [CrossRef] [PubMed]
- Klopfstein, S.; Kropf, C.; Quicke, D.L. An evaluation of phylogenetic informativeness profiles and the molecular phylogeny of Diplazontinae (Hymenoptera, Ichneumonidae). Syst. Biol. 2010, 59, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.P.; Lopez-Giraldez, F. Optimal selection of gene and ingroup taxon sampling for resolving phylogenetic relationships. Syst. Biol. 2010, 59, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.P.; Leuenberger, C. Taxon sampling and the optimal rates of evolution for phylogenetic inference. Syst. Biol. 2011, 60, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Buckley, T.R. Model selection and model averaging in phylogenetics: Advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; Ashton, N.W. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.S.; Wiegert, K.E.; Triemer, R.E. Characterization of Euglenaformis gen. nov. and the chloroplast genome of Euglenaformis [Euglena] proxima (Euglenophyta). Phycologia 2014, 53, 66–73. [Google Scholar] [CrossRef]
- Finn, R.D.; Mistry, J.; Schuster Böckler, B.; Griffiths Jones, S.; Hollich, V.; Lassmann, T.; Moxon, S.; Marshall, M.; Khanna, A.; Durbin, R. Pfam: Clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Huai, J.L.; Wang, M.; He, J.G.; Zheng, J.; Dong, Z.G.; Lv, H.K.; Zhao, J.F.; Wang, G.Y. Cloning and characterization of the SnRK2 gene family from Zea mays. Plant Cell Rep. 2008, 27, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Kaittanis, C.; Saski, C.; Lee, S.B.; Tomkins, J.; Alverson, A.J.; Daniell, H. Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: Effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol. Biol. 2006, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Song, W.N. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, 36869. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [PubMed]
- Olsen, C.; Qaadri, K. Geneious R7: A Bioinformatics Platform for Biologists. In Proceedings of the Plant and Animal Genome XXII Conference, San Diego, CA, USA, 10–15 January 2014.
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; Carlson, J.E.; Boore, J.L.; Jansen, R.K. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: Implications for the phylogenetic relationships of magnoliids. BMC Evol. Biol. 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993, 36, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Pamilo, P.; Bianchi, N.O. Evolution of the Zfx and Zfy genes: Rates and interdependence between the genes. Mol. Biol. Evol. 1993, 10, 271–281. [Google Scholar] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, H.C.; Lin, I.P.; Chow, T.Y.; Chen, H.H.; Chen, W.H.; Cheng, C.H.; Lin, C.Y.; Liu, S.M.; Chang, C.C. The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): Comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 2006, 23, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Race, H.L.; Herrmann, R.G.; Martin, W. Why have organelles retained genomes? Trends Genet. 1999, 15, 364–370. [Google Scholar] [CrossRef]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.C.; Wilhelm, S.W.; Gobler, C.J.; Bullerjahn, G.; Jacobs, M.A.; McKay, J.; Sims, E.H.; Gillett, W.G.; Zhou, Y.; Haugen, E. Analyses of the complete chloroplast genome sequences of two members of the pelagophyceae: Aureococcus anophagefferens CCMP1984 and Aureoumbra lagunensis CCMP15071. J. Phycol. 2010, 46, 602–615. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repeat Number | Size (bp) | Repeat Unit | Location |
|---|---|---|---|
| 1 | 30 | AAAAAAGAAAAAATCAACACGAGCAGTAAAA(×2) 1 | rpoC2 (CDS 2) |
| 2 | 36 | TTGGACGATTTAGAATACGAAACTACATTGGACAAT(×2) | ycf2 (CDS) |
| 3 | 132 | AAGTATTATTTTCAATGGAAAAAAGCATTCAAAAGATACTATATTGAATTCATAAAAACATTGAATAAGTATTATTTTGAATGGAAAAAAGTATTATTTTGATTCTGTATTAAATTCATAAAAACATTGAAT(×2) | ycf2 (CDS) |
| 4 | 66 | AAGTATTATTTTGAATGGAAAAAAGTATTAAAAGATTCTGTATTGAATTCATAAAAACATTGAAT(×4) | ycf2 (CDS) |
| 5 | 94 | TTACGAGCAATAATGAAACAAAACTTGCCAAATACAATGATGACATTATATAATGATACATAGAGATATTGTGTTGCGTTGTTTACAAAACATG(×2) | IGS 3 (rpl20, ycf1) |
| 6 | 104 | CAAAACTTGCCAAATACAATGATGACATTATATAATGATACATAGAGATATTGTGTTGCGTTGTTTACAAAACATGTTACGAGCAATAATGAAACAAAACTTGT(×2) | IGS (rpl20, ycf1) |
| 7 | 119 | ACAAAACTTGACAAAACTTGCCAAATACAATGATGACATTCTATAATGATAAATAGAGATATTGTGTTGCGTTGTTTAAATGTTACGAGCAATAATGAAACAAAACTTGTCAAAACTG(×2) | IGS (rpl20, ycf1) |
| 8 | 185 | GGAAAAACAAAAAGAACAAATTGAAAGAATAAGATGCTTAAAATTGACTAATAATATTTTTTTTAATGCAACAAAAATTATTTTAAATACCACTACCACAGGAGGGATATGATCACCACTTTTGCATTGTCTTGGCTACAAAGATGTAGCCCAATAATATTGTTTGGTTTCTATTATGGTTTTTT(×2) | IGS (rpl20, ycf1), ycf1 (CDS) |
| 9 | 30 | GAAAAGAAAAGAGAAAAGAACAAGAAGCAT | ycf1 (CDS) |
| 10 | 66 | ATGAATGAGGCAAAGGATACAAAAATAGACTCCATAACTTCGTCTCAAATGGACTCTTTTTGTAGC(×2) | ycf1 (CDS) |
| 11 | 44 | TTATTATCTCTTCTAAAATTATTTTGAAAGATCTGATTCAATGG(×2) | ycf1, IGS (ycf1, tmp) |
| 12 | 44 | CTCTTCTAAAATTATTTTGAAAGATCTGATTCAATGGTTATAAC(×2) | ycf1, IGS (ycf1, tmp) |
| 13 | 33 | TTTGTTTCAATATTTTCAGAATCTTTGTTTTCC(×3) | accD (CDS) |
| NO. | Taxon | Family | Gneus | Accession Number | NO. | Taxon | Family | Gneus | Accession Number | NO. | Taxon | Family | Gneus | Accession Number |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Selaginella moellendorffii | Selaginellaceae | Selaginella | NC_013086 | 16 | Pinus thunbergii | Pinaceae | Pinus | NC_001631 | 31 | Calycanthus floridus var. glaucus | Calycanthaceae | Calycanthus | NC_004993 |
| 2 | Cycas revoluta | Cycadaceae | Cycas | NC_020319 | 17 | Pinus massoniana | Pinaceae | Pinus | NC_021439 | 32 | Liriodendron tulipifera | Magnoliaceae | Liriodendron | NC_008326 |
| 3 | Cycas taitungensis | Cycadaceae | Cycas | NC_009618 | 18 | Pinus taeda | Pinaceae | Pinus | NC_021440 | 33 | Magnolia grandiflora voucher NJ016 | Magnoliaceae | Magnolia | NC_020318 |
| 4 | Ginkgo biloba | Ginkgoaceae | Ginkgo | NC_016986 | 19 | Taxus mairei voucher | Taxaceae | Taxus | NC_020321 | 34 | Piper cenocladum | Piperaceae | Piper | NC_008457 |
| 5 | Agathis dammara | Araucariaceae | Agathis | NC_023119 | 20 | Cucumis sativus | Cucurbitaceae | Cucumis | NC_007144 | 35 | Acorus americanus | Acoraceae | Acorus | NC_010093 |
| 6 | Cephalotaxus wilsoniana | Cephalotaxaceae | Cephalotaxus | NC_016063 | 21 | Lotus japonicus | Fabaceae | Lotus | NC_002694 | 36 | Phalaenopsis aphrodite subsp. formosana | Orchidaceae | Phalaenopsis | NC_007499 |
| 7 | Calocedrus formosana | Cupressaceae | Calocedrus | NC_023121 | 22 | Medicago truncatula | Fabaceae | Medicago | NC_003119 | 37 | Phyllostachys propinqua | Gramineae | Phyllostachys | NC_016699 |
| 8 | Cryptomeria japonica | Cupressaceae | Cryptomeria | NC_010548 | 23 | Populus alba | Salicaceae | Populus | NC_008235 | 38 | Oryza sativa Japonica Group | Gramineae | Oryza | NC_001320 |
| 9 | Cunninghamia lanceolata | Cupressaceae | Cunninghamia | NC_021437 | 24 | Populus trichocarpa | Salicaceae | Populus | NC_009143 | 39 | Phyllostachys edulis | Gramineae | Phyllostachys | NC_015817 |
| 10 | Taiwania flousiana | Cupressaceae | Taiwania | NC_021441 | 25 | Gossypium hirsutum | Malvaceae | Gossypium | NC_007944 | 40 | Saccharum hybrid cultivar NCo 310 | Gramineae | Saccharum | NC_006084 |
| 11 | Taiwania cryptomerioides | Cupressaceae | Taiwania | NC_016065 | 26 | Eucalyptus globulus subsp. globulus | Myrtaceae | Eucalyptus | NC_008115 | 41 | Triticum aestivum | Gramineae | Triticeae | NC_002762 |
| 12 | Cathaya argyrophylla | Pinaceae | Cathaya | NC_014589 | 27 | Ranunculus macranthus | Ranunculaceae | Ranunculus | NC_008796 | 42 | Zea mays | Gramineae | Zea | NC_001666 |
| 13 | Cedrus deodara | Pinaceae | Cedrus | NC_014575 | 28 | Nicotiana tabacum | Solanaceae | Nicotiana | NC_001879 | 43 | Typha latifolia | Typhaceae | Typha | NC_013823 |
| 14 | Keteleeria davidiana | Pinaceae | Keteleeria | NC_011930 | 29 | Vitis vinifera | Vitaceae | Vitis | NC_007957 | 44 | Amborella trichopoda | Amborellaceae | Amborella | NC_005086 |
| 15 | Picea abies | Pinaceae | Picea | NC_021456 | 30 | Drimys granadensis | Winteraceae | Drimys | NC_008456 | 45 | Nymphaea alba | Nymphaeaceae | Nymphaea | NC_006050 |
| Photosynthetic Electron Transport and Related Processes (I) | Subunits of Photosystem I | psaA, psaB, psaC, psaI, psaJ, psaM |
| Subunits of Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of Cytochrome | petA, petB, petD, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | |
| Large subunit of Rubisco | rbcL | |
| Gene Expression (II) | DNA dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 |
| Small/Large subunits of Ribosome | rps2, rps3, rps4, rps7, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19, rpl2, rpl14, rpl16, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| Other (III) | ccsA, cemA, clpP, matK, ycf3, ycf4 |
| Category | Category ID | Fields |
|---|---|---|
| gene function | I | Photosynthetic Electron Transport and Related Processes |
| II | Gene Expression | |
| III | Other | |
| selection force (dN/dS) | A | dN/dS ≤ 0.25 |
| B | 0.25 < dN/dS ≤ 0.5 | |
| C | 0.5 < dN/dS | |
| substitution rate (Ts + Tv) | a | Ts + Tv ≤ 0.25 |
| b | 0.25 < Ts + Tv ≤ 0.5 | |
| c | 0.5 < Ts + Tv |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, W.; Chen, J.; Hao, Z.; Shi, J. Comparative Analysis of the Chloroplast Genomic Information of Cunninghamia lanceolata (Lamb.) Hook with Sibling Species from the Genera Cryptomeria D. Don, Taiwania Hayata, and Calocedrus Kurz. Int. J. Mol. Sci. 2016, 17, 1084. https://doi.org/10.3390/ijms17071084
Zheng W, Chen J, Hao Z, Shi J. Comparative Analysis of the Chloroplast Genomic Information of Cunninghamia lanceolata (Lamb.) Hook with Sibling Species from the Genera Cryptomeria D. Don, Taiwania Hayata, and Calocedrus Kurz. International Journal of Molecular Sciences. 2016; 17(7):1084. https://doi.org/10.3390/ijms17071084
Chicago/Turabian StyleZheng, Weiwei, Jinhui Chen, Zhaodong Hao, and Jisen Shi. 2016. "Comparative Analysis of the Chloroplast Genomic Information of Cunninghamia lanceolata (Lamb.) Hook with Sibling Species from the Genera Cryptomeria D. Don, Taiwania Hayata, and Calocedrus Kurz" International Journal of Molecular Sciences 17, no. 7: 1084. https://doi.org/10.3390/ijms17071084