
The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment


Abstract
:
{kind=link}
{kind=link}
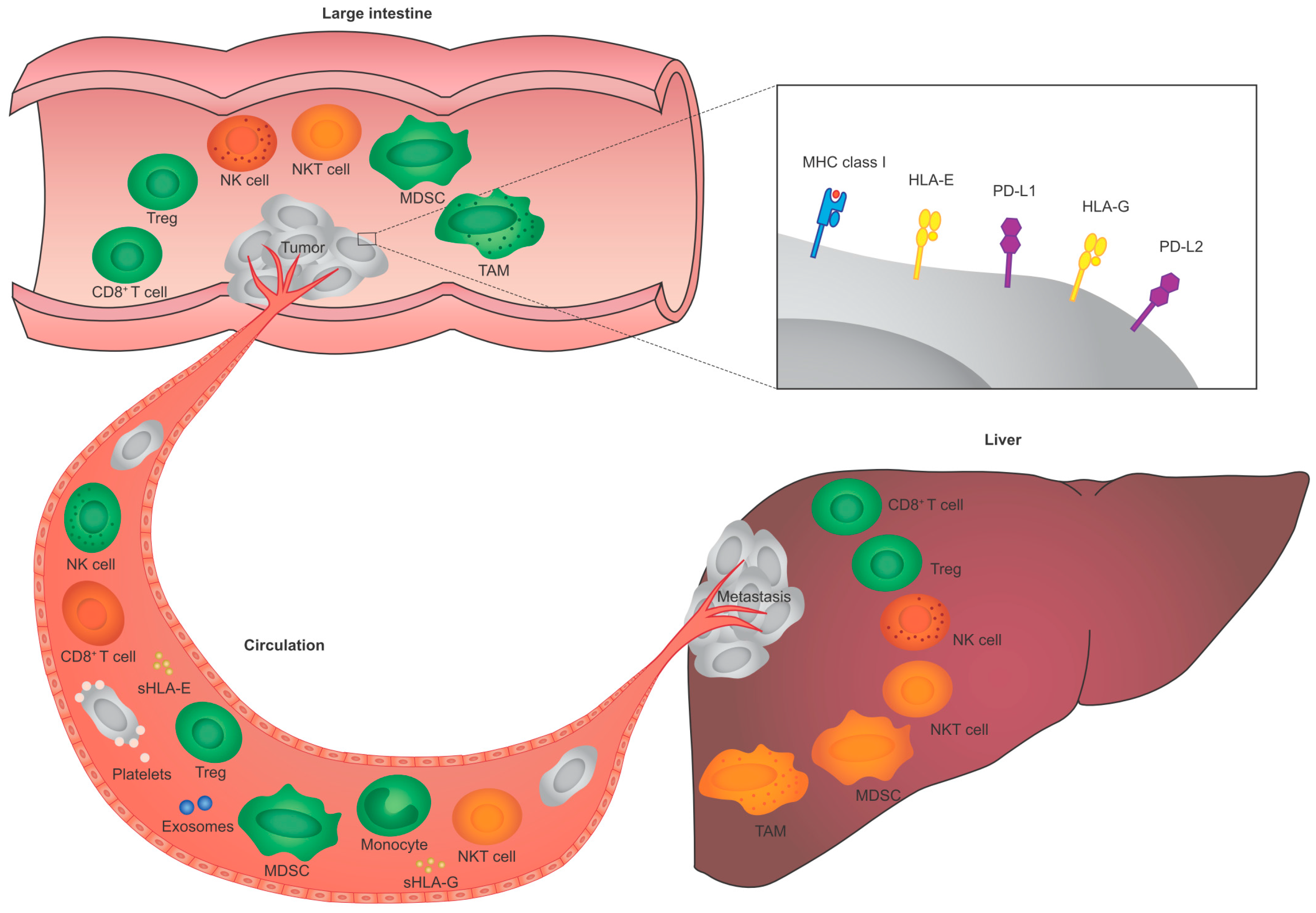
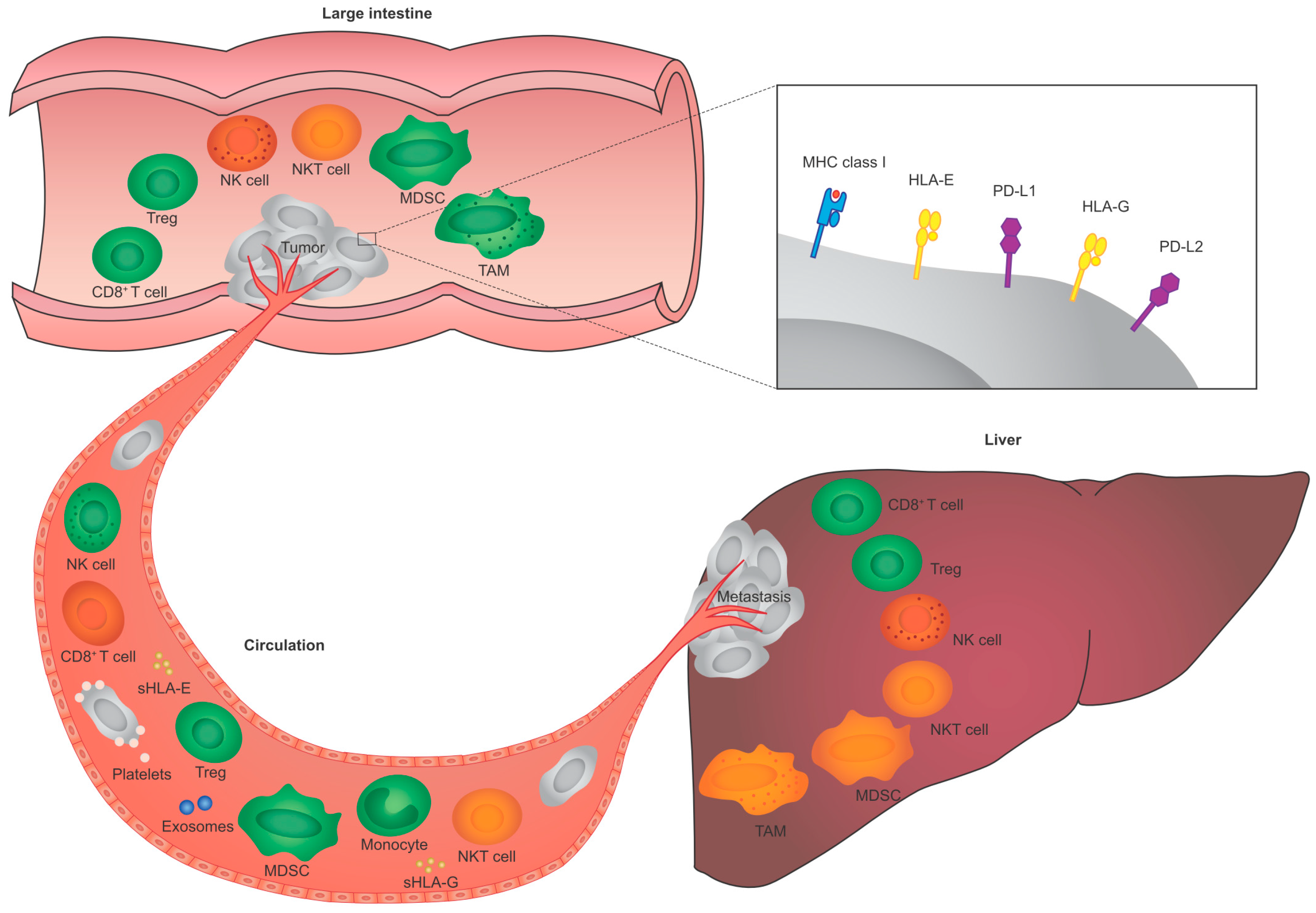{kind=link}
1. Introduction
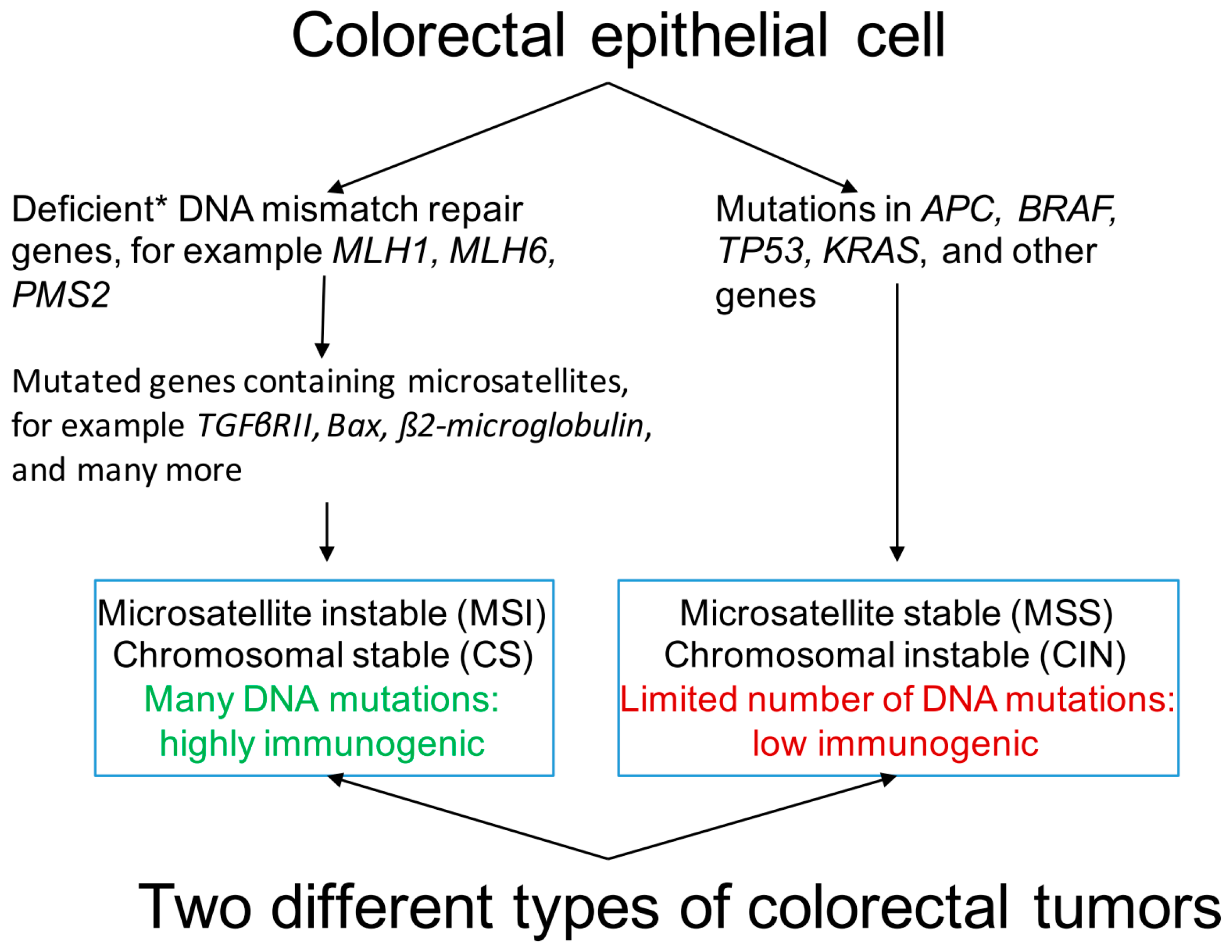
2. Genetic and Epigenetic Makeup of Colorectal Cancer (CRC)
3. Immunogenicity of CRC
4. Tumor-Immune System Interactions in CRC
4.1. Tumor-Immune System Interactions in Primary Tumors
4.2. Tumor-Immune System Interactions in the Circulation
4.3. Tumor-Immune System Interactions in Liver Metastases
5. Role of Immune Checkpoints in CRC
6. Current Treatment Options and CRC Immunogenicity
7. Importance of CRC Immunogenicity for Novel Immune-Based Therapeutic Strategies
8. Perspective
9. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Chimeric antigen receptor |
| CEA | Carcinoembryonic antigen |
| CIMP | CpG island methylator phenotype |
| CIN | Chromosomal instability |
| CRC | Colorectal cancer |
| CTLA-4 | Cytotoxic T-lymphocyte-associated antigen 4 |
| DC | Dendritic cell |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transition |
| EpCAM | Epithelial cell adhesion molecule |
| HER2 | Human epidermal growth factor receptor 2 |
| HLA | Human leukocyte antigen |
| IHP | Isolated hepatic perfusion |
| MDSC | Myeloid-derived suppressor cell |
| MHC | Major histocompatibility complex |
| MMR | Mismatch repair |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| MUC1 | Mucin 1 |
| NK | Natural killer |
| NKT | Natural killer T |
| PD-1 | Programmed death 1 |
| PD-L1 | Programmed death-ligand 1 |
| PD-L2 | Programmed death-ligand 2 |
| sHLA | Soluble HLA |
| TAM | Tumor-associated macrophage |
| TIL | Tumor-infiltrating lymphocyte |
| TP53 | Tumor protein 53 |
| Treg | Regulatory T cell |
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, H.J.; van Cutsem, E.; Stein, A.; Valentini, V.; Glimelius, B.; Haustermans, K.; Nordlinger, B.; van de Velde, C.J.; Balmana, J.; Regula, J.; et al. ESMO consensus guidelines for management of patients with colon and rectal cancer. A personalized approach to clinical decision making. Ann. Oncol. 2012, 23, 2479–2516. [Google Scholar] [CrossRef] [PubMed]
- Pardieck, I.N.; Jawahier, P.A.; Swets, M.; van de Velde, C.J.; Kuppen, P.J. Novel avenues in immunotherapies for colorectal cancer. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 1997, 94, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9–14. [Google Scholar] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. Apc mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, M.H.; Vasen, H.F. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit. Rev. Oncol. Hematol. 2007, 61, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS ONE 2010, 5, e15661. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Morikawa, T.; Kuchiba, A.; Imamura, Y.; Qian, Z.R.; Nishihara, R.; Liao, X.; Waldron, L.; Hoshida, Y.; Huttenhower, C.; et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012, 61, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Pernot, S.; Terme, M.; Voron, T.; Colussi, O.; Marcheteau, E.; Tartour, E.; Taieb, J. Colorectal cancer and immunity: What we know and perspectives. World J. Gastroenterol. 2014, 20, 3738–3750. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Flamini, G.; Curigliano, G.; Ratto, C.; Astone, A.; Ferretti, G.; Nucera, P.; Sofo, L.; Sgambato, A.; Boninsegna, A.; Crucitti, F.; et al. Prognostic significance of cytoplasmic p53 overexpression in colorectal cancer. An immunohistochemical analysis. Eur. J. Cancer 1996, 32A, 802–806. [Google Scholar] [CrossRef]
- Houbiers, J.G.; van der Burg, S.H.; van de Watering, L.M.; Tollenaar, R.A.; Brand, A.; van de Velde, C.J.; Melief, C.J. Antibodies against p53 are associated with poor prognosis of colorectal cancer. Br. J. Cancer 1995, 72, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Menon, A.G.; Redeker, A.; Bonnet, M.C.; Drijfhout, J.W.; Tollenaar, R.A.; van de Velde, C.J.; Moingeon, P.; Kuppen, P.J.; Offringa, R.; et al. Induction of p53-specific immune responses in colorectal cancer patients receiving a recombinant ALVAC-p53 candidate vaccine. Clin. Cancer Res. 2002, 8, 1019–1027. [Google Scholar] [PubMed]
- Menon, A.G.; Kuppen, P.J.; van der Burg, S.H.; Offringa, R.; Bonnet, M.C.; Harinck, B.I.; Tollenaar, R.A.; Redeker, A.; Putter, H.; Moingeon, P.; et al. Safety of intravenous administration of a canarypox virus encoding the human wild-type p53 gene in colorectal cancer patients. Cancer Gene Ther. 2003, 10, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.M.; Banerjea, A.; Feakins, R.; Li, S.R.; Bustin, S.A.; Dorudi, S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br. J. Surg. 2004, 91, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Banerjea, A.; Hands, R.E.; Powar, M.P.; Bustin, S.A.; Dorudi, S. Microsatellite and chromosomal stable colorectal cancers demonstrate poor immunogenicity and early disease recurrence. Colorectal. Dis. 2009, 11, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Drescher, K.M.; Sharma, P.; Lynch, H.T. Current hypotheses on how microsatellite instability leads to enhanced survival of lynch syndrome patients. Clin. Dev. Immunol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Becker, C.; Benner, A.; Woerner, S.M.; Gebert, J.; Ferrone, S.; von Knebel Doeberitz, M. Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Cancer Res. 2005, 65, 6418–6424. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; McCuaig, S.; Franchini, F.; Powrie, F. Emerging cytokine networks in colorectal cancer. Nat. Rev. Immunol. 2015, 15, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pagès, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol 2009, 27, 5944–5951. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.H.; Pagès, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.G.; Janssen-van Rhijn, C.M.; Morreau, H.; Putter, H.; Tollenaar, R.A.; van de Velde, C.J.; Fleuren, G.J.; Kuppen, P.J. Immune system and prognosis in colorectal cancer: A detailed immunohistochemical analysis. Lab. Investig 2004, 84, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Beckhove, P.; Op den Winkel, J.; Autenrieth, D.; Wagner, P.; Nummer, D.; Specht, S.; Antolovic, D.; Galindo, L.; Schmitz-Winnenthal, F.H.; et al. Tumor infiltrating T lymphocytes in colorectal cancer: Tumor-selective activation and cytotoxic activity in situ. Ann. Surg. 2006, 244, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Reissfelder, C.; Stamova, S.; Gossmann, C.; Braun, M.; Bonertz, A.; Walliczek, U.; Grimm, M.; Rahbari, N.N.; Koch, M.; Saadati, M.; et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J. Clin. Investig. 2015, 125, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Koch, M.A. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat. Rev. Immunol. 2011, 11, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Louafi, S.; Bardier, A.; Charlotte, F.; Vaillant, J.C.; Ménégaux, F.; Rosenzwajg, M.; Lemoine, F.; Klatzmann, D.; Taieb, J. Identification of CD8+CD25+FOXP3+ suppressive T cells in colorectal cancer tissue. Gut 2009, 58, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.; Edin, S.; Wikberg, M.L.; Öberg, Å.; Palmqvist, R. The intratumoural subsite and relation of CD8+ and FOXP3+ T lymphocytes in colorectal cancer provide important prognostic clues. Br. J. Cancer 2014, 110, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Bonertz, A.; Weitz, J.; Pietsch, D.H.; Rahbari, N.N.; Schlude, C.; Ge, Y.; Juenger, S.; Vlodavsky, I.; Khazaie, K.; Jaeger, D.; et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J. Clin. Investig. 2009, 119, 3311–3321. [Google Scholar] [CrossRef] [PubMed]
- Sandel, M.H.; Speetjens, F.M.; Menon, A.G.; Albertsson, P.A.; Basse, P.H.; Hokland, M.; Nagelkerke, J.F.; Tollenaar, R.A.; van de Velde, C.J.; Kuppen, P.J. Natural killer cells infiltrating colorectal cancer and MHC class I expression. Mol. Immunol. 2005, 42, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Zeestraten, E.C.; Reimers, M.S.; Saadatmand, S.; Goossens-Beumer, I.J.; Dekker, J.W.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.; Kuppen, P.J. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br. J. Cancer 2014, 110, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Braun, M.; Kahlert, C.; Spille, A.; Quack, C.; Rahbari, N.; Koch, M.; Weitz, J.; Kloor, M.; Zoernig, I.; et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin. Cancer Res. 2011, 17, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Algarra, I.; García-Lora, A.; Cabrera, T.; Ruiz-Cabello, F.; Garrido, F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: Implications for tumor immune escape. Cancer Immunol. Immunother. 2004, 53, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J. Exp. Med. 1999, 189, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Le Bouteiller, P. HLA-G in human early pregnancy: Control of uterine immune cell activation and likely vascular remodeling. Biomed. J. 2015, 38, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.M.; Bianchini, M.; Von Euw, E.M.; Barrio, M.M.; Bravo, A.I.; Furman, D.; Domenichini, E.; Macagno, C.; Pinsky, V.; Zucchini, C.; et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int. J. Oncol. 2008, 32, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.Y.; Lv, Y.G.; Wang, L.; Shi, S.J.; Yang, F.; Zheng, G.X.; Wen, W.H.; Yang, A.G. Predictive value of HLA-G and HLA-E in the prognosis of colorectal cancer patients. Cell Immunol. 2015, 293, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Benevolo, M.; Mottolese, M.; Tremante, E.; Rollo, F.; Diodoro, M.G.; Ercolani, C.; Sperduti, I.; Lo Monaco, E.; Cosimelli, M.; Giacomini, P. High expression of HLA-E in colorectal carcinoma is associated with a favorable prognosis. J. Transl. Med. 2011, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, Y.; Oshika, Y.; Nakamura, M.; Tokunaga, T.; Hatanaka, H.; Abe, Y.; Yamazaki, H.; Kijima, H.; Ueyama, Y.; Tamaoki, N. Increased expression of human histocompatibility leukocyte antigen-G in colorectal cancer cells. Int. J. Mol. Med. 1998, 2, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Kanno, R.; Ito, T.; Higashino, K.; Taniguchi, M. Predominant expression of invariant Vα 14+ TCR α chain in NK1.1+ T cell populations. Int. Immunol. 1995, 7, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. NKT cells in immunoregulation of tumor immunity: A new immunoregulatory axis. Trends Immunol. 2007, 28, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, T.; Onodera, H.; Tsuruyama, T.; Mori, A.; Nagayama, S.; Hiai, H.; Imamura, M. Increased intratumor Vα24-positive natural killer T cells: A prognostic factor for primary colorectal carcinomas. Clin. Cancer Res. 2005, 11, 7322–7327. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Watanabe, M. Myeloid-derived suppressor cells and therapeutic strategies in cancer. Mediators Inflamm. 2015, 2015, 159269. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.L.; Zhou, X.; Xue, Y.F.; Wang, K.; Shen, Y.F.; Mao, J.J.; Guo, H.F.; Miao, Z.N. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J. Gastroenterol. 2012, 18, 3303–3309. [Google Scholar] [PubMed]
- OuYang, L.Y.; Wu, X.J.; Ye, S.B.; Zhang, R.X.; Li, Z.L.; Liao, W.; Pan, Z.Z.; Zheng, L.M.; Zhang, X.S.; Wang, Z.; et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J. Transl. Med. 2015, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.A.; Palmqvist, R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Herrera, A.; Domínguez, G.; Silva, J.; García, V.; García, J.M.; Gómez, I.; Soldevilla, B.; Muñoz, C.; Provencio, M.; et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013, 104, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Luo, X.; Lin, Y.; Tang, X.; Ling, L.; Wang, L.; Jiang, Y. A higher frequency of CD14+ CD169+ monocytes/macrophages in patients with colorectal cancer. PLoS ONE 2015, 10, e0141817. [Google Scholar] [CrossRef] [PubMed]
- Harouaka, R.A.; Nisic, M.; Zheng, S.Y. Circulating tumor cell enrichment based on physical properties. J. Lab. Autom. 2013, 18, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Reimers, M.S.; Bastiaannet, E.; Langley, R.E.; van Eijk, R.; van Vlierberghe, R.L.; Lemmens, V.E.; van Herk-Sukel, M.P.; van Wezel, T.; Fodde, R.; Kuppen, P.J.; et al. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern. Med. 2014, 174, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Rocca, Y.S.; Roberti, M.P.; Arriaga, J.M.; Amat, M.; Bruno, L.; Pampena, M.B.; Huertas, E.; Loria, F.S.; Pairola, A.; Bianchini, M.; et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun. 2013, 19, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.P.; Zhu, Y.; Zhang, J.J.; Xu, Z.K.; Qian, Z.Y.; Dai, C.C.; Jiang, K.R.; Wu, J.L.; Gao, W.T.; Li, Q.; et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J. Transl. Med. 2013, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Park, G.M.; Lee, S.; Park, B.; Kim, E.; Shin, J.; Cho, K.; Ahn, K. Soluble HLA-G generated by proteolytic shedding inhibits NK-mediated cell lysis. Biochem. Biophys. Res. Commun. 2004, 313, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Coupel, S.; Moreau, A.; Hamidou, M.; Horejsi, V.; Soulillou, J.P.; Charreau, B. Expression and release of soluble HLA-E is an immunoregulatory feature of endothelial cell activation. Blood 2007, 109, 2806–2814. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.; Oger, R.; Vignard, V.; Percier, J.M.; Fregni, G.; Périer, A.; Caignard, A.; Charreau, B.; Bernardeau, K.; Khammari, A.; et al. Serum soluble HLA-E in melanoma: A new potential immune-related marker in cancer. PLoS ONE 2011, 6, e21118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.; Yie, S.M.; Liu, J.; Ye, S.R.; Xia, D.; Gao, E. Plasma soluble HLA-G is a potential biomarker for diagnosis of colorectal, gastric, esophageal and lung cancer. Tissue Antigens 2011, 78, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Ferretti, E.; Castriconi, R.; Dondero, A.; Petretto, A.; Bottino, C.; Pistoia, V. Soluble HLA-G dampens CD94/NKG2A expression and function and differentially modulates chemotaxis and cytokine and chemokine secretion in CD56bright and CD56dim NK cells. Blood 2011, 118, 5840–5850. [Google Scholar] [CrossRef] [PubMed]
- Shwetank; Date, O.S.; Carbone, E.; Manjunath, R. Inhibition of ERK and proliferation in NK cell lines by soluble HLA-E released from japanese encephalitis virus infected cells. Immunol. Lett. 2014, 162, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Diamond, M.P.; Al-Hendy, A. The emerging role of extracellular vesicle-derived miRNAs: Implication in cancer progression and stem cell related diseases. J. Clin. Epigenet. 2016, 2, 13. [Google Scholar] [PubMed]
- Matsumura, T.; Sugimachi, K.; Iinuma, H.; Takahashi, Y.; Kurashige, J.; Sawada, G.; Ueda, M.; Uchi, R.; Ueo, H.; Takano, Y.; et al. Exosomal microRNA in serum is a novel biomarker of recurrence in human colorectal cancer. Br. J. Cancer 2015, 113, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Scheibenbogen, C.; Schaller, G.; Leigh, B.; Schmittel, A.; Letsch, A.; Thiel, E.; Keilholz, U. Differences in T-cell immunity toward tumor-associated antigens in colorectal cancer and breast cancer patients. Int. J. Cancer 2003, 105, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.L.; Pratap, S.E.; Bates, G.J.; Singh, B.; Mortensen, N.J.; George, B.D.; Warren, B.F.; Piris, J.; Roncador, G.; Fox, S.B.; et al. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun. 2007, 7, 7. [Google Scholar] [PubMed]
- Liu, Z.; Huang, Q.; Liu, G.; Dang, L.; Chu, D.; Tao, K.; Wang, W. Presence of FOXP3(+)Treg cells is correlated with colorectal cancer progression. Int. J. Clin. Exp. Med. 2014, 7, 1781–1785. [Google Scholar] [PubMed]
- Zhang, B.; Wang, Z.; Wu, L.; Zhang, M.; Li, W.; Ding, J.; Zhu, J.; Wei, H.; Zhao, K. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS ONE 2013, 8, e0057114. [Google Scholar] [CrossRef] [PubMed]
- Oberg, A.; Samii, S.; Stenling, R.; Lindmark, G. Different occurrence of CD8+, CD45R0+, and CD68+ immune cells in regional lymph node metastases from colorectal cancer as potential prognostic predictors. Int. J. Colorectal. Dis. 2002, 17, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, H.; Luan, Y.; Zhang, J.; Xing, Q.; Dong, S.; Wu, X.; Liu, M.; Wang, S. Accumulation of FOXP3+ T regulatory cells in draining lymph nodes correlates with disease progression and immune suppression in colorectal cancer patients. Clin. Cancer Res. 2010, 16, 4105–4112. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.; Koch, M.; Nummer, D.; Palm, S.; Galindo, L.; Autenrieth, D.; Rahbari, N.; Schmitz-Winnenthal, F.H.; Schirrmacher, V.; Büchler, M.W.; et al. Detection and functional analysis of tumor infiltrating T-lymphocytes (TIL) in liver metastases from colorectal cancer. Ann. Surg. Oncol. 2008, 15, 2310–2317. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Pillarisetty, V.; Bamboat, Z.M.; Shia, J.; Hedvat, C.; Gonen, M.; Jarnagin, W.; Fong, Y.; Blumgart, L.; D’Angelica, M.; et al. T cell infiltrate predicts long-term survival following resection of colorectal cancer liver metastases. Ann. Surg. Oncol. 2009, 16, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011, 71, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, S.; Katz, S.C.; Shia, J.; Jarnagin, W.R.; Kingham, T.P.; Allen, P.J.; Fong, Y.; D’Angelica, M.I.; DeMatteo, R.P. Tumor MHC class I expression improves the prognostic value of T-cell density in resected colorectal liver metastases. Cancer Immunol. Res. 2014, 2, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Bamboat, Z.M.; Maker, A.V.; Shia, J.; Pillarisetty, V.G.; Yopp, A.C.; Hedvat, C.V.; Gonen, M.; Jarnagin, W.R.; Fong, Y.; et al. Regulatory T cell infiltration predicts outcome following resection of colorectal cancer liver metastases. Ann. Surg. Oncol. 2013, 20, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.G.; Tollenaar, R.A.; van de Velde, C.J.; Putter, H.; Janssen-van Rhijn, C.M.; Keijzer, R.; Fleuren, G.J.; Kuppen, P.J. P53 and HLA class-I expression are not down-regulated in colorectal cancer liver metastases. Clin. Exp. Metastasis 2004, 21, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Swets, M.; König, M.H.; Zaalberg, A.; Dekker-Ensink, N.G.; Gelderblom, H.; van de Velde, C.J.; van den Elsen, P.J.; Kuppen, P.J. HLA-G and classical HLA class I expression in primary colorectal cancer and associated liver metastases. Hum. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Radaeva, S.; Park, O. Liver natural killer and natural killer T cells: Immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 2009, 86, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Gulubova, M.; Manolova, I.; Kyurkchiev, D.; Julianov, A.; Altunkova, I. Decrease in intrahepatic CD56+ lymphocytes in gastric and colorectal cancer patients with liver metastases. APMIS 2009, 117, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Pugh, S.A.; Harrison, R.J.; Primrose, J.N.; Khakoo, S.I. T cells but not NK cells are associated with a favourable outcome for resected colorectal liver metastases. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Lassen, M.G.; Lukens, J.R.; Dolina, J.S.; Brown, M.G.; Hahn, Y.S. Intrahepatic IL-10 maintains NKG2A + LY49- liver NK cells in a functionally hyporesponsive state. J. Immunol. 2010, 184, 2693–2701. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Ham, B.; Wang, N.; D’Costa, Z.; Fernandez, M.C.; Bourdeau, F.; Auguste, P.; Illemann, M.; Eefsen, R.L.; Høyer-Hansen, G.; Vainer, B.; et al. TNF receptor-2 facilitates an immunosuppressive microenvironment in the liver to promote the colonization and growth of hepatic metastases. Cancer Res. 2015, 75, 5235–5247. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.L.; Li, H.K.; Zhou, H.Y.; Zhang, T.; Li, Q. Correlations of tumor-associated macrophage subtypes with liver metastases of colorectal cancer. Asian Pac. J. Cancer Prev. 2013, 14, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Sharma, P.K.; Krishnan, G.; Lockhart, A.C. Immune checkpoints and immunotherapy for colorectal cancer. Gastroenterol. Rep. 2015, 3, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Smits, E.; Lardon, F.; Pauwels, P.; Deschoolmeester, V. Immune checkpoint modulation in colorectal cancer: What’s new and what to expect. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Yee, C.; Lee, K.M. The effect of radiation on the immune response to cancers. Int. J. Mol. Sci. 2014, 15, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Formenti, S.C. Role of T lymphocytes in tumor response to radiotherapy. Front. Oncol. 2012, 2, 95. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Van Gijn, W.; Marijnen, C.A.; Nagtegaal, I.D.; Kranenbarg, E.M.; Putter, H.; Wiggers, T.; Rutten, H.J.; Påhlman, L.; Glimelius, B.; van de Velde, C.J.; et al. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer: 12-year follow-up of the multicentre, randomised controlled TME trial. Lancet Oncol. 2011, 12, 575–582. [Google Scholar] [CrossRef]
- Maas, M.; Beets-Tan, R.G.; Lambregts, D.M.; Lammering, G.; Nelemans, P.J.; Engelen, S.M.; van Dam, R.M.; Jansen, R.L.; Sosef, M.; Leijtens, J.W.; et al. Wait-and-see policy for clinical complete responders after chemoradiation for rectal cancer. J. Clin. Oncol. 2011, 29, 4633–4640. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; Ruby, J.A.; Goodman, K.A.; Saltz, L.B.; Guillem, J.G.; Weiser, M.R.; Temple, L.K.; Nash, G.M.; Paty, P.B. Nonoperative management of rectal cancer with complete clinical response after neoadjuvant therapy. Ann. Surg. 2012, 256, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Jones, R.; Tang, J.M.; Parmar, C.; Fenwick, S.; Malik, H.; Poston, G. Ablative therapies for colorectal liver metastases: A systematic review. Colorectal. Dis. 2011, 13, e252–e265. [Google Scholar] [CrossRef] [PubMed]
- Vahrmeijer, A.L.; van Dierendonck, J.H.; Keizer, H.J.; Beijnen, J.H.; Tollenaar, R.A.; Pijl, M.E.; Marinelli, A.; Kuppen, P.J.; van Bockel, J.H.; Mulder, G.J.; et al. Increased local cytostatic drug exposure by isolated hepatic perfusion: A phase I clinical and pharmacologic evaluation of treatment with high dose melphalan in patients with colorectal cancer confined to the liver. Br. J. Cancer 2000, 82, 1539–1546. [Google Scholar] [PubMed]
- Van Duijnhoven, F.H.; Jannsen-van Rhijn, C.J.; Terpstra, O.T.; Kuppen, P.J. Local Therapy of Colorectal Liver Metastases and Formation of Antibodies. “Local Ablative Therapies for Colorectal Liver Metastases and the Immune System”, Ph.D. Thesis, University of Leiden, The Netherlands, 22 June 2005. [Google Scholar]
- Hansler, J.; Wissniowski, T.T.; Schuppan, D.; Witte, A.; Bernatik, T.; Hahn, E.G.; Strobel, D. Activation and dramatically increased cytolytic activity of tumor specific T lymphocytes after radio-frequency ablation in patients with hepatocellular carcinoma and colorectal liver metastases. World J. Gastroenterol. 2006, 12, 3716–3721. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.J.; Wissniowski, T.T.; Naguib, N.N.; Hammerstingl, R.M.; Mack, M.G.; Münch, S.; Ocker, M.; Strobel, D.; Hahn, E.G.; Hänsler, J. Activation of tumor-specific T lymphocytes after laser-induced thermotherapy in patients with colorectal liver metastases. Cancer Immunol. Immunother. 2009, 58, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Desrichard, A.; Snyder, A.; Chan, T.A. Cancer neoantigens and applications for immunotherapy. Clin. Cancer Res. 2016, 22, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Speetjens, F.M.; Zeestraten, E.C.; Kuppen, P.J.; Melief, C.J.; Van der Burg, S.H. Colorectal cancer vaccines in clinical trials. Expert Rev. Vaccines 2011, 10, 899–921. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Point, G.R.; Cunetta, M.; Thorn, M.; Guha, P.; Espat, N.J.; Boutros, C.; Hanna, N.; Junghans, R.P. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016, 23, 142–148. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vries, N.L.; Swets, M.; Vahrmeijer, A.L.; Hokland, M.; Kuppen, P.J.K. The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment. Int. J. Mol. Sci. 2016, 17, 1030. https://doi.org/10.3390/ijms17071030
De Vries NL, Swets M, Vahrmeijer AL, Hokland M, Kuppen PJK. The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment. International Journal of Molecular Sciences. 2016; 17(7):1030. https://doi.org/10.3390/ijms17071030
Chicago/Turabian StyleDe Vries, Natasja L., Marloes Swets, Alexander L. Vahrmeijer, Marianne Hokland, and Peter J. K. Kuppen. 2016. "The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment" International Journal of Molecular Sciences 17, no. 7: 1030. https://doi.org/10.3390/ijms17071030