
Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis—Masters of Survival and Clonality?


Abstract
:
1. Introduction
2. MSCs: Nomenclature
3. MSPCs: Phenotypic Characterization and “Plasticity”
Abnormal Phenotype of MSPCs in MDS and AML
4. MSPCs: Cell of Origin
4.1. Evidence for Mesodermal Origin of MSPCs
4.1.1. EndoMT as a Potential Source for MSPCs
(Circulating) Endothelial Cells in MDS and AML
4.2. Neuro-Ectodermal Origin of MSPC-Like Cells
4.2.1. Pericytes and Endoneural (Myo)fibroblasts as Potential Sources of MSPCs
4.2.2. Neural Phenotype Plasticity of MSPCs—Common Neural-Crest Origin with Sympathetic Neurons?
MSPCs and Neuro-Epithelial Markers in MDS and AML
A Potential Role for Sympathetic Nerves in the BM
4.2.3. EMT as Potential Source of MSPCs
TWIST in MDS and AML
4.3. MSPCs: Evidence for Multiple Developmental Origins
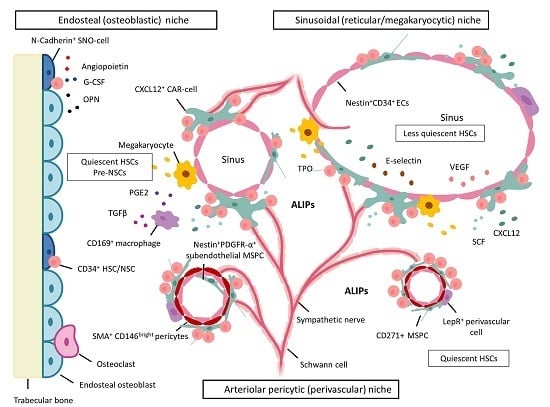
5. BM Microenvironment and Stem Cell Niche Concepts
5.1. Dysplastic and Leukemic Niches
NSCs: Competition for the Stem Cell Niche—Spatial Localization in Mice
5.2. Stem Cell Niche: Soluble Components in Normal Hematopoiesis
Stem Cell Niche: Soluble Components in MDS and AML
5.3. Stem Cell Niche: Signaling Pathways in Normal Hematopoiesis
Stem Cell Niche: Signaling Pathways in MDS and AML
6. MSPCs and Their Progeny: Key Cellular Niche Components
6.1. MSPCs: Spatial Localization in Vivo in Normal and Dysplastic Hematopoiesis
6.2. MSPCs: Dichotomous Effects on Erythropoiesis
6.3. Osteoblasts Support and Regulate HSCs
Osteoblasts in MDS and AML
6.4. Other Niche Cells that Regulate HSCs
7. MSPCs: Immunomodulation
7.1. Immunosuppressive Effects of MSPCs on Immune Cells
7.1.1. MSPCs: Mechanisms of Immunosuppression
MSPC-Mediated Immunosuppression via Secretion of Soluble Factors
MSPC-Mediated Immunosuppression via Expression of Cell Surface Molecules
7.2. MSPC-Mediated Mechanisms of Immune Evasion in Malignancy
7.3. MSPC-Mediated Mechanisms of Immune Evasion in MDS and AML
7.4. MSPCs: Immunosuppressive Licensing
7.5. MSPCs: TLR Signaling Balances Immunosuppressive versus Pro-Inflammatory Licensing
TLR Signaling in MDS and AML
7.6. MSPCs: Autophagy and Phagocytosis
Autophagy in MDS and AML
8. MSPCs in MDS and AML: Aberrant Function
8.1. Dysplastic/Leukemic Niche: Altered Gene Expression
8.2. NSCs: Bidirectional Crosstalk with MSPCs—“Reprogramming” to Diseased MSPCs
8.3. NSCs: Bidirectional Crosstalk with ECs
8.4. NSCs: Bidirectional Crosstalk with Other Cells
9. MSPCs in MDS and AML: Clone-Derived or Clone-Induced?
10. Malignancy-Inducing Microenvironment?
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| αSMA | α-smooth muscle actin |
| Akt | protein kinase B |
| ALIPs | abnormal localization of immature precursors |
| AML | acute myeloid leukemia |
| APC | antigen-presenting cells |
| APCregs | regulatory antigen-presenting cells |
| BFU-E | blast forming unit-erythroid |
| BM | bone marrow |
| Bmp4 | bone morphogenic protein |
| CAFs | cancer-associated fibroblasts |
| CAR | CXCL12-abundant reticular |
| CCL | chemokine (c-c motif) ligand |
| CECs | circulating ECs |
| CFU-F | fibroblast colony-forming unit |
| cPEC | circulating progenitors of ECs |
| CXCL12 | chemokine (C-X-C motif) ligand 12 |
| CXCR4 | chemokine (C-X-C motif) receptor 4 |
| DC | dendritic cell |
| EC | endothelial cell |
| EMT | epithelial-to-mesenchymal transition |
| EndoMT | endothelial-to-mesenchymal transition |
| G-CSF | granulocyte colony-stimulating factor |
| HGF | hepatocyte growth factor |
| HLA | human leukocyte antigen |
| HLA-DR | human leukocyte antigen-D-related |
| HSC | hematopoietic stem cell |
| HSPC | hematopoietic stem/progenitor cell |
| IDO | indoleamine 2,3-dioxygenase |
| IFN | interferon |
| IL | interleukin |
| IL1RA | interleukin 1 receptor antagonist |
| ISCT | International Society for Cellular Therapy |
| LFA-1 | lymphocyte function-associated antigen-1 |
| LPM | lateral plate mesoderm |
| LSC | leukemic stem cell |
| MCP1 | monocyte chemotactic protein 1 |
| M-CSF | macrophage colony-stimulating factor |
| MDS | myelodysplastic syndromes |
| MDSC | myeloid-derived suppressor cells |
| MSC | mesenchymal stem cell |
| MSPC | mesenchymal stem and progenitor cells |
| NCSC | neural crest-derived stem cell |
| NKC | natural killer cell |
| NKT | natural killer T-cell |
| NSC | neoplastic stem cell |
| OPN | osteopontin |
| PDGFRα | platelet-derived growth factor-α |
| PD-L1 | programmed death ligand 1 |
| PGE2 | prostaglandin E2 |
| PI3K | phosphoinositide 3-kinase |
| PTH | parathyroid hormone |
| PTH-R | parathyroid hormone receptor |
| PTN | pleotrophin |
| SC | stem cells |
| SCF | stem-cell factor |
| SDF1 | stromal cell-derived factor-1 |
| SNO | spindle-shaped N-cadherin+ osteoblastic |
| SSEA-4 | stage-specific embryonic antigen-4 |
| TGFβ | tumor growth factor β |
| TH | T-helper |
| TLR | toll-like receptor |
| TNFα | tumor necrosis factor α |
| Tregs | regulatory T-cells |
| VEGF | vascular endothelial growth factor |
| VLA-4 | very late antigen-4 |
| Wnt | wingless-Int |
| Φ | macrophage |
References
- Bryder, D.; Rossi, D.J.; Weissman, I.L. Hematopoietic stem cells: The paradigmatic tissue-specific stem cell. Am. J. Pathol. 2006, 169, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Wognum, A.W.; Szilvassy, S.J. Hematopoietic stem and progenitor cells. Document #29068, Version 6.0.0. Available online: http://www.stemcell.com/~/media/Technical%20Resources/F/B/7/E/9/MR019HematopoiesisOnline_29784WEB.pdf (accessed on 7 April 2016).
- Pleyer, L.; Neureiter, D.; Faber, V.; Greil, R. Myelodysplastic syndromes (MDS). In Chronic Myeloid Neoplasias and Clonal Overlap Syndromes: Epidemiology, Pathophysiology and Treatment Options; Greil, R., Pleyer, L., Neureiter, D., Faber, V., Eds.; Springer-Verlag: Vienna, Austria, 2010; pp. 153–222. [Google Scholar]
- Ma, X.; Does, M.; Raza, A.; Mayne, S.T. Myelodysplastic syndromes: Incidence and survival in the United States. Cancer 2007, 109, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Burgstaller, S.; Girschikofsky, M.; Linkesch, W.; Stauder, R.; Pfeilstocker, M.; Schreder, M.; Tinchon, C.; Sliwa, T.; Lang, A.; et al. Azacitidine in 302 patients with WHO-defined acute myeloid leukemia: Results from the Austrian Azacitidine Registry of the AGMT-Study Group. Ann. Hematol. 2014, 93, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; Sill, H.; Schlick, K.; Thaler, J.; Halter, B.; Machherndl-Spandl, S.; Zebisch, A.; et al. Azacitidine front-line in 339 patients with myelodysplastic syndromes and acute myeloid leukaemia: Comparison of French-American-British and World Health Organization classifications. J. Hematol. Oncol. 2016, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Passegue, E.; Jamieson, C.H.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? PNAS 2003, 100 (Suppl. 1), 11842–11849. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Dai, B.; Gentles, A.J.; Majeti, R.; Feinberg, A.P. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat. Commun. 2015, 6, 8489. [Google Scholar] [CrossRef] [PubMed]
- Sarry, J.E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Strzelecki, A.C.; Cavelier, C.; Recher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J. Clin. Investig. 2011, 121, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y.; Weissman, I.L. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Dayyani, F.; Conley, A.P.; Strom, S.S.; Stevenson, W.; Cortes, J.E.; Borthakur, G.; Faderl, S.; O’Brien, S.; Pierce, S.; Kantarjian, H.; et al. Cause of death in patients with lower-risk myelodysplastic syndrome. Cancer 2010, 116, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Rodriguez, V.; Narboni, G.; Bodey, G.P.; Luna, M.A.; Freireich, E.J. Causes of death in adults with acute leukemia. Medicine 1976, 55, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; André, F.; Tesniere, A.; Kroemer, G. The anticancer immune response: Indispensable for therapeutic success? J. Clin. Investig. 2008, 118, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Riether, C.; Schurch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Balderman, S.R.; Calvi, L.M. Biology of BM failure syndromes: Role of microenvironment and niches. Hematol. Am. Soc. Hematol. Educ. Program. 2014, 2014, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Bulycheva, E.; Rauner, M.; Medyouf, H.; Theurl, I.; Bornhauser, M.; Hofbauer, L.C.; Platzbecker, U. Myelodysplasia is in the niche: Novel concepts and emerging therapies. Leukemia 2015, 29, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Deeg, H.J. Marrow stroma in MDS: Culprit or bystander? Leuk. Res. 2002, 26, 687–688. [Google Scholar] [CrossRef]
- Fei, C.; Guo, J.; Zhao, Y.; Gu, S.; Zhao, S.; Li, X.; Chang, C. Notch-Hes pathway mediates the impaired osteogenic differentiation of bone marrow mesenchymal stromal cells from myelodysplastic syndromes patients through the down-regulation of Runx2. Am. J. Transl. Res. 2015, 7, 1939–1951. [Google Scholar] [PubMed]
- Greenberger, J.S.; FitzGerald, T.J.; Anklesaria, P. Recent studies of the hematopoietic microenvironment in long-term bone marrow cultures. Immunol. Res. 1989, 8, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Lemischka, I.R. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes Dev. 1990, 4, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Lemischka, I.R. Microenvironmental regulation of hematopoietic stem cells. Stem Cells 1997, 15 (Suppl. 1), 63–68. [Google Scholar] [CrossRef] [PubMed]
- Mayani, H. Composition and function of the hemopoietic microenvironment in human myeloid leukemia. Leukemia 1996, 10, 1041–1047. [Google Scholar] [PubMed]
- Pavlaki, K.; Pontikoglou, C.G.; Demetriadou, A.; Batsali, A.K.; Damianaki, A.; Simantirakis, E.; Kontakis, M.; Galanopoulos, A.; Kotsianidis, I.; Kastrinaki, M.C.; et al. Impaired proliferative potential of bone marrow mesenchymal stromal cells in patients with myelodysplastic syndromes is associated with abnormal WNT signaling pathway. Stem Cells Dev. 2014, 23, 1568–1581. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H. Niche contributions to oncogenesis: Emerging concepts and implications for the hematopoietic system. Haematologica 2011, 96, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, A.; Deeg, H.J. A novel role for the marrow microenvironment in initiating and sustaining hematopoietic disease. Expert Opin. Biol. Ther. 2009, 9, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, A.; Awaya, N.; Bryant, E.; Torok-Storb, B. The stromal component of the marrow microenvironment is not derived from the malignant clone in MDS. Blood 2006, 108, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; Kiss, J.; Varkonyi, J.; Vas, V.; Farkas, P.; Paloczi, K.; Uher, F. Inappropriate Notch activity and limited mesenchymal stem cell plasticity in the bone marrow of patients with myelodysplastic syndromes. Pathol. Oncol. Res. 2007, 13, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Link, D.C. The hematopoietic stem cell niche in homeostasis and disease. Blood 2015, 126, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Flores-Figueroa, E.; Gutiérrez-Espindola, G.; Montesinos, J.J.; Arana-Trejo, R.M.; Mayani, H. In vitro characterization of hematopoietic microenvironment cells from patients with myelodysplastic syndrome. Leuk. Res. 2002, 26, 677–686. [Google Scholar] [CrossRef]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Tauro, S.; Hepburn, M.D.; Bowen, D.T.; Pippard, M.J. Assessment of stromal function, and its potential contribution to deregulation of hematopoiesis in the myelodysplastic syndromes. Haematologica 2001, 86, 1038–1045. [Google Scholar] [PubMed]
- Zhang, W.; Knieling, G.; Vohwinkel, G.; Martinez, T.; Kuse, R.; Hossfeld, D.K.; Duhrsen, U. Origin of stroma cells in long-term bone marrow cultures from patients with acute myeloid leukemia. Ann. Hematol. 1999, 78, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Walenda, T.; Bork, S.; Horn, P.; Wein, F.; Saffrich, R.; Diehlmann, A.; Eckstein, V.; Ho, A.D.; Wagner, W. Co-culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. J. Cell. Mol. Med. 2010, 14, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.S.; Choi, Y.; Kim, H.S.; Kim, H.O. Comparison of molecular profiles of human mesenchymal stem cells derived from bone marrow, umbilical cord blood, placenta and adipose tissue. Int. J. Mol. Med. 2016, 37, 115–125. [Google Scholar] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Michienzi, S.; di, C.S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.C.; Phinney, D.G.; Lacey, M.R.; Barrilleaux, B.L.; Meyertholen, K.E.; O’Connor, K.C. In vitro high-capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells 2010, 28, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Feldmann, R.E., Jr.; Seckinger, A.; Maurer, M.H.; Wein, F.; Blake, J.; Krause, U.; Kalenka, A.; Burgers, H.F.; Saffrich, R.; et al. The heterogeneity of human mesenchymal stem cell preparations—Evidence from simultaneous analysis of proteomes and transcriptomes. Exp. Hematol. 2006, 34, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.J.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Sensebe, L. Mesenchymal stromal cells: Misconceptions and evolving concepts. Cytotherapy 2013, 15, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. Clarification of the nomenclature for MSC: The international society for cellular therapy position statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed]
- DeRuiter, M.C.; Poelmann, R.E.; VanMunsteren, J.C.; Mironov, V.; Markwald, R.R.; Gittenberger-de Groot, A.C. Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro. Circ. Res. 1997, 80, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.H.; Joyner, C.J.; Triffitt, J.T.; Owen, M.E. Adipocytic cells cultured from marrow have osteogenic potential. J. Cell Sci. 1991, 99, 131–139. [Google Scholar] [PubMed]
- Dennis, J.E.; Charbord, P. Origin and differentiation of human and murine stroma. Stem Cells 2002, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.E.; Carbillet, J.P.; Caplan, A.I.; Charbord, P. The STRO-1+ marrow cell population is multipotential. Cells Tissues Organs 2002, 170, 73–82. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, D.C.; Ferreira, M.R.; Franzen, J.; Weidner, C.I.; Frobel, J.; Zenke, M.; Costa, I.G.; Wagner, W. Epigenetic classification of human mesenchymal stromal cells. Stem Cell Rep. 2016, 6, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Blasi, A.; Martino, C.; Balducci, L.; Saldarelli, M.; Soleti, A.; Navone, S.E.; Canzi, L.; Cristini, S.; Invernici, G.; Parati, E.A.; et al. Dermal fibroblasts display similar phenotypic and differentiation capacity to fat-derived mesenchymal stem cells, but differ in anti-inflammatory and angiogenic potential. Vasc. Cell 2011, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Harichandan, A.; Buhring, H.J. Prospective isolation of human MSC. Best Pract. Res. Clin. Haematol. 2011, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Buhring, H.J.; Battula, V.L.; Treml, S.; Schewe, B.; Kanz, L.; Vogel, W. Novel markers for the prospective isolation of human MSC. Ann. N. Y. Acad. Sci. 2007, 1106, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Buhring, H.J.; Treml, S.; Cerabona, F.; de, Z.P.; Kanz, L.; Sobiesiak, M. Phenotypic characterization of distinct human bone marrow-derived MSC subsets. Ann. N. Y. Acad. Sci. 2009, 1176, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Lacombe, J.; Hanoun, M.; Mizoguchi, T.; Bruns, I.; Kunisaki, Y.; Frenette, P.S. PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013, 210, 1351–1367. [Google Scholar] [CrossRef] [PubMed]
- Quirici, N.; Soligo, D.; Bossolasco, P.; Servida, F.; Lumini, C.; Deliliers, G.L. Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Exp. Hematol. 2002, 30, 783–791. [Google Scholar] [CrossRef]
- Samsonraj, R.M.; Rai, B.; Sathiyanathan, P.; Puan, K.J.; Rotzschke, O.; Hui, J.H.; Raghunath, M.; Stanton, L.W.; Nurcombe, V.; Cool, S.M. Establishing criteria for human mesenchymal stem cell potency. Stem Cells 2015, 33, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal stem cell: Keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramaniyan, K.; Lehnen, D.; Ghazanfari, R.; Sobiesiak, M.; Harichandan, A.; Mortha, E.; Petkova, N.; Grimm, S.; Cerabona, F.; de, Z.P.; et al. Phenotypic and functional heterogeneity of human bone marrow- and amnion-derived MSC subsets. Ann. N. Y. Acad. Sci. 2012, 1266, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ghazanfari, R.; Zacharaki, D.; Ditzel, N.; Isern, J.; Ekblom, M.; Mendez-Ferrer, S.; Kassem, M.; Scheding, S. Low/negative expression of PDGFR-α identifies the candidate primary mesenchymal stromal cells in adult human bone marrow. Stem Cell Rep. 2014, 3, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M.; Galipeau, J.; Shi, Y.; Tarte, K.; Sensebe, L. Immunological characterization of multipotent mesenchymal stromal cells—The international society for cellular therapy (ISCT) working proposal. Cytotherapy 2013, 15, 1054–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, A. Mesenchymal stromal cells: New directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Sarugaser, R.; Hanoun, L.; Keating, A.; Stanford, W.L.; Davies, J.E. Human mesenchymal stem cells self-renew and differentiate according to a deterministic hierarchy. PLoS ONE 2009, 4, e6498. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Villar, O.; Garcia, J.L.; Sanchez-Guijo, F.M.; Robledo, C.; Villaron, E.M.; Hernandez-Campo, P.; Lopez-Holgado, N.; Diez-Campelo, M.; Barbado, M.V.; Perez-Simon, J.A.; et al. Both expanded and uncultured mesenchymal stem cells from MDS patients are genomically abnormal, showing a specific genetic profile for the 5q- syndrome. Leukemia 2009, 23, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Gul-Uludag, H.; Valencia-Serna, J.; Kucharski, C.; Marquez-Curtis, L.A.; Jiang, X.; Larratt, L.; Janowska-Wieczorek, A.; Uludag, H. Polymeric nanoparticle-mediated silencing of CD44 receptor in CD34 acute myeloid leukemia cells. Leuk. Res. 2014, 38, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Aanei, C.M.; Flandrin, P.; Eloae, F.Z.; Carasevici, E.; Guyotat, D.; Wattel, E.; Campos, L. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev. 2012, 21, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.S.; Wei, M.; Ma, W.L.; Meng, W.; Zheng, W.L. Knockdown of CD44 enhances chemosensitivity of acute myeloid leukemia cells to ADM and Ara-C. Tumour. Biol. 2014, 35, 3933–3940. [Google Scholar] [CrossRef] [PubMed]
- Quere, R.; Andradottir, S.; Brun, A.C.; Zubarev, R.A.; Karlsson, G.; Olsson, K.; Magnusson, M.; Cammenga, J.; Karlsson, S. High levels of the adhesion molecule CD44 on leukemic cells generate acute myeloid leukemia relapse after withdrawal of the initial transforming event. Leukemia 2011, 25, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Spitzer, T.R.; Stowell, C.P. The concentration of CD44 is increased in hematopoietic stem cell grafts of patients with acute myeloid leukemia, plasma cell myeloma, and non-Hodgkin lymphoma. Arch. Pathol. Lab. Med. 2010, 134, 1033–1038. [Google Scholar] [PubMed]
- Huang, X.; Li, D.; Li, T.; Zhao, B.O.; Chen, X. Prognostic value of the expression of phosphatase and tensin homolog and CD44 in elderly patients with refractory acute myeloid leukemia. Oncol. Lett. 2015, 10, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huang, H.; Wu, J.; Lu, R.; Wu, Y.; Jiang, X.; Yuan, Q.; Chen, Y. Bone marrow stromal cells protect acute myeloid leukemia cells from anti-CD44 therapy partly through regulating PI3K/Akt-p27(Kip1) axis. Mol. Carcinog. 2015, 54, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Wang, Y.J.; Lo, C.C.; Ragu, C.; Bullinger, L.; Sykes, S.M.; Ferraro, F.; Shterental, S.; Lin, C.P.; Gilliland, D.G.; et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood 2011, 118, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 7 April 2016).
- Casucci, M.; Nicolis di, R.B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef] [PubMed]
- Aanei, C.M.; Eloae, F.Z.; Flandrin-Gresta, P.; Tavernier, E.; Carasevici, E.; Guyotat, D.; Campos, L. Focal adhesion protein abnormalities in myelodysplastic mesenchymal stromal cells. Exp. Cell Res. 2011, 317, 2616–2629. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Z.; Li, Q.; Li, W.; You, Y.; Zou, P. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PLoS ONE 2012, 7, e45675. [Google Scholar] [CrossRef] [PubMed]
- Fei, C.; Zhao, Y.; Guo, J.; Gu, S.; Li, X.; Chang, C. Senescence of bone marrow mesenchymal stromal cells is accompanied by activation of p53/p21 pathway in myelodysplastic syndromes. Eur. J. Haematol. 2014, 93, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.C.; Kurzer, J.H.; Greenberg, P.L.; Gratzinger, D. Mesenchymal stromal cell density is increased in higher grade myelodysplastic syndromes and independently predicts survival. Am. J. Clin. Pathol. 2014, 142, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Cao, B.; Crisan, M.; Sun, B.; Li, G.; Logar, A.; Yap, S.; Pollett, J.B.; Drowley, L.; Cassino, T.; et al. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nat. Biotechnol. 2007, 25, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Era, T.; Nakao, K.; Kondo, S.; Kasuga, M.; Smith, A.G.; Nishikawa, S. Neuroepithelial cells supply an initial transient wave of MSC differentiation. Cell 2007, 129, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Corselli, M.; Chen, C.W.; Sun, B.; Yap, S.; Rubin, J.P.; Peault, B. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells Dev. 2012, 21, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Majesky, M.W.; Dong, X.R.; Hoglund, V.; Mahoney, W.M., Jr.; Daum, G. The adventitia: A dynamic interface containing resident progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Park, T.S.; Murray, I.R.; Zimmerlin, L.; Lazzari, L.; Huard, J.; Peault, B. Cellular kinetics of perivascular MSC precursors. Stem Cells Int. 2013, 2013, 983059. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Corselli, M.; Peault, B.; Huard, J. Human blood-vessel-derived stem cells for tissue repair and regeneration. J. Biomed. Biotechnol. 2012, 2012, 597439. [Google Scholar] [CrossRef] [PubMed]
- Tavian, M.; Zheng, B.; Oberlin, E.; Crisan, M.; Sun, B.; Huard, J.; Peault, B. The vascular wall as a source of stem cells. Ann. N. Y. Acad. Sci. 2005, 1044, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G. The developmental basis of mesenchymal stem/stromal cells (MSCs). BMC. Dev. Biol. 2015, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Vodyanik, M.A.; Yu, J.; Zhang, X.; Tian, S.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 2010, 7, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Slukvin, I.I.; Vodyanik, M. Endothelial origin of mesenchymal stem cells. Cell Cycle 2011, 10, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Chapuli, R.; Carmona, R.; Guadix, J.A.; Macias, D.; Perez-Pomares, J.M. The origin of the endothelial cells: An evo-devo approach for the invertebrate/vertebrate transition of the circulatory system. Evol. Dev. 2005, 7, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Choi, K. The hemangioblast: A common progenitor of hematopoietic and endothelial cells. J. Hematother. Stem Cell Res. 2002, 11, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Ciraci, E.; Della, B.S.; Salvucci, O.; Rofani, C.; Segarra, M.; Bason, C.; Molinari, A.; Maric, D.; Tosato, G.; Berardi, A.C. Adult human circulating CD34−Lin−CD45−CD133− cells can differentiate into hematopoietic and endothelial cells. Blood 2011, 118, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Lancrin, C.; Sroczynska, P.; Stephenson, C.; Allen, T.; Kouskoff, V.; Lacaud, G. The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature 2009, 457, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Gunsilius, E.; Duba, H.C.; Petzer, A.L.; Kahler, C.M.; Grunewald, K.; Stockhammer, G.; Gabl, C.; Dirnhofer, S.; Clausen, J.; Gastl, G. Evidence from a leukaemia model for maintenance of vascular endothelium by bone-marrow-derived endothelial cells. Lancet 2000, 355, 1688–1691. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sanchez, V.; Takata, N.; Yokomizo, T.; Yamanaka, Y.; Kataoka, H.; Hoppe, P.S.; Schroeder, T.; Nishikawa, S. Circulation-independent differentiation pathway from extraembryonic mesoderm toward hematopoietic stem cells via hemogenic angioblasts. Cell Rep. 2014, 8, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Basak, G.W.; Yasukawa, S.; Alfaro, A.; Halligan, S.; Srivastava, A.S.; Min, W.P.; Minev, B.; Carrier, E. Human embryonic stem cells hemangioblast express HLA-antigens. J. Transl. Med. 2009, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Itoh, H.; Hirashima, M.; Ogawa, M.; Nishikawa, S.; Yurugi, T.; Naito, M.; Nakao, K.; Nishikawa, S. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 2000, 408, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Kuprijanov, V.V. Vascular endothelium (review). I. General morphology. 2B: Phylogenesis of the vascular endothelium. Gegenbaurs Morphol. Jahrb. 1990, 136, 201–217. [Google Scholar] [PubMed]
- Green, A.R. Haemangioblast origin of chronic myeloid leukaemia? Lancet 2000, 355, 1659–1660. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.; Boxberger, S.; Jorgensen, B.; Feldmann, S.; Ehninger, G.; Bornhauser, M.; Werner, C. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells 2004, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M. Transition of mesenchymal stem/stromal cells to endothelial cells. Stem Cell Res. Ther. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Chute, J.P.; Muramoto, G.G.; Salter, A.B.; Meadows, S.K.; Rickman, D.W.; Chen, B.; Himburg, H.A.; Chao, N.J. Transplantation of vascular endothelial cells mediates the hematopoietic recovery and survival of lethally irradiated mice. Blood 2007, 109, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.B.; Meadows, S.K.; Muramoto, G.G.; Himburg, H.; Doan, P.; Daher, P.; Russell, L.; Chen, B.; Chao, N.J.; Chute, J.P. Endothelial progenitor cell infusion induces hematopoietic stem cell reconstitution in vivo. Blood 2009, 113, 2104–2107. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Johnson, S.A.; Shelley, W.C.; Yoder, M.C. Hematopoietic stem cell repopulating ability can be maintained in vitro by some primary endothelial cells. Exp. Hematol. 2004, 32, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Reale, A.; Melaccio, A.; Lamanuzzi, A.; Saltarella, I.; Dammacco, F.; Vacca, A.; Ria, R. Functional and biological role of endothelial precursor cells in tumour progression: A new potential therapeutic target in haematological malignancies. Stem Cells Int. 2016, 2016, 7954580. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Masuda, H.; Takahashi, T.; Kalka, C.; Pastore, C.; Silver, M.; Kearne, M.; Magner, M.; Isner, J.M. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 1999, 85, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Buckstein, R.; Kerbel, R.; Cheung, M.; Shaked, Y.; Chodirker, L.; Lee, C.R.; Lenis, M.; Davidson, C.; Cussen, M.A.; Reis, M.; et al. Lenalidomide and metronomic melphalan for CMML and higher risk MDS: A phase 2 clinical study with biomarkers of angiogenesis. Leuk. Res. 2014, 38, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Cortelezzi, A.; Fracchiolla, N.S.; Mazzeo, L.M.; Silvestris, I.; Pomati, M.; Somalvico, F.; Bertolini, F.; Mancuso, P.; Pruneri, G.C.; Gianelli, U.; et al. Endothelial precursors and mature endothelial cells are increased in the peripheral blood of myelodysplastic syndromes. Leuk. Lymphoma 2005, 46, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Della Porta, M.G.; Malcovati, L.; Rigolin, G.M.; Rosti, V.; Bonetti, E.; Travaglino, E.; Boveri, E.; Galli, A.; Boggi, S.; Ciccone, M.; et al. Immunophenotypic, cytogenetic and functional characterization of circulating endothelial cells in myelodysplastic syndromes. Leukemia 2008, 22, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Sudhoff, T.; Germing, U.; Aul, C. Levels of circulating endothelial adhesion molecules in patients with myelodysplastic syndromes. Int. J. Oncol. 2002, 20, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Teofili, L.; Martini, M.; Nuzzolo, E.R.; Capodimonti, S.; Iachininoto, M.G.; Cocomazzi, A.; Fabiani, E.; Voso, M.T.; Larocca, L.M. Endothelial progenitor cell dysfunction in myelodysplastic syndromes: Possible contribution of a defective vascular niche to myelodysplasia. Neoplasia 2015, 17, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Agliano, A.; Martin-Padura, I.; Mancuso, P.; Marighetti, P.; Rabascio, C.; Pruneri, G.; Shultz, L.D.; Bertolini, F. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int. J. Cancer 2008, 123, 2222–2227. [Google Scholar] [CrossRef] [PubMed]
- Rigolin, G.M.; Mauro, E.; Ciccone, M.; Fraulini, C.; Sofritti, O.; Castoldi, G.; Cuneo, A. Neoplastic circulating endothelial-like cells in patients with acute myeloid leukaemia. Eur. J. Haematol. 2007, 78, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Wierzbowska, A.; Robak, T.; Krawczynska, A.; Wrzesien-Kus, A.; Pluta, A.; Cebula, B.; Smolewski, P. Circulating endothelial cells in patients with acute myeloid leukemia. Eur. J. Haematol. 2005, 75, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Wierzbowska, A.; Robak, T.; Krawczynska, A.; Pluta, A.; Wrzesien-Kus, A.; Cebula, B.; Robak, E.; Smolewski, P. Kinetics and apoptotic profile of circulating endothelial cells as prognostic factors for induction treatment failure in newly diagnosed acute myeloid leukemia patients. Ann. Hematol. 2008, 87, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.; Oyan, A.M.; Ersvaer, E.; Kalland, K.H.; Lassalle, P.; Gjertsen, B.T.; Bruserud, O. Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators. Br. J. Haematol. 2009, 144, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Pezeshkian, B.; Donnelly, C.; Tamburo, K.; Geddes, T.; Madlambayan, G.J. Leukemia mediated endothelial cell activation modulates leukemia cell susceptibility to chemotherapy through a positive feedback loop mechanism. PLoS ONE 2013, 8, e60823. [Google Scholar] [CrossRef] [PubMed]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Cote, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, R.J.; Azadniv, M.; Guo, N.; Acklin, J.; Lacagnina, K.; Coppage, M.; Liesveld, J.L. Phenotypic, genotypic, and functional characterization of normal and acute myeloid leukemia-derived marrow endothelial cells. Exp. Hematol. 2016, 44, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Drusbosky, L.; Gars, E.; Trujillo, A.; McGee, C.; Meacham, A.; Wise, E.; Scott, E.W.; Cogle, C.R. Endothelial cell derived angiocrine support of acute myeloid leukemia targeted by receptor tyrosine kinase inhibition. Leuk. Res. 2015, 39, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Streubel, B.; Chott, A.; Huber, D.; Exner, M.; Jager, U.; Wagner, O.; Schwarzinger, I. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N. Engl. J. Med. 2004, 351, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Saint-Jeannet, J.P. Induction of the neural crest and the opportunities of life on the edge. Dev. Biol. 2004, 275, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Korn, J.; Christ, B.; Kurz, H. Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. J. Comp. Neurol. 2002, 442, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Etchevers, H.C.; Vincent, C.; Le Douarin, N.M.; Couly, G.F. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development 2001, 128, 1059–1068. [Google Scholar] [PubMed]
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Pericytes at the intersection between tissue regeneration and pathology. Clin. Sci. Lond. 2015, 128, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Rom, S.; Ramirez, S.H.; Persidsky, Y. Emerging roles of pericytes in the regulation of the neurovascular unit in health and disease. J. Neuroimmune Pharmacol. 2014, 9, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Sa-Pereira, I.; Brites, D.; Brito, M.A. Neurovascular unit: A focus on pericytes. Mol. Neurobiol. 2012, 45, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Zimmerlin, L.; Park, T.S.; Donnenberg, V.S.; Zambidis, E.T.; Donnenberg, A.D. Pericytes: A ubiquitous source of multipotent adult tissue stem cells. In Stem Cells in Aesthetic Procedures: Art, Science, and Clinical Techniques; Shiffman, A.M., di Giuseppe, A., Bassetto, F., Eds.; Springer: Berlin, Germany, 2014; pp. 135–148. [Google Scholar]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Brachvogel, B.; Moch, H.; Pausch, F.; Schlotzer-Schrehardt, U.; Hofmann, C.; Hallmann, R.; von der, M.K.; Winkler, T.; Poschl, E. Perivascular cells expressing annexin A5 define a novel mesenchymal stem cell-like population with the capacity to differentiate into multiple mesenchymal lineages. Development 2005, 132, 2657–2668. [Google Scholar] [CrossRef] [PubMed]
- Covas, D.T.; Panepucci, R.A.; Fontes, A.M.; Silva, W.A., Jr.; Orellana, M.D.; Freitas, M.C.; Neder, L.; Santos, A.R.; Peres, L.C.; Jamur, M.C.; et al. Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Exp. Hematol. 2008, 36, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Gokcinar-Yagci, B.; Uckan-Cetinkaya, D.; Celebi-Saltik, B. Pericytes: Properties, functions and applications in tissue engineering. Stem Cell Rev. 2015, 11, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Tormin, A.; Li, O.; Brune, J.C.; Walsh, S.; Schutz, B.; Ehinger, M.; Ditzel, N.; Kassem, M.; Scheding, S. CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 2011, 117, 5067–5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A.I. All MSCs are pericytes? Cell Stem Cell 2008, 3, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Rieske, P.; Krynska, B.; Azizi, S.A. Human fibroblast-derived cell lines have characteristics of embryonic stem cells and cells of neuro-ectodermal origin. Differentiation 2005, 73, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Mukouyama, Y.S.; Mosher, J.T.; Jaegle, M.; Crone, S.A.; Dormand, E.L.; Lee, K.F.; Meijer, D.; Anderson, D.J.; Morrison, S.J. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development 2004, 131, 5599–5612. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Lee, H.L.; Hong, C.; Wang, C.Y. Single CD271 marker isolates mesenchymal stem cells from human dental pulp. Int. J. Oral Sci. 2015, 7, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, G.; Giuffrida, R.; Lo, F.D.; Parrinello, N.L.; Forte, S.; Gulino, R.; Colarossi, C.; Schinocca, L.R.; Giuffrida, R.; Cardile, V.; et al. Potential effect of CD271 on human mesenchymal stromal cell proliferation and differentiation. Int. J. Mol. Sci. 2015, 16, 15609–15624. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; White, P.M.; Zock, C.; Anderson, D.J. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell 1999, 96, 737–749. [Google Scholar] [CrossRef]
- Poloni, A.; Maurizi, G.; Rosini, V.; Mondini, E.; Mancini, S.; Discepoli, G.; Biasio, S.; Battaglini, G.; Felicetti, S.; Berardinelli, E.; et al. Selection of CD271+ cells and human AB serum allows a large expansion of mesenchymal stromal cells from human bone marrow. Cytotherapy 2009, 11, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Churchman, S.M.; Ponchel, F.; Boxall, S.A.; Cuthbert, R.; Kouroupis, D.; Roshdy, T.; Giannoudis, P.V.; Emery, P.; McGonagle, D.; Jones, E.A. Transcriptional profile of native CD271+ multipotential stromal cells: Evidence for multiple fates, with prominent osteogenic and Wnt pathway signaling activity. Arthritis Rheum. 2012, 64, 2632–2643. [Google Scholar] [CrossRef] [PubMed]
- Flores-Figueroa, E.; Varma, S.; Montgomery, K.; Greenberg, P.L.; Gratzinger, D. Distinctive contact between CD34+ hematopoietic progenitors and CXCL12+ CD271+ mesenchymal stromal cells in benign and myelodysplastic bone marrow. Lab. Investig. 2012, 92, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Zimmerman, L.B.; McKay, R.D. CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60, 585–595. [Google Scholar] [CrossRef]
- Isern, J.; Garcia-Garcia, A.; Martin, A.M.; Arranz, L.; Martin-Perez, D.; Torroja, C.; Sanchez-Cabo, F.; Mendez-Ferrer, S. The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. Elife 2014, 3, e03696. [Google Scholar] [CrossRef] [PubMed]
- Wislet-Gendebien, S.; Laudet, E.; Neirinckx, V.; Alix, P.; Leprince, P.; Glejzer, A.; Poulet, C.; Hennuy, B.; Sommer, L.; Shakhova, O.; et al. Mesenchymal stem cells and neural crest stem cells from adult bone marrow: Characterization of their surprising similarities and differences. Cell. Mol. Life Sci. 2012, 69, 2593–2608. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Namiki, J.; Shibata, S.; Mastuzaki, Y.; Okano, H. The neural stem/progenitor cell marker nestin is expressed in proliferative endothelial cells, but not in mature vasculature. J. Histochem. Cytochem. 2010, 58, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Hagio, M.; Ishiwata, T. Nestin: A novel angiogenesis marker and possible target for tumor angiogenesis. World J. Gastroenterol. 2013, 19, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Kishaba, Y.; Matsubara, D.; Niki, T. Heterogeneous expression of nestin in myofibroblasts of various human tissues. Pathol. Int. 2010, 60, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Krupkova, O., Jr.; Loja, T.; Zambo, I.; Veselska, R. Nestin expression in human tumors and tumor cell lines. Neoplasma 2010, 57, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.E.; Bowman, E.P.; Wagers, A.J.; Butcher, E.C.; Weissman, I.L. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J. Exp. Med. 2002, 195, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Tavor, S.; Petit, I.; Porozov, S.; Avigdor, A.; Dar, A.; Leider-Trejo, L.; Shemtov, N.; Deutsch, V.; Naparstek, E.; Nagler, A.; et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004, 64, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ema, H.; Karlsson, G.; Yamaguchi, T.; Miyoshi, H.; Shioda, S.; Taketo, M.M.; Karlsson, S.; Iwama, A.; Nakauchi, H. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 2011, 147, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ramos, J.; Song, S.; Cardozo-Pelaez, F.; Hazzi, C.; Stedeford, T.; Willing, A.; Freeman, T.B.; Saporta, S.; Janssen, W.; Patel, N.; et al. Adult bone marrow stromal cells differentiate into neural cells in vitro. Exp. Neurol. 2000, 164, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Woodbury, D.; Schwarz, E.J.; Prockop, D.J.; Black, I.B. Adult rat and human bone marrow stromal cells differentiate into neurons. J. Neurosci. Res. 2000, 61, 364–370. [Google Scholar] [CrossRef]
- Tzeng, H.H.; Hsu, C.H.; Chung, T.H.; Lee, W.C.; Lin, C.H.; Wang, W.C.; Hsiao, C.Y.; Leu, Y.W.; Wang, T.H. Cell signaling and differential protein expression in neuronal differentiation of bone marrow mesenchymal stem cells with hypermethylated Salvador/Warts/Hippo (SWH) pathway genes. PLoS ONE 2015, 10, e0145542. [Google Scholar] [CrossRef] [PubMed]
- Bossolasco, P.; Cova, L.; Calzarossa, C.; Rimoldi, S.G.; Borsotti, C.; Deliliers, G.L.; Silani, V.; Soligo, D.; Polli, E. Neuro-glial differentiation of human bone marrow stem cells in vitro. Exp. Neurol. 2005, 193, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.S.; Park, J.B.; Kim, H.S.; Kim, D.S.; Park, D.J.; Kang, S.J. Neuron-like differentiation of bone marrow-derived mesenchymal stem cells. Yonsei Med. J. 2011, 52, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Liu, Z.L.; Yao, X.Q.; Yang, Z.J.; Xu, R.X. Neural differentiation ability of mesenchymal stromal cells from bone marrow and adipose tissue: A comparative study. Cytotherapy 2012, 14, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Widera, D.; Heimann, P.; Zander, C.; Imielski, Y.; Heidbreder, M.; Heilemann, M.; Kaltschmidt, C.; Kaltschmidt, B. Schwann cells can be reprogrammed to multipotency by culture. Stem Cells Dev. 2011, 20, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Apostolova, G.; Widera, D.; Mittelbronn, M.; Dechant, G.; Kaltschmidt, B.; Rohrer, H. Alternative generation of CNS neural stem cells and PNS derivatives from neural crest-derived peripheral stem cells. Stem Cells 2015, 33, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Maisel, M.; Storch, A. Epigenetic conversion of human adult bone mesodermal stromal cells into neuroectodermal cell types for replacement therapy of neurodegenerative disorders. Expert Opin. Biol. Ther. 2006, 6, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Coste, C.; Neirinckx, V.; Gothot, A.; Wislet, S.; Rogister, B. Are neural crest stem cells the missing link between hematopoietic and neurogenic niches? Front. Cell Neurosci. 2015, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Abe-Suzuki, S.; Kurata, M.; Abe, S.; Onishi, I.; Kirimura, S.; Nashimoto, M.; Murayama, T.; Hidaka, M.; Kitagawa, M. CXCL12+ stromal cells as bone marrow niche for CD34+ hematopoietic cells and their association with disease progression in myelodysplastic syndromes. Lab. Investig. 2014, 94, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Afrin, F.; Satija, N.; Tripathi, R.P.; Gangenahalli, G.U. Stromal-derived factor-1/CXCR4 signaling: Indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev. 2011, 20, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Fuhler, G.M.; Drayer, A.L.; Olthof, S.G.; Schuringa, J.J.; Coffer, P.J.; Vellenga, E. Reduced activation of protein kinase B, Rac, and F-actin polymerization contributes to an impairment of stromal cell derived factor-1 induced migration of CD34+ cells from patients with myelodysplasia. Blood 2008, 111, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Morita, Y.; Hanamoto, H.; Tatsumi, Y.; Maeda, Y.; Kanamaru, A. CD34+ progenitors from MDS patients are unresponsive to SDF-1, despite high levels of SDF-1 in bone marrow plasma. Leukemia 2004, 18, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Oh, Y.S.; Song, I.C.; Kim, S.W.; Lee, H.J.; Yun, H.J.; Kim, S.; Jo, D.Y. Endogenous stromal cell-derived factor-1 (CXCL12) supports autonomous growth of acute myeloid leukemia cells. Leuk. Res. 2013, 37, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Conneally, E.; Cashman, J.; Petzer, A.; Eaves, C. Expansion in vitro of transplantable human cord blood stem cells demonstrated using a quantitative assay of their lympho-myeloid repopulating activity in nonobese diabetic-scid/scid mice. Proc. Natl. Acad. Sci. USA 1997, 94, 9836–9841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Patel, S.; Abdelouahab, H.; Wittner, M.; Willekens, C.; Shen, S.; Betems, A.; Joulin, V.; Opolon, P.; Bawa, O.; et al. CXCR4 inhibitors selectively eliminate CXCR4-expressing human acute myeloid leukemia cells in NOG mouse model. Cell Death. Dis. 2012, 3, e396. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.S.; Zeng, Z.; Mu, H.; Wang, Z.; Konoplev, S.; McQueen, T.; Protopopova, M.; Cortes, J.; Marszalek, J.R.; Peng, S.B.; et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015, 126, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.; Battista, M.; Shi, P.A.; Isola, L.; Frenette, P.S. Mobilized hematopoietic stem cell yield depends on species-specific circadian timing. Cell Stem Cell 2008, 3, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Chow, A.; Merad, M.; Frenette, P.S. Circadian rhythms influence hematopoietic stem cells. Curr. Opin. Hematol. 2009, 16, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fong, C.; Chen, Y.; Cai, G.; Yang, M. β2- and β3-, but not beta1-adrenergic receptors are involved in osteogenesis of mouse mesenchymal stem cells via cAMP/PKA signaling. Arch. Biochem. Biophys. 2010, 496, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Battista, M.; Frenette, P.S. Cooperation of β2- and β3-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann. N. Y. Acad. Sci. 2010, 1192, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Wang, L.; Zhao, Y.; Cao, J.; Wang, T.; Liu, P.; Zhang, Y.; Yang, X.; Cheng, X.; Liu, B.; et al. Sympathetic denervation-induced MSC mobilization in distraction osteogenesis associates with inhibition of MSC migration and osteogenesis by norepinephrine/adrb3. PLoS ONE 2014, 9, e105976. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, R.; Mendez-Ferrer, S. Autonomic regulation of hematopoiesis and cancer. Haematologica 2013, 98, 1663–1666. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Sanchez-Aguilera, A.; Martin-Perez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntion, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Evans, K.W.; Hollier, B.G.; Shi, Y.; Marini, F.C.; Ayyanan, A.; Wang, R.Y.; Brisken, C.; Guerra, R.; Andreeff, M.; et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 2010, 28, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.Y.; Liu, X.Y.; Wang, N.; Wang, L.N.; Yang, B.X.; Ren, Q.; Liang, H.Y.; Ma, X.T. Twist-1, a novel regulator of hematopoietic stem cell self-renewal and myeloid lineage development. Stem Cells 2014, 32, 3173–3182. [Google Scholar] [PubMed]
- Merindol, N.; Riquet, A.; Szablewski, V.; Eliaou, J.F.; Puisieux, A.; Bonnefoy, N. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood Cancer J. 2014, 4, e206. [Google Scholar] [CrossRef] [PubMed]
- Isenmann, S.; Arthur, A.; Zannettino, A.C.; Turner, J.L.; Shi, S.; Glackin, C.A.; Gronthos, S. TWIST family of basic helix-loop-helix transcription factors mediate human mesenchymal stem cell growth and commitment. Stem Cells 2009, 27, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Cakouros, D.; Raices, R.M.; Gronthos, S.; Glackin, C.A. Twist-ing cell fate: Mechanistic insights into the role of twist in lineage specification/differentiation and tumorigenesis. J. Cell. Biochem. 2010, 110, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Norozi, F.; Ahmadzadeh, A.; Shahjahani, M.; Shahrabi, S.; Saki, N. Twist as a new prognostic marker in hematological malignancies. Clin. Transl. Oncol. 2016, 18, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, P.; Shen, H.; Yu, J.; Liu, Q.; Du, J. Prognostic role of Twist or Snail in various carcinomas: A systematic review and meta-analysis. Eur. J. Clin. Investig. 2014, 44, 1072–1094. [Google Scholar] [CrossRef] [PubMed]
- Cosset, E.; Hamdan, G.; Jeanpierre, S.; Voeltzel, T.; Sagorny, K.; Hayette, S.; Mahon, F.X.; Dumontet, C.; Puisieux, A.; Nicolini, F.E.; et al. Deregulation of TWIST-1 in the CD34+ compartment represents a novel prognostic factor in chronic myeloid leukemia. Blood 2011, 117, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Marcondes, A.M.; Gooley, T.A.; Deeg, H.J. The helix-loop-helix transcription factor TWIST is dysregulated in myelodysplastic syndromes. Blood 2010, 116, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Mhyre, A.J.; Marcondes, A.M.; Spaulding, E.Y.; Deeg, H.J. Stroma-dependent apoptosis in clonal hematopoietic precursors correlates with expression of PYCARD. Blood 2009, 113, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.; Lucas, D.M.; Matkovic, J.J.; Bennett, K.L.; Liyanarachchi, S.; Young, D.C.; Rassenti, L.; Kipps, T.J.; Grever, M.R.; Byrd, J.C.; et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J. Clin. Oncol. 2005, 23, 3877–3885. [Google Scholar] [CrossRef] [PubMed]
- Thathia, S.H.; Ferguson, S.; Gautrey, H.E.; van Otterdijk, S.D.; Hili, M.; Rand, V.; Moorman, A.V.; Meyer, S.; Brown, R.; Strathdee, G. Epigenetic inactivation of TWIST2 in acute lymphoblastic leukemia modulates proliferation, cell survival and chemosensitivity. Haematologica 2012, 97, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, W.; Cui, J.; Yao, H.; Zhou, H.; Ge, Y.; Xiao, L.; Hu, X.; Liu, B.H.; Yang, J.; et al. Regulation of p21 by TWIST2 contributes to its tumor-suppressor function in human acute myeloid leukemia. Oncogene 2015, 34, 3000–3010. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; You, J.Y.; Gau, J.P.; Huang, C.E.; Chen, Y.Y.; Tsai, Y.H.; Chou, H.J.; Lung, J.; Yang, M.H. Favorable clinical outcome and unique characteristics in association with Twist1 overexpression in de novo acute myeloid leukemia. Blood Cancer J. 2015, 5, e339. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Guo, D.; Zhao, Y.Y.; Dong, C.Y.; Liu, X.Y.; Yang, B.X.; Wang, S.W.; Wang, L.; Liu, Q.G.; Ren, Q.; et al. TWIST-1 promotes cell growth, drug resistance and progenitor clonogenic capacities in myeloid leukemia and is a novel poor prognostic factor in acute myeloid leukemia. Oncotarget 2015, 6, 20977–20992. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Chen, H.C.; Zhang, D.; Fu, J. Twist: A molecular target in cancer therapeutics. Tumour. Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Percio, S.; Coltella, N.; Grisanti, S.; Bernardi, R.; Pattini, L. A HIF-1 network reveals characteristics of epithelial-mesenchymal transition in acute promyelocytic leukemia. Genome Med. 2014, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Piquer-Gil, M.; Garcia-Verdugo, J.M.; Zipancic, I.; Sanchez, M.J.; Alvarez-Dolado, M. Cell fusion contributes to pericyte formation after stroke. J. Cereb. Blood Flow Metab. 2009, 29, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, S.; Bianchi, F.; Galletti, M.; Olivi, E.; Alviano, F.; Galie, N.; Ventura, C. Occurring of in vitro functional vasculogenic pericytes from human circulating early endothelial precursor cell culture. Stem Cells Int. 2015, 2015, 943671. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; DiRocco, D.P.; Humphreys, B.D. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J. Pathol. 2013, 231, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Flores, L.; Gutierrez, R.; Garcia, M.P.; Saez, F.J.; Díaz-Flores, L., Jr.; Valladares, F.; Madrid, J.F. CD34+ stromal cells/fibroblasts/fibrocytes/telocytes as a tissue reserve and a principal source of mesenchymal cells. Location, morphology, function and role in pathology. Histol. Histopathol. 2014, 29, 831–870. [Google Scholar] [PubMed]
- Butler, J.M.; Nolan, D.J.; Vertes, E.L.; Varnum-Finney, B.; Kobayashi, H.; Hooper, A.T.; Seandel, M.; Shido, K.; White, I.A.; Kobayashi, M.; et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell 2010, 6, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M. Osteolineage cells and regulation of the hematopoietic stem cell. Best Pract. Res. Clin. Haematol. 2013, 26, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Lucas, D.; Hidalgo, A.; Mendez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; van Rooijen, N.; et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Lo, C.C.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, C.M.; Calvi, L.M. Minireview: Complexity of hematopoietic stem cell regulation in the bone marrow microenvironment. Mol. Endocrinol. 2014, 28, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Butler, J.M.; O’Donnell, R.; Kobayashi, M.; Ding, B.S.; Bonner, B.; Chiu, V.K.; Nolan, D.J.; Shido, K.; Benjamin, L.; et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat. Cell Biol. 2010, 12, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Lawal, R.A.; Calvi, L.M. The niche as a target for hematopoietic manipulation and regeneration. Tissue Eng. Part B Rev. 2011, 17, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Lymperi, S.; Ersek, A.; Ferraro, F.; Dazzi, F.; Horwood, N.J. Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood 2011, 117, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.; Wakkach, A.; Blin-Wakkach, C. Role of osteoclasts in the hematopoietic stem cell niche formation. Cell Cycle 2012, 11, 2045–2046. [Google Scholar] [CrossRef] [PubMed]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Sadahira, Y.; Mori, M. Role of the macrophage in erythropoiesis. Pathol. Int. 1999, 49, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.N.; Calvi, L.M. Concise review: Current concepts in bone marrow microenvironmental regulation of hematopoietic stem and progenitor cells. Stem Cells 2013, 31, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Reilly, M.J.; Emerson, S.G. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 1996, 87, 518–524. [Google Scholar] [PubMed]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar] [PubMed]
- Turksen, K. (Ed.) Stem Cell Biology and Regenerative Medicine: Tissue-Specific Stem Cell Niche; Humana Press: New York, NY, USA, 2015.
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Doan, P.L.; Chute, J.P. The vascular niche: Home for normal and malignant hematopoietic stem cells. Leukemia 2012, 26, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Scadden, D.T. A hostel for the hostile: The bone marrow niche in hematologic neoplasms. Haematologica 2015, 100, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of mesenchymal stem cells. Cell Death. Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Nor, J.E. The perivascular niche and self-renewal of stem cells. Front. Physiol. 2015, 6, 367. [Google Scholar] [CrossRef] [PubMed]
- Guerrouahen, B.S.; Al-Hijji, I.; Tabrizi, A.R. Osteoblastic and vascular endothelial niches, their control on normal hematopoietic stem cells, and their consequences on the development of leukemia. Stem Cells Int. 2011, 2011, 375857. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Li, L. The stem cell niches in bone. J. Clin. Investig. 2006, 116, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Nagasawa, T. Bone marrow niches for hematopoietic stem cells and immune cells. Inflamm. Allergy Drug Targets 2012, 11, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takubo, K.; Fujioka, M.; Suda, T. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem. Biophys. Res. Commun. 2014, 454, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takubo, K.; Kobayashi, H.; Suzuki-Inoue, K.; Suda, T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J. Exp. Med. 2015, 212, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.M.; Chen, Z.X.; Cen, J.N.; He, J.; Jiao, X.L.; Pan, J.L.; Qiu, Q.C.; Dai, L.; Liu, D.D. Osteoblasts from patients with myelodysplastic syndrome express multiple cytokines and support hematopoietic progenitor cell survival in vitro. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2008, 16, 78–83. [Google Scholar] [PubMed]
- Fei, C.; Zhao, Y.; Gu, S.; Guo, J.; Zhang, X.; Li, X.; Chang, C. Impaired osteogenic differentiation of mesenchymal stem cells derived from bone marrow of patients with lower-risk myelodysplastic syndromes. Tumour. Biol. 2014, 35, 4307–4316. [Google Scholar] [CrossRef] [PubMed]
- Schajnovitz, A.; Scadden, D.T. Bone’s dark side: Mutated osteoblasts implicated in leukemia. Cell Res. 2014, 24, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Nybakken, G.; Gratzinger, D. Myelodysplastic syndrome macrophages have aberrant iron storage and heme oxygenase-1 expression. Leuk. Lymphoma 2016. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Swerdlow, S.H.; TenEyck, S.P.; Boyiadzis, M.; Felgar, R.E. Natural killer cell (NK) subsets and NK-like T-cell populations in acute myeloid leukemias and myelodysplastic syndromes. Cytom. B Clin. Cytom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, A.M.; Mhyre, A.J.; Stirewalt, D.L.; Kim, S.H.; Dinarello, C.A.; Deeg, H.J. Dysregulation of IL-32 in myelodysplastic syndrome and chronic myelomonocytic leukemia modulates apoptosis and impairs NK function. Proc. Natl. Acad. Sci. USA 2008, 105, 2865–2870. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Shimizu, K.; Klimek, V.; Geller, M.D.; Nimer, S.D.; Dhodapkar, M.V. Severe and selective deficiency of interferon-γ-producing invariant natural killer T cells in patients with myelodysplastic syndromes. Br. J. Haematol. 2003, 122, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, K.; Morii, T.; Nieda, M.; Tsukaguchi, N.; Amano, I.; Tanaka, H.; Yagi, H.; Narita, N.; Kimura, H. The peripheral blood Valpha24+ NKT cell numbers decrease in patients with haematopoietic malignancy. Leuk. Res. 2005, 29, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Bouchliou, I.; Miltiades, P.; Nakou, E.; Spanoudakis, E.; Goutzouvelidis, A.; Vakalopoulou, S.; Garypidou, V.; Kotoula, V.; Bourikas, G.; Tsatalas, C.; et al. Th17 and Foxp3+ T regulatory cell dynamics and distribution in myelodysplastic syndromes. Clin. Immunol. 2011, 139, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Fozza, C.; Longinotti, M. The role of T-cells in the pathogenesis of myelodysplastic syndromes: Passengers and drivers. Leuk. Res. 2013, 37, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, A.W.; Epling-Burnette, P.K. Effector memory regulatory T-cell expansion marks a pivotal point of immune escape in myelodysplastic syndromes. Oncoimmunology 2013, 2, e22654. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Zheng, Y.; Li, X.; Lu, S.; Li, H.; Chen, F.; Chen, D.; Shao, Y.; Shi, J.; Feng, S. Differential expression profile of Th1/Th17/Th2-related chemokines and their receptors in patients with acquired bone marrow failure syndromes. Hum. Immunol. 2013, 74, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.L.; Zhang, L.; Hou, Y.; Yu, S.; Liu, X.G.; Huang, X.Y.; Sun, Y.X.; Tian, T.; He, N.; Ma, D.X.; et al. Th22 cells as well as Th17 cells expand differentially in patients with early-stage and late-stage myelodysplastic syndrome. PLoS ONE 2012, 7, e51339. [Google Scholar] [CrossRef] [PubMed]
- Sand, K.E.; Rye, K.P.; Mannsaker, B.; Bruserud, O.; Kittang, A.O. Expression patterns of chemokine receptors on circulating T cells from myelodysplastic syndrome patients. Oncoimmunology 2013, 2, e23138. [Google Scholar] [CrossRef] [PubMed]
- Davison, G.M.; Novitzky, N.; Abdulla, R. Monocyte derived dendritic cells have reduced expression of co-stimulatory molecules but are able to stimulate autologous T-cells in patients with MDS. Hematol. Oncol. Stem Cell Ther. 2013, 6, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoff, N.; Bontkes, H.J.; Westers, T.M.; de Gruijl, T.D.; Kordasti, S.; van de Loosdrecht, A.A. Dendritic cells in myelodysplastic syndromes: From pathogenesis to immunotherapy. Immunotherapy 2013, 5, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, H.J.; Osei, E.; Schweitzer, K.; Blidaru, G.; Edinger, A.; Schlegelmilch, J.; Awadallah, A.; Goyal, T. CD1c+ myeloid dendritic cells in myeloid neoplasia. Cytom. B Clin. Cytom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, R.A.; Wobus, M.; List, C.; Wehner, R.; Schönefeldt, C.; Brocard, B.; Mohr, B.; Rauner, M.; Schmitz, M.; Stiehler, M.; et al. Mesenchymal stromal cells from patients with myelodyplastic syndrome display distinct functional alterations that are modulated by lenalidomide. Haematologica 2013, 98, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Tennant, G.B.; Walsh, V.; Truran, L.N.; Edwards, P.; Mills, K.I.; Burnett, A.K. Abnormalities of adherent layers grown from bone marrow of patients with myelodysplasia. Br. J. Haematol. 2000, 111, 853–862. [Google Scholar] [PubMed]
- Li, X.; Marcondes, A.M.; Ragoczy, T.; Telling, A.; Deeg, H.J. Effect of intravenous coadministration of human stroma cell lines on engraftment of long-term repopulating clonal myelodysplastic syndrome cells in immunodeficient mice. Blood Cancer J. 2013, 3, e113. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deeg, H.J. Murine xenogeneic models of myelodysplastic syndrome: An essential role for stroma cells. Exp. Hematol. 2014, 42, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, S.; Yuan, J.; Yang, Y.; Li, J.; Wu, X.; Freund, M.; Pollok, K.; Hanenberg, H.; Goebel, W.S.; et al. Mesenchymal stem/progenitor cells promote the reconstitution of exogenous hematopoietic stem cells in Fancg-/- mice in vivo. Blood 2009, 113, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, Y.; Matsushita, H.; Yahata, T.; Yumino, S.; Tanaka, Y.; Miyachi, H.; Ogawa, Y.; Kawada, H.; Ito, M.; Ando, K. Establishment of a xenograft model of human myelodysplastic syndromes. Haematologica 2011, 96, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Kerbauy, D.M.; Lesnikov, V.; Torok-Storb, B.; Bryant, E.; Deeg, H.J. Engraftment of distinct clonal MDS-derived hematopoietic precursors in NOD/SCID-β2-microglobulin-deficient mice after intramedullary transplantation of hematopoietic and stromal cells. Blood 2004, 104, 2202–2203. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Durig, J.; Gobel, M.; Hanoun, M.; Klein-Hitpass, L.; Duhrsen, U. Functional abnormalities and changes in gene expression in fibroblasts and macrophages from the bone marrow of patients with acute myeloid leukemia. Int. J. Hematol. 2015, 102, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Acheampong, D.O.; Wang, Y.; Xie, W.; Wang, M.; Zhang, J. Tumoral NKG2D alters cell cycle of acute myeloid leukemic cells and reduces NK cell-mediated immune surveillance. Immunol Res 2016, 64, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Borrego, D.; Moreno-Lafont, M.C.; Vazquez-Sanchez, E.A.; Gutierrez-Hoya, A.; Lopez-Santiago, R.; Montiel-Cervantes, L.A.; Ramirez-Saldana, M.; Vela-Ojeda, J. Overexpression of CD158 and NKG2A Inhibitory Receptors and Underexpression of NKG2D and NKp46 Activating Receptors on NK cells in Acute Myeloid Leukemia. Arch. Med. Res. 2016, 47, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.S.; Granjeaud, S.; Gondois-Rey, F.; Harbi, S.; Orlanducci, F.; Blaise, D.; Vey, N.; Arnoulet, C.; Fauriat, C.; Olive, D. Increased NK Cell Maturation in Patients with Acute Myeloid Leukemia. Front. Immunol. 2015, 6, 564. [Google Scholar] [CrossRef] [PubMed]
- Najera Chuc, A.E.; Cervantes, L.A.; Retiguin, F.P.; Ojeda, J.V.; Maldonado, E.R. Low number of invariant NKT cells is associated with poor survival in acute myeloid leukemia. J. Cancer Res. Clin. Oncol. 2012, 138, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xu, Y. Clinical significance of Treg cell frequency in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Memarian, A.; Nourizadeh, M.; Masoumi, F.; Tabrizi, M.; Emami, A.H.; Alimoghaddam, K.; Hadjati, J.; Mirahmadian, M.; Jeddi-Tehrani, M. Upregulation of CD200 is associated with Foxp3+ regulatory T cell expansion and disease progression in acute myeloid leukemia. Tumour Biol. 2013, 34, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Yu, S.; Liu, L.; Xue, F.; Yuan, C.; Wang, M.; Ji, C.; Ma, D. The Profile of T Helper Subsets in Bone Marrow Microenvironment Is Distinct for Different Stages of Acute Myeloid Leukemia Patients and Chemotherapy Partly Ameliorates These Variations. PLoS ONE 2015, 10, e0131761. [Google Scholar] [CrossRef] [PubMed]
- Musuraca, G.; De, M.S.; Napolitano, R.; Papayannidis, C.; Guadagnuolo, V.; Fabbri, F.; Cangini, D.; Ceccolini, M.; Giannini, M.B.; Lucchesi, A.; et al. IL-17/IL-10 double-producing T cells: New link between infections, immunosuppression and acute myeloid leukemia. J. Transl. Med. 2015, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ye, A.; Bi, L.; Wu, J.; Yu, K.; Zhang, S. Th17 cells and interleukin-17 increase with poor prognosis in patients with acute myeloid leukemia. Cancer Sci. 2014, 105, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, C.; Zhang, L.; Shan, B.; Tian, T.; Hu, Y.; Shao, L.; Sun, Y.; Ji, C.; Ma, D. Elevated Th22 cells correlated with Th17 cells in peripheral blood of patients with acute myeloid leukemia. Int. J. Mol. Sci. 2014, 15, 1927–1945. [Google Scholar] [CrossRef] [PubMed]
- Derolf, A.R.; Laane, E.; Bjorklund, E.; Saft, L.; Bjorkholm, M.; Porwit, A. Dendritic cells in bone marrow at diagnosis and after chemotherapy in adult patients with acute myeloid leukaemia. Scand. J. Immunol. 2014, 80, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Zhang, Z.F.; Ju, Y.; Li, L.; Zhang, B.C.; Liu, B. Increase in myeloid-derived suppressor cells (MDSCs) associated with minimal residual disease (MRD) detection in adult acute myeloid leukemia. Int. J. Hematol. 2015, 102, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H. Disease progression in myelodysplastic syndromes: Do mesenchymal cells pave the way? Cell Stem Cell 2014, 14, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Cogle, C.R.; Saki, N.; Khodadi, E.; Li, J.; Shahjahani, M.; Azizidoost, S. Bone marrow niche in the myelodysplastic syndromes. Leuk. Res. 2015, 39, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Cogle, C.R.; Bosse, R.C.; Brewer, T.; Migdady, Y.; Shirzad, R.; Kampen, K.R.; Saki, N. Acute myeloid leukemia in the vascular niche. Cancer Lett. 2015. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.L.; Campbell, C.J.; Hopkins, C.I.; Fiebig-Comyn, A.; Russell, J.; Ulemek, J.; Foley, R.; Leber, B.; Xenocostas, A.; Collins, T.J.; et al. Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J. Exp. Med. 2014, 211, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Glait-Santar, C.; Desmond, R.; Feng, X.; Bat, T.; Chen, J.; Heuston, E.; Mizukawa, B.; Mulloy, J.C.; Bodine, D.M.; Larochelle, A.; et al. Functional niche competition between normal hematopoietic stem and progenitor cells and myeloid leukemia cells. Stem Cells 2015, 33, 3635–3642. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Scadden, D.T.; Preffer, F.I. The hematopoietic stem cell niche—Home for friend and foe? Cytom. B Clin. Cytom. 2013, 84, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Walenda, T.; Stiehl, T.; Braun, H.; Frobel, J.; Ho, A.D.; Schroeder, T.; Goecke, T.W.; Rath, B.; Germing, U.; Marciniak-Czochra, A.; et al. Feedback signals in myelodysplastic syndromes: Increased self-renewal of the malignant clone suppresses normal hematopoiesis. PLoS Comput. Biol. 2014, 10, e1003599. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, J.J.; Clay, D.; Bourin, P.; Herodin, F.; Dupuy, C.; Jasmin, C.; Le Bousse-Kerdiles, M.C. Stromal cell-derived factor 1 regulates primitive hematopoiesis by suppressing apoptosis and by promoting G(0)/G(1) transition in CD34+ cells: Evidence for an autocrine/paracrine mechanism. Blood 2002, 99, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, A.; Desterke, C.; Rodenburger, E.; Clay, D.; Guerton, B.; Boutin, L.; Bennaceur-Griscelli, A.; Pierre-Louis, O.; Uzan, G.; Abecassis, L.; et al. A crosstalk between stromal cell-derived factor-1 and transforming growth factor-β controls the quiescence/cycling switch of CD34+ progenitors through FoxO3 and mammalian target of rapamycin. Stem Cells 2008, 26, 3150–3161. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Frenette, P.S. This niche is a maze; an amazing niche. Cell Stem Cell 2013, 12, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Avecilla, S.T.; Hattori, K.; Heissig, B.; Tejada, R.; Liao, F.; Shido, K.; Jin, D.K.; Dias, S.; Zhang, F.; Hartman, T.E.; et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat. Med. 2004, 10, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, A.T.; Vellenga, E.; Schuringa, J.J. Convergence of hypoxia and TGFbeta pathways on cell cycle regulation in human hematopoietic stem/progenitor cells. PLoS ONE 2014, 9, e93494. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.; Copley, M.; Benz, C.; Dykstra, B.; Bowie, M.; Eaves, C. Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin. Cancer Res. 2008, 14, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhao, Y.; Zhang, Y.; Ruan, Z. The Macrophage Polarization Regulates MSC Osteoblast Differentiation in vitro. Ann. Clin. Lab. Sci. 2016, 46, 65–71. [Google Scholar] [PubMed]
- Anthony, B.A.; Link, D.C. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol. 2014, 35, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.H.; Oh, S.H.; Park, C.J.; Lee, B.R.; Kim, Y.J.; Cho, Y.U.; Jang, S.; Lee, J.H.; Kim, N.; Park, S.H.; et al. VLA-4 and CXCR4 expression levels show contrasting prognostic impact (favorable and unfavorable, respectively) in acute myeloid leukemia. Ann. Hematol. 2015, 94, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, F.; Cutini, I.; Gianfaldoni, G.; Bencini, S.; Scappini, B.; Pancani, F.; Ponziani, V.; Bonetti, M.I.; Biagiotti, C.; Longo, G.; et al. CXCR4 expression accounts for clinical phenotype and outcome in acute myeloid leukemia. Cytom. B Clin. Cytom. 2014, 86, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, E.J.; Pavic, B.; Lowenberg, B.; Ploemacher, R.E. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 2004, 104, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Landry, B.; Gul-Uludag, H.; Plianwong, S.; Kucharski, C.; Zak, Z.; Parmar, M.B.; Kutsch, O.; Jiang, H.; Brandwein, J.; Uludag, H. Targeting CXCR4/SDF-1 axis by lipopolymer complexes of siRNA in acute myeloid leukemia. J. Control. Release 2016, 224, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, H.; Yang, Y.; Meng, J.; Liu, J.; Wang, C.; Xu, H. A designed peptide targeting CXCR4 displays anti-acute myelocytic leukemia activity in vitro and in vivo. Sci. Rep. 2014, 4, 6610. [Google Scholar] [CrossRef] [PubMed]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009, 113, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Zaitseva, L.; Murray, M.Y.; Shafat, M.S.; Lawes, M.J.; MacEwan, D.J.; Bowles, K.M.; Rushworth, S.A. Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget 2014, 5, 9930–9938. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.M.; Calvi, L.M. Notch signaling and the bone marrow hematopoietic stem cell niche. Bone 2010, 46, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.G.; Calvi, L.M. Notch signaling in the malignant bone marrow microenvironment: Implications for a niche-based model of oncogenesis. Ann. N. Y. Acad. Sci. 2015, 1335, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Rattis, F.M.; DiMascio, L.N.; Congdon, K.L.; Pazianos, G.; Zhao, C.; Yoon, K.; Cook, J.M.; Willert, K.; Gaiano, N.; et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat. Immunol. 2005, 6, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Milner, L.A.; Deng, Y.; Iwata, M.; Banta, A.; Graf, L.; Marcovina, S.; Friedman, C.; Trask, B.J.; Hood, L.; et al. The human homolog of rat Jagged1 expressed by marrow stroma inhibits differentiation of 32D cells through interaction with Notch1. Immunity 1998, 8, 43–55. [Google Scholar] [CrossRef]
- Huang, J.C.; Basu, S.K.; Zhao, X.; Chien, S.; Fang, M.; Oehler, V.G.; Appelbaum, F.R.; Becker, P.S. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015, 5, e302. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Koo, B.K.; Jeong, H.W.; Yoon, M.J.; Song, R.; Shin, J.; Jeong, D.C.; Kim, S.H.; Kong, Y.Y. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood 2008, 112, 4628–4638. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Sutphin, R.M.; Hall, M.G.; Golfman, L.S.; Fang, W.; Nolo, R.M.; Akers, L.J.; Hammitt, R.A.; McMurray, J.S.; Kornblau, S.M.; et al. Notch activation inhibits AML growth and survival: A potential therapeutic approach. J. Exp. Med. 2013, 210, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Thanendrarajan, S.; Schmidt-Wolf, I.G. Wnt/ss-catenin: A new therapeutic approach to acute myeloid leukemia. Leuk. Res. Treat. 2011, 2011, 428960. [Google Scholar] [CrossRef] [PubMed]
- Minke, K.S.; Staib, P.; Puetter, A.; Gehrke, I.; Gandhirajan, R.K.; Schlosser, A.; Schmitt, E.K.; Hallek, M.; Kreuzer, K.A. Small molecule inhibitors of WNT signaling effectively induce apoptosis in acute myeloid leukemia cells. Eur. J. Haematol. 2009, 82, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Levesque, J.P. A niche in a dish: Pericytes support HSC. Blood 2013, 121, 2816–2818. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Mosialou, I.; Manavalan, S.J.; Rathinam, C.V.; Friedman, R.A.; Teruya-Feldstein, J.; Bhagat, G.; Berman, E.; Kousteni, S. FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia 2016, 30, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Falconi, G.; Fabiani, E.; Fianchi, L.; Criscuolo, M.; Raffaelli, C.S.; Bellesi, S.; Hohaus, S.; Voso, M.T.; D’Alo, F.; Leone, G. Impairment of PI3K/AKT and WNT/beta-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes. Exp. Hematol. 2016, 44, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Suzuki, M.; Niwa, Y.; Hiraga, J.; Nagasaka, T.; Ito, M.; Nakamura, S.; Tomita, A.; Abe, A.; Kiyoi, H.; et al. Clinical significance of nuclear non-phosphorylated beta-catenin in acute myeloid leukaemia and myelodysplastic syndrome. Br. J. Haematol. 2008, 140, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fan, R.; Wang, X.Q.; Wu, D.P.; Lin, G.W.; Xu, Y.; Li, W.Y. Methylation of Wnt antagonist genes: A useful prognostic marker for myelodysplastic syndrome. Ann. Hematol. 2013, 92, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Cao, J.; Li, H.; Liu, P.; Xu, S.; Zhou, R.; Yao, Z.; Guo, X. Bone marrow fibrosis with fibrocytic and immunoregulatory responses induced by beta-catenin activation in osteoprogenitors. Bone 2016, 84, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Sykes, S.M.; Al-Shahrour, F.; Shterental, S.; Paktinat, M.; Lo, C.C.; Jesneck, J.L.; Ebert, B.L.; Williams, D.A.; Gilliland, D.G. The APC(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood 2010, 115, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Masala, E.; Valencia, A.; Buchi, F.; Nosi, D.; Spinelli, E.; Gozzini, A.; Sassolini, F.; Sanna, A.; Zecchi, S.; Bosi, A.; et al. Hypermethylation of Wnt antagonist gene promoters and activation of Wnt pathway in myelodysplastic marrow cells. Leuk. Res. 2012, 36, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D.; Hooker, C.; McDevitt, M.A.; Karp, J.E.; Smith, B.D.; Mohammad, H.P.; Ye, Y.; Herman, J.G.; Carraway, H.E. Acute myeloid leukemia is characterized by Wnt pathway inhibitor promoter hypermethylation. Leuk. Lymphoma 2010, 51, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, A.; Rostami, S.; Chahardouli, B.; Alizad, G.N.; Ghotaslou, A.; Nadali, F. Study of SFRP1 and SFRP2 methylation status in patients with de novo acute myeloblastic leukemia. Int. J. Hematol. Oncol. Stem Cell Res. 2015, 9, 15–21. [Google Scholar] [PubMed]
- Martin, V.; Valencia, A.; Agirre, X.; Cervera, J.; San Jose-Eneriz, E.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Sanz, M.A.; Herrera, C.; Torres, A.; et al. Epigenetic regulation of the non-canonical Wnt pathway in acute myeloid leukemia. Cancer Sci. 2010, 101, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Roman-Gomez, J.; Cervera, J.; Such, E.; Barragan, E.; Bolufer, P.; Moscardo, F.; Sanz, G.F.; Sanz, M.A. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia 2009, 23, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Hu, C.; Mei, C.; Ren, Z.; Vera, J.C.; Zhuang, Z.; Jin, J.; Tong, H. Sequential combination of decitabine and idarubicin synergistically enhances anti-leukemia effect followed by demethylating Wnt pathway inhibitor promoters and downregulating Wnt pathway nuclear target. J. Transl. Med. 2014, 12, 167. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Greil, R. Digging deep into “dirty” drugs—Modulation of the methylation machinery. Drug Metab. Rev. 2015, 47, 252–279. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Chatterjee, S.; Basak, P.; Das, P.; Pereira, J.A.; Dutta, R.K.; Chaklader, M.; Chaudhuri, S.; Law, S. The bone marrow stem stromal imbalance—A key feature of disease progression in case of myelodysplastic mouse model. J. Stem Cells 2010, 5, 49–64. [Google Scholar] [PubMed]
- Mishima, S.; Nagai, A.; Abdullah, S.; Matsuda, C.; Taketani, T.; Kumakura, S.; Shibata, H.; Ishikura, H.; Kim, S.U.; Masuda, J. Effective ex vivo expansion of hematopoietic stem cells using osteoblast-differentiated mesenchymal stem cells is CXCL12 dependent. Eur. J. Haematol. 2010, 84, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.H.; Kwon, K.R. Concise review: Multiple niches for hematopoietic stem cell regulations. Stem Cells 2010, 28, 1243–1249. [Google Scholar] [PubMed]
- Koh, S.H.; Choi, H.S.; Park, E.S.; Kang, H.J.; Ahn, H.S.; Shin, H.Y. Co-culture of human CD34+ cells with mesenchymal stem cells increases the survival of CD34+ cells against the 5-aza-deoxycytidine- or trichostatin A-induced cell death. Biochem. Biophys. Res. Commun. 2005, 329, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Fonseca, A.V.; Alakel, N.; Fierro, F.A.; Muller, K.; Bornhauser, M.; Ehninger, G.; Corbeil, D.; Ordemann, R. Hematopoietic stem cells in co-culture with mesenchymal stromal cells—Modeling the niche compartments in vitro. Haematologica 2010, 95, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Arai, F.; Iwasaki, H.; Hosokawa, K.; Kobayashi, I.; Gomei, Y.; Matsumoto, Y.; Yoshihara, H.; Suda, T. Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood 2010, 116, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Semerad, C.L.; Christopher, M.J.; Liu, F.; Short, B.; Simmons, P.J.; Winkler, I.; Levesque, J.P.; Chappel, J.; Ross, F.P.; Link, D.C. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood 2005, 106, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Emerson, S.G. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J. Exp. Med. 1994, 179, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Visnjic, D.; Kalajzic, Z.; Rowe, D.W.; Katavic, V.; Lorenzo, J.; Aguila, H.L. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004, 103, 3258–3264. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T. New niches for B cells. Nat. Immunol. 2008, 9, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T. The chemokine CXCL12 and regulation of HSC and B lymphocyte development in the bone marrow niche. Adv. Exp. Med. Biol. 2007, 602, 69–75. [Google Scholar] [PubMed]
- Van Pel, M.; Fibbe, W.E.; Schepers, K. The human and murine hematopoietic stem cell niches: Are they comparable? Ann. N. Y. Acad. Sci. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Iancu-Rubin, C.; Mosoyan, G.; Wang, J.; Kraus, T.; Sung, V.; Hoffman, R. Stromal cell-mediated inhibition of erythropoiesis can be attenuated by Sotatercept (ACE-011), an activin receptor type II ligand trap. Exp. Hematol. 2013, 41, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Lazar-Karsten, P.; Dorn, I.; Meyer, G.; Lindner, U.; Driller, B.; Schlenke, P. The influence of extracellular matrix proteins and mesenchymal stem cells on erythropoietic cell maturation. Vox Sang. 2011, 101, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.B.; Sworder, B.; Bouladoux, N.; Roy, C.N.; Uchida, A.M.; Grigg, M.; Robey, P.G.; Belkaid, Y. Stromal-derived IL-6 alters the balance of myeloerythroid progenitors during Toxoplasma gondii infection. J. Leukoc. Biol. 2012, 92, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Bergua, J.M.; Campos, C.; Gayoso, I.; Arcos, M.J.; Banas, H.; Morgado, S.; Casado, J.G.; Solana, R.; Tarazona, R. Cytokine profiles in acute myeloid leukemia patients at diagnosis: Survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine 2013, 61, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Frigon, N.L., Jr.; Young, A.L.; Yu, A.L.; Mathews, L.S.; Vaughan, J.; Vale, W.; Yu, J. Effect of activin A on globin gene expression in purified human erythroid progenitors. Blood 1992, 79, 773–781. [Google Scholar] [PubMed]
- Shav-Tal, Y.; Zipori, D. The role of activin a in regulation of hemopoiesis. Stem Cells 2002, 20, 493–500. [Google Scholar] [PubMed]
- Yu, J.; Shao, L.; Vaughan, J.; Vale, W.; Yu, A.L. Characterization of the potentiation effect of activin on human erythroid colony formation in vitro. Blood 1989, 73, 952–960. [Google Scholar] [PubMed]
- Gibson, D.P.; DeGowin, R.L.; Knapp, S.A. Effect of X irradiation on release of prostaglandin E from marrow stromal cells in culture. Radiat. Res. 1982, 89, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Nocka, K.H.; Ottman, O.G.; Pelus, L.M. The role of marrow accessory cell populations in the augmentation of human erythroid progenitor cell (BFU-E) proliferation by prostaglandin E. Leuk. Res. 1989, 13, 527–534. [Google Scholar] [CrossRef]
- DeGowin, R.L.; Gibson, D.P. Prostaglandin-mediated enhancement of erythroid colonies by marrow stromal cells (MSC). Exp. Hematol. 1981, 9, 274–280. [Google Scholar] [PubMed]
- Werts, E.D.; DeGowin, R.L.; Knapp, S.K.; Gibson, D.P. Characterization of marrow stromal (fibroblastoid) cells and their association with erythropoiesis. Exp. Hematol. 1980, 8, 423–433. [Google Scholar] [PubMed]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Stier, S.; Ko, Y.; Forkert, R.; Lutz, C.; Neuhaus, T.; Grunewald, E.; Cheng, T.; Dombkowski, D.; Calvi, L.M.; Rittling, S.R.; et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med. 2005, 201, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J.; Hay, E.; Saidak, Z. Integrin and cadherin signaling in bone: Role and potential therapeutic targets. Trends Endocrinol. Metab. 2014, 25, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Huber, B.C.; Grabmaier, U.; Brunner, S. Impact of parathyroid hormone on bone marrow-derived stem cell mobilization and migration. World J. Stem Cells 2014, 6, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Fulzele, K.; Krause, D.S.; Panaroni, C.; Saini, V.; Barry, K.J.; Liu, X.; Lotinun, S.; Baron, R.; Bonewald, L.; Feng, J.Q.; et al. Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood 2013, 121, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.T.; Kim, J.H.; Kang, E.J.; Lee, S.W.; Park, M.C.; Park, Y.B.; Lee, S.K. Osteopontin might be involved in bone remodelling rather than in inflammation in ankylosing spondylitis. Rheumatology 2008, 47, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Reinholt, F.P.; Hultenby, K.; Oldberg, A.; Heinegard, D. Osteopontin—A possible anchor of osteoclasts to bone. PNAS 1990, 87, 4473–4475. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.X.; Denhardt, D.T. Osteopontin: Role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008, 19, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.A.; Thomas, D.; Barry, E.F.; Kok, C.H.; McClure, B.J.; Tsykin, A.; To, L.B.; Brown, A.; Lewis, I.D.; Herbert, K.; et al. Expression profiling of a hemopoietic cell survival transcriptome implicates osteopontin as a functional prognostic factor in AML. Blood 2009, 114, 4859–4870. [Google Scholar] [CrossRef] [PubMed]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Liersch, R.; Gerss, J.; Schliemann, C.; Bayer, M.; Schwoppe, C.; Biermann, C.; Appelmann, I.; Kessler, T.; Lowenberg, B.; Buchner, T.; et al. Osteopontin is a prognostic factor for survival of acute myeloid leukemia patients. Blood 2012, 119, 5215–5220. [Google Scholar] [CrossRef] [PubMed]
- Ludin, A.; Itkin, T.; Gur-Cohen, S.; Mildner, A.; Shezen, E.; Golan, K.; Kollet, O.; Kalinkovich, A.; Porat, Z.; D’Uva, G.; et al. Monocytes-macrophages that express alpha-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat. Immunol. 2012, 13, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Alakel, N.; Jing, D.; Muller, K.; Bornhauser, M.; Ehninger, G.; Ordemann, R. Direct contact with mesenchymal stromal cells affects migratory behavior and gene expression profile of CD133+ hematopoietic stem cells during ex vivo expansion. Exp. Hematol. 2009, 37, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Pistoia, V.; Moretta, L. Mesenchymal stem cells: A new strategy for immunosuppression? Trends Immunol. 2007, 28, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wehner, R.; Bornhauser, M.; Wassmuth, R.; Bachmann, M.; Schmitz, M. Immunomodulatory properties of mesenchymal stromal cells and their therapeutic consequences for immune-mediated disorders. Stem Cells Dev. 2010, 19, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Mattar, P.; Bieback, K. Comparing the immunomodulatory properties of bone marrow, adipose tissue, and birth-associated tissue mesenchymal stromal vells. Front. Immunol. 2015, 6, 560. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Wolf, A.M. Mesenchymal stem cells as cellular immunosuppressants. Lancet 2008, 371, 1553–1554. [Google Scholar] [CrossRef]
- Najar, M.; Raicevic, G.; Crompot, E.; Fayyad-Kazan, H.; Bron, D.; Toungouz, M.; Lagneaux, L. The immunomodulatory potential of mesenchymal stromal cells: A story of a regulatory network. J. Immunother. 2016, 39, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Raicevic, G.; Fayyad-Kazan, H.; Bron, D.; Toungouz, M.; Lagneaux, L. Mesenchymal stromal cells and immunomodulation: A gathering of regulatory immune cells. Cytotherapy 2016, 18, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.; Saldanha-Araujo, F. Mechanisms of T-cell immunosuppression by mesenchymal stromal cells: What do we know so far? BioMed Res. Int. 2014, 2014, 216806. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.X.; Zhang, Y.; Li, X.S.; Wu, Y.; Yu, X.D.; Tang, P.H.; Mao, N. Osteoblasts derived from mesenchymal stem cells harbor immunoregulatory effect. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2005, 13, 50–53. [Google Scholar] [PubMed]
- Liu, H.; Kemeny, D.M.; Heng, B.C.; Ouyang, H.W.; Melendez, A.J.; Cao, T. The immunogenicity and immunomodulatory function of osteogenic cells differentiated from mesenchymal stem cells. J. Immunol. 2006, 176, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Tammik, C.; Rosendahl, K.; Zetterberg, E.; Ringden, O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol. 2003, 31, 890–896. [Google Scholar] [CrossRef]
- radier, A.; Passweg, J.; Villard, J.; Kindler, V. Human bone marrow stromal cells and skin fibroblasts inhibit natural killer cell proliferation and cytotoxic activity. Cell Transplant. 2011, 20, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Haniffa, M.A.; Wang, X.N.; Holtick, U.; Rae, M.; Isaacs, J.D.; Dickinson, A.M.; Hilkens, C.M.; Collin, M.P. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. J. Immunol. 2007, 179, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Poloni, A.; Maurizi, G.; Ciarlantini, M.; Medici, M.; Mattiucci, D.; Mancini, S.; Maurizi, A.; Falconi, M.; Olivieri, A.; Leoni, P. Interaction between human mature adipocytes and lymphocytes induces T-cell proliferation. Cytotherapy 2015, 17, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J. Are mesenchymal stromal cells immune cells? Arthritis Res. Ther. 2015, 17, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romieu-Mourez, R.; Coutu, D.L.; Galipeau, J. The immune plasticity of mesenchymal stromal cells from mice and men: Concordances and discrepancies. Front. Biosci. 2012, 4, 824–837. [Google Scholar]
- Chinnadurai, R.; Ng, S.; Velu, V.; Galipeau, J. Challenges in animal modelling of mesenchymal stromal cell therapy for inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 4779–4787. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Su, J.; Zhang, L.; Zhao, X.; Ling, W.; L’huillie, A.; Zhang, J.; Lu, Y.; Roberts, A.I.; Ji, W.; et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells 2009, 27, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, E.; Kelchtermans, H.; Mitera, T.; Geboes, L.; Matthys, P. Discrepancy between the in vitro and in vivo effects of murine mesenchymal stem cells on T-cell proliferation and collagen-induced arthritis. Arthritis Res. Ther. 2010, 12, R31. [Google Scholar] [CrossRef] [PubMed]
- Romieu-Mourez, R.; Francois, M.; Boivin, M.N.; Stagg, J.; Galipeau, J. Regulation of MHC class II expression and antigen processing in murine and human mesenchymal stromal cells by IFN-gamma, TGF-beta, and cell density. J. Immunol. 2007, 179, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J.; Krampera, M.; Barrett, J.; Dazzi, F.; Deans, R.J.; DeBruijn, J.; Dominici, M.; Fibbe, W.E.; Gee, A.P.; Gimble, J.M.; et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 2016, 18, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.M.; Ritter, T.; Ceredig, R.; Griffin, M.D. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res. Ther. 2011, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Engela, A.U.; Baan, C.C.; Litjens, N.H.; Franquesa, M.; Betjes, M.G.; Weimar, W.; Hoogduijn, M.J. Mesenchymal stem cells control alloreactive CD8+ CD28− T cells. Clin. Exp. Immunol. 2013, 174, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M.; Glennie, S.; Dyson, J.; Scott, D.; Laylor, R.; Simpson, E.; Dazzi, F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 2003, 101, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, X.; Kuang, X.; Liao, Y.; Li, H.; Luo, D. Mesenchymal stem cells suppress CD8+ T cell-mediated activation by suppressing natural killer group 2, member D protein receptor expression and secretion of prostaglandin E2, indoleamine 2,3-dioxygenase and transforming growth factor-beta. Clin. Exp. Immunol. 2014, 178, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.; Laranjeira, P.; Mendes, S.; Velada, I.; Leite, C.; Andrade, P.; Santos, F.; Henriques, A.; Graos, M.; Cardoso, C.M.; et al. Mesenchymal stem cells from umbilical cord matrix, adipose tissue and bone marrow exhibit different capability to suppress peripheral blood B, natural killer and T cells. Stem Cell Res. Ther. 2013, 4, 125. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ozaki, K.; Oh, I.; Meguro, A.; Hatanaka, K.; Nagai, T.; Muroi, K.; Ozawa, K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 2007, 109, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, S.; Pene, J.; Moquet-Torcy, G.; Jorgensen, C.; Yssel, H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J. Immunol. 2010, 185, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Noel, D.; Fernandez, X.; Khoury, M.; Figueroa, F.; Carrion, F.; Jorgensen, C.; Djouad, F. Mesenchymal stem cells repress Th17 molecular program through the PD-1 pathway. PLoS ONE 2012, 7, e45272. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Kurte, M.; Bravo-Alegria, J.; Contreras, R.; Nova-Lamperti, E.; Tejedor, G.; Noel, D.; Jorgensen, C.; Figueroa, F.; Djouad, F.; et al. Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res. Ther. 2013, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Djouad, F.; Toupet, K.; Bony, C.; Franquesa, M.; Hoogduijn, M.J.; Jorgensen, C.; Noel, D. Mesenchymal stem cell-derived interleukin I receptor antagonist promotes macrophage polarization and inhibits B cell differentiation. Stem Cells 2015, 34, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Fleury-Cappellesso, S.; Gadelorge, M.; Dietrich, G.; Bourin, P.; Fournie, J.J.; Poupot, R. A regulatory crosstalk between Vgamma9Vdelta2 T lymphocytes and mesenchymal stem cells. Eur. J. Immunol. 2009, 39, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Petrini, I.; Pacini, S.; Petrini, M.; Fazzi, R.; Trombi, L.; Galimberti, S. Mesenchymal cells inhibit expansion but not cytotoxicity exerted by gamma-delta T cells. Eur. J. Clin. Investig. 2009, 39, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Prigione, I.; Benvenuto, F.; Bocca, P.; Battistini, L.; Uccelli, A.; Pistoia, V. Reciprocal interactions between human mesenchymal stem cells and gammadelta T cells or invariant natural killer T cells. Stem Cells 2009, 27, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qu, Y.H.; Wu, Y.F.; Liu, L.; Lin, X.H.; Huang, K.; Wei, J. Bone marrow mesenchymal stem cells suppressing activation of allogeneic cytokine-induced killer/natural killer cells either by direct or indirect interaction. Cell Biol. Int. 2015, 39, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulou, P.A.; Perez, S.A.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006, 24, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Becchetti, S.; Mingari, M.C.; Moretta, L. Mesenchymal stem cell-natural killer cell interactions: Evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 2006, 107, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, S.; Morbelli, S.; Morando, S.; Massollo, M.; Marini, C.; Bertoni, A.; Frassoni, F.; Bartolome, S.T.; Sambuceti, G.; Traggiai, E.; et al. Mesenchymal stem cells impair in vivo T-cell priming by dendritic cells. PNAS 2011, 108, 17384–17389. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, P.; Gomes, J.; Pedreiro, S.; Pedrosa, M.; Martinho, A.; Antunes, B.; Ribeiro, T.; Santos, F.; Domingues, R.; Abecasis, M.; et al. Human bone marrow-derived mesenchymal stromal cells differentially inhibit cytokine production by peripheral blood monocytes subpopulations and myeloid dendritic cells. Stem Cells Int. 2015, 2015, 819084. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Kruisselbrink, A.B.; Lurvink, E.; Willemze, R.; Fibbe, W.E. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J. Immunol. 2006, 177, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Fazekasova, H.; Lam, E.W.; Soeiro, I.; Lombardi, G.; Dazzi, F. Mesenchymal stem cells inhibit dendritic cell differentiation and function by preventing entry into the cell cycle. Transplantation 2007, 83, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Abdelrazik, H.; Becchetti, F.; Moretta, L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: Central role of MSC-derived prostaglandin E2. Blood 2009, 113, 6576–6583. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Moretta, L. Interactions between mesenchymal stem cells and dendritic cells. Adv. Biochem. Eng. Biotechnol. 2013, 130, 199–208. [Google Scholar] [PubMed]
- Zhang, B.; Liu, R.; Shi, D.; Liu, X.; Chen, Y.; Dou, X.; Zhu, X.; Lu, C.; Liang, W.; Liao, L.; et al. Mesenchymal stem cells induce mature dendritic cells into a novel Jagged-2-dependent regulatory dendritic cell population. Blood 2009, 113, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Wehner, R.; Wehrum, D.; Bornhauser, M.; Zhao, S.; Schakel, K.; Bachmann, M.P.; Platzbecker, U.; Ehninger, G.; Rieber, E.P.; Schmitz, M. Mesenchymal stem cells efficiently inhibit the proinflammatory properties of 6-sulfo LacNAc dendritic cells. Haematologica 2009, 94, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Che, N.; Li, X.; Zhou, S.; Liu, R.; Shi, D.; Lu, L.; Sun, L. Umbilical cord mesenchymal stem cells suppress B-cell proliferation and differentiation. Cell Immunol. 2012, 274, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Rosado, M.M.; Bernardo, M.E.; Scarsella, M.; Conforti, A.; Giorda, E.; Biagini, S.; Cascioli, S.; Rossi, F.; Guzzo, I.; Vivarelli, M.; et al. Inhibition of B-cell proliferation and antibody production by mesenchymal stromal cells is mediated by T cells. Stem Cells Dev. 2015, 24, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Asari, S.; Itakura, S.; Ferreri, K.; Liu, C.P.; Kuroda, Y.; Kandeel, F.; Mullen, Y. Mesenchymal stem cells suppress B-cell terminal differentiation. Exp. Hematol. 2009, 37, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate B-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Franquesa, M.; Hoogduijn, M.J.; Bestard, O.; Grinyo, J.M. Immunomodulatory effect of mesenchymal stem cells on B cells. Front. Immunol. 2012, 3, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinucci, A.; Rizzetto, L.; Pieri, L.; Nosi, D.; Romagnoli, P.; Biagioli, T.; Mazzanti, B.; Saccardi, R.; Beltrame, L.; Massacesi, L.; et al. Inhibition of immune synapse by altered dendritic cell actin distribution: A new pathway of mesenchymal stem cell immune regulation. J. Immunol. 2010, 185, 5102–5110. [Google Scholar] [CrossRef] [PubMed]
- Franquesa, M.; Mensah, F.K.; Huizinga, R.; Strini, T.; Boon, L.; Lombardo, E.; DelaRosa, O.; Laman, J.D.; Grinyo, J.M.; Weimar, W.; et al. Human adipose tissue-derived mesenchymal stem cells abrogate plasmablast formation and induce regulatory B cells independently of T helper cells. Stem Cells 2015, 33, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Bianchi, G.; Bertolotto, M.; Montecucco, F.; Busca, A.; Dallegri, F.; Ottonello, L.; Pistoia, V. Human mesenchymal stem cells inhibit neutrophil apoptosis: A model for neutrophil preservation in the bone marrow niche. Stem Cells 2008, 26, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Cahill, E.F.; Tobin, L.M.; Carty, F.; Mahon, B.P.; English, K. Jagged-1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res. Ther. 2015, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Castro-Manrreza, M.E.; Mayani, H.; Monroy-Garcia, A.; Flores-Figueroa, E.; Chavez-Rueda, K.; Legorreta-Haquet, V.; Santiago-Osorio, E.; Montesinos, J.J. Human mesenchymal stromal cells from adult and neonatal sources: A comparative in vitro analysis of their immunosuppressive properties against T cells. Stem Cells Dev. 2014, 23, 1217–1232. [Google Scholar] [CrossRef] [PubMed]
- Crop, M.J.; Baan, C.C.; Korevaar, S.S.; Ijzermans, J.N.; Weimar, W.; Hoogduijn, M.J. Human adipose tissue-derived mesenchymal stem cells induce explosive T-cell proliferation. Stem Cells Dev. 2010, 19, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Di Ianni, M.; Del Papa, B.; De Ioanni, M.; Moretti, L.; Bonifacio, E.; Cecchini, D.; Sportoletti, P.; Falzetti, F.; Tabilio, A. Mesenchymal cells recruit and regulate T regulatory cells. Exp. Hematol. 2008, 36, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Engela, A.U.; Baan, C.C.; Peeters, A.M.; Weimar, W.; Hoogduijn, M.J. Interaction between adipose tissue-derived mesenchymal stem cells and regulatory T-cells. Cell Transplant. 2013, 22, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Engela, A.U.; Hoogduijn, M.J.; Boer, K.; Litjens, N.H.; Betjes, M.G.; Weimar, W.; Baan, C.C. Human adipose-tissue derived mesenchymal stem cells induce functional de-novo regulatory T cells with methylated FOXP3 gene DNA. Clin. Exp. Immunol. 2013, 173, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Jitschin, R.; Johansson, C.C.; Okita, R.; Kiessling, R.; Le Blanc, K. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood 2011, 117, 4826–4835. [Google Scholar] [CrossRef] [PubMed]
- Prevosto, C.; Zancolli, M.; Canevali, P.; Zocchi, M.R.; Poggi, A. Generation of CD4+ or CD8+ regulatory T cells upon mesenchymal stem cell-lymphocyte interaction. Haematologica 2007, 92, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Baban, B.; Isales, C.M.; Shi, X.M. Crosstalk between bone marrow-derived mesenchymal stem cells and regulatory T cells through a glucocorticoid-induced leucine zipper/developmental endothelial locus-1-dependent mechanism. FASEB J. 2015, 29, 3954–3963. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zheng, H.; Chen, X.; Peng, Y.; Huang, W.; Li, X.; Li, G.; Xia, W.; Sun, Q.; Xiang, A.P. Human mesenchymal stromal cells enhance the immunomodulatory function of CD8+CD28− regulatory T cells. Cell Mol. Immunol. 2015, 12, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Yi, S.; Wang, G.; Cheng, J.; Zhang, Y.; Chen, W.; Tai, Y.; Chen, S.; Chen, G.; Liu, W.; et al. Umbilical cord-derived mesenchymal stem cells instruct dendritic cells to acquire tolerogenic phenotypes through the IL-6-mediated upregulation of SOCS1. Stem Cells Dev. 2014, 23, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Paczesny, S.; Lauret, E.; Poirault, S.; Bordigoni, P.; Mekhloufi, F.; Hequet, O.; Bertrand, Y.; Ou-Yang, J.P.; Stoltz, J.F.; et al. Human mesenchymal stem cells license adult CD34+ hemopoietic progenitor cells to differentiate into regulatory dendritic cells through activation of the Notch pathway. J. Immunol. 2008, 180, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.G.; Xu, W.; Sun, L.; You, Y.; Li, F.; Li, Q.B.; Zou, P. Immunomodulatory function of regulatory dendritic cells induced by mesenchymal stem cells. Immunol. Investig. 2012, 41, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Hao, H.; Tong, C.; Cheng, Y.; Liu, J.; Pang, Y.; Si, Y.; Guo, Y.; Zang, L.; Mu, Y.; et al. Human umbilical cord-derived mesenchymal stem cells elicit macrophages into an anti-inflammatory phenotype to alleviate insulin resistance in type 2 diabetic rats. Stem Cells 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Barminko, J.A.; Nativ, N.I.; Schloss, R.; Yarmush, M.L. Fractional factorial design to investigate stromal cell regulation of macrophage plasticity. Biotechnol. Bioeng. 2014, 111, 2239–2251. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.I.; Kim, M.R.; Jeong, H.Y.; Jeong, H.C.; Jeong, M.H.; Yoon, S.H.; Kim, Y.S.; Ahn, Y. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Exp. Mol. Med. 2014, 46, e70. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. 2012, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Beyth, S.; Borovsky, Z.; Mevorach, D.; Liebergall, M.; Gazit, Z.; Aslan, H.; Galun, E.; Rachmilewitz, J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood 2005, 105, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Gur-Wahnon, D.; Borovsky, Z.; Beyth, S.; Liebergall, M.; Rachmilewitz, J. Contact-dependent induction of regulatory antigen-presenting cells by human mesenchymal stem cells is mediated via STAT3 signaling. Exp. Hematol. 2007, 35, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Yen, B.L.; Yen, M.L.; Hsu, P.J.; Liu, K.J.; Wang, C.J.; Bai, C.H.; Sytwu, H.K. Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-Met and STAT3. Stem Cell Rep. 2013, 1, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.W.; Chen, H.Y.; Wang, L.T.; Wang, F.H.; Fang, L.W.; Lai, H.Y.; Chen, H.H.; Lu, J.; Hung, M.S.; Cheng, Y.; et al. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J. Immunol. 2013, 190, 5065–5077. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chan, K.H.; Lai, W.H.; Siu, C.W.; Kwan, S.C.; Tse, H.F.; Wing-Lok, H.P.; Wing-Man, H.J. Human mesenchymal stem cells upregulate CD1dhighCD5+ regulatory B cells in experimental autoimmune encephalomyelitis. Neuroimmunomodulation 2013, 20, 294–303. [Google Scholar] [CrossRef] [PubMed]
- in, Y.; Zhou, Z.; Zhang, F.; Wang, Y.; Shen, B.; Liu, Y.; Guo, Y.; Fan, Y.; Qiu, J. Induction of regulatory B-cells by mesenchymal stem cells is affected by SDF-1alpha-CXCR7. Cell. Physiol. Biochem. 2015, 37, 117–130. [Google Scholar]
- Burr, S.P.; Dazzi, F.; Garden, O.A. Mesenchymal stromal cells and regulatory T cells: The Yin and Yang of peripheral tolerance? Immunol. Cell Biol. 2013, 91, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Deregibus, M.C.; Camussi, G. The secretome of mesenchymal stromal cells: Role of extracellular vesicles in immunomodulation. Immunol. Lett. 2015, 168, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, M.J.; Shih, H.; Schafer, R.; Pittenger, M.F. Unraveling the mesenchymal stromal cells’ paracrine immunomodulatory effects. Transfus. Med. Rev. 2016, 30, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J.; Popp, F.; Verbeek, R.; Masoodi, M.; Nicolaou, A.; Baan, C.; Dahlke, M.H. The immunomodulatory properties of mesenchymal stem cells and their use for immunotherapy. Int. Immunopharmacol. 2010, 10, 1496–1500. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Luciano, R.; Pascucci, L.; Goffredo, B.M.; Giorda, E.; Scapaticci, M.; Fierabracci, A.; Muraca, M. Immunoregulatory effects of mesenchymal stem cell-derived extracellular vesicles on T lymphocytes. Cell Transplant. 2015, 24, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.E.; Maitra, B.; Szekely, E.; Koc, O.N. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive T cells. Exp. Hematol. 2005, 33, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Meisel, R.; Brockers, S.; Heseler, K.; Degistirici, O.; Bülle, H.; Woite, C.; Stuhlsatz, S.; Schwippert, W.; Jager, M.; Sorg, R.; et al. Human but not murine multipotent mesenchymal stromal cells exhibit broad-spectrum antimicrobial effector function mediated by indoleamine 2,3-dioxygenase. Leukemia 2011, 25, 648–654. [Google Scholar] [CrossRef] [PubMed]
- English, K.; Ryan, J.M.; Tobin, L.; Murphy, M.J.; Barry, F.P.; Mahon, B.P. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin. Exp. Immunol. 2009, 156, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Zoso, A.; Mazza, E.M.; Bicciato, S.; Mandruzzato, S.; Bronte, V.; Serafini, P.; Inverardi, L. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur. J. Immunol. 2014, 44, 3307–3319. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed]
- Koorella, C.; Nair, J.R.; Murray, M.E.; Carlson, L.M.; Watkins, S.K.; Lee, K.P. Novel regulation of CD80/CD86-induced phosphatidylinositol 3-kinase signaling by NOTCH1 protein in interleukin-6 and indoleamine 2,3-dioxygenase production by dendritic cells. J. Biol. Chem. 2014, 289, 7747–7762. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Egilmez, N.K. Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol. Investig. 2012, 41, 738–764. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Lee, R.H.; Bazhanov, N.; Oh, J.Y.; Prockop, D.J. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-kappaB signaling in resident macrophages. Blood 2011, 118, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Melief, S.M.; Geutskens, S.B.; Fibbe, W.E.; Roelofs, H. Multipotent stromal cells skew monocytes towards an anti-inflammatory interleukin-10-producing phenotype by production of interleukin-6. Haematologica 2013, 98, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Melief, S.M.; Schrama, E.; Brugman, M.H.; Tiemessen, M.M.; Hoogduijn, M.J.; Fibbe, W.E.; Roelofs, H. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells 2013, 31, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Glennie, S.; Soeiro, I.; Dyson, P.J.; Lam, E.W.; Dazzi, F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005, 105, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Wobus, M.; Benath, G.; Ferrer, R.A.; Wehner, R.; Schmitz, M.; Hofbauer, L.C.; Rauner, M.; Ehninger, G.; Bornhäuser, M.; Platzbecker, U. Impact of lenalidomide on the functional properties of human mesenchymal stromal cells. Exp. Hematol. 2012, 40, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Tatara, R.; Ozaki, K.; Kikuchi, Y.; Hatanaka, K.; Oh, I.; Meguro, A.; Matsu, H.; Sato, K.; Ozawa, K. Mesenchymal stromal cells inhibit Th17 but not regulatory T-cell differentiation. Cytotherapy 2011, 13, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.X.; Zhang, Y.; Liu, B.; Zhang, S.X.; Wu, Y.; Yu, X.D.; Mao, N. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood 2005, 105, 4120–4126. [Google Scholar] [CrossRef] [PubMed]
- Zhi-Gang, Z.; Wei-Ming, L.; Zhi-Chao, C.; Yong, Y.; Ping, Z. Immunosuppressive properties of mesenchymal stem cells derived from bone marrow of patient with hematological malignant diseases. Leuk. Lymphoma 2008, 49, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Djouad, F.; Charbonnier, L.M.; Bouffi, C.; Louis-Plence, P.; Bony, C.; Apparailly, F.; Cantos, C.; Jorgensen, C.; Noel, D. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells 2007, 25, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ge, W.; Li, C.; You, S.; Liao, L.; Han, Q.; Deng, W.; Zhao, R.C. Effects of mesenchymal stem cells on differentiation, maturation, and function of human monocyte-derived dendritic cells. Stem Cells Dev. 2004, 13, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chen, X.; Liu, Q.; Xu, D.; Zheng, H.; Liu, L.; Liu, Q.; Liu, M.; Fan, Z.; Sun, J.; et al. Alteration of naive and memory B-cell subset in chronic graft-versus-host disease patients after treatment with mesenchymal stromal cells. Stem Cells Transl. Med. 2014, 3, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Romieu-Mourez, R.; Francois, M.; Boivin, M.N.; Bouchentouf, M.; Spaner, D.E.; Galipeau, J. Cytokine modulation of TLR expression and activation in mesenchymal stromal cells leads to a proinflammatory phenotype. J. Immunol. 2009, 182, 7963–7973. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Cao, W.; Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, M.K.; Keane-Moore, M.; Buyaner, D.; Hardy, W.B.; Moorman, M.A.; McIntosh, K.R.; Mosca, J.D. Characterization and functionality of cell surface molecules on human mesenchymal stem cells. J. Biomed. Sci. 2003, 10, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Quaedackers, M.E.; Baan, C.C.; Weimar, W.; Hoogduijn, M.J. Cell contact interaction between adipose-derived stromal cells and allo-activated T lymphocytes. Eur. J. Immunol. 2009, 39, 3436–3446. [Google Scholar] [CrossRef] [PubMed]
- Regateiro, F.S.; Cobbold, S.P.; Waldmann, H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin. Exp. Immunol. 2013, 171, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Augello, A.; Tasso, R.; Negrini, S.M.; Amateis, A.; Indiveri, F.; Cancedda, R.; Pennesi, G. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur. J. Immunol. 2005, 35, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Xue, Q.; Chen, Y.J.; Yu, G.H.; Qing, M.D.; Shen, Y.; Wang, M.Y.; Shi, Q.; Zhang, X.G. Different roles of PD-L1 and FasL in immunomodulation mediated by human placenta-derived mesenchymal stem cells. Hum. Immunol. 2013, 74, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sung, B.; Gupta, S.C. Inflammation and Cancer, 1st ed.; Aggarwal, B.B., Sung, B., Gupta, S.C., Eds.; Springer-Verlag: Berlin, Germany, 2014; pp. 1–490. [Google Scholar]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Spaeth, E.; Watson, K.; Burks, J.; Lu, H.; Klopp, A.; Andreeff, M.; Marini, F.C. Origins of the tumor microenvironment: Quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE 2012, 7, e30563. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sun, R.; Origuchi, M.; Kanehira, M.; Takahata, T.; Itoh, J.; Umezawa, A.; Kijima, H.; Fukuda, S.; Saijo, Y. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol. Med. 2011, 17, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, M.; Ramasamy, R.; Tan, B.C.; Seow, H.F. Human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSC) inhibit the proliferation of K562 (human erythromyeloblastoid leukaemic cell line). Cell Biol. Int. 2012, 36, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Sarmadi, V.H.; Tong, C.K.; Vidyadaran, S.; Abdullah, M.; Seow, H.F.; Ramasamy, R. Mesenchymal stem cells inhibit proliferation of lymphoid origin haematopoietic tumour cells by inducing cell cycle arrest. Med. J. Malaysia 2010, 65, 209–214. [Google Scholar] [PubMed]
- Ramasamy, R.; Lam, E.W.; Soeiro, I.; Tisato, V.; Bonnet, D.; Dazzi, F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: Impact on in vivo tumor growth. Leukemia 2007, 21, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Johann, P.D.; Muller, I. Multipotent Mesenchymal Stromal Cells: Possible Culprits in Solid Tumors? Stem Cells Int. 2015, 2015, 914632. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFalpha. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Razmkhah, M.; Jaberipour, M.; Erfani, N.; Habibagahi, M.; Talei, A.R.; Ghaderi, A. Adipose derived stem cells (ASCs) isolated from breast cancer tissue express IL-4, IL-10 and TGF-beta1 and upregulate expression of regulatory molecules on T cells: Do they protect breast cancer cells from the immune response? Cell Immunol. 2011, 266, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.G.; Li, W.M.; Chen, Z.C.; You, Y.; Zou, P. Immunosuppressive properties of mesenchymal stem cells derived from bone marrow of patients with chronic myeloid leukemia. Immunol. Investig. 2008, 37, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, J.J.; Mora-Garcia, M.L.; Mayani, H.; Flores-Figueroa, E.; Garcia-Rocha, R.; Fajardo-Orduna, G.R.; Castro-Manrreza, M.E.; Weiss-Steider, B.; Monroy-Garcia, A. In vitro evidence of the presence of mesenchymal stromal cells in cervical cancer and their role in protecting cancer cells from cytotoxic T cell activity. Stem Cells Dev. 2013, 22, 2508–2519. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.; Zhang, J.; Yuan, Z.; Ren, G.; Zhang, L.; Chen, X.; Rabson, A.B.; Roberts, A.I.; Wang, Y.; Shi, Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014, 74, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Maby-El, H.H.; Me-Thomas, P.; Pangault, C.; Tribut, O.; DeVos, J.; Jean, R.; Bescher, N.; Monvoisin, C.; Dulong, J.; Lamy, T.; et al. Functional alteration of the lymphoma stromal cell niche by the cytokine context: Role of indoleamine-2,3 dioxygenase. Cancer Res. 2009, 69, 3228–3237. [Google Scholar] [CrossRef] [PubMed]
- Johann, P.D.; Vaegler, M.; Gieseke, F.; Mang, P.; rmeanu-Ebinger, S.; Kluba, T.; Handgretinger, R.; Muller, I. Tumour stromal cells derived from paediatric malignancies display MSC-like properties and impair NK cell cytotoxicity. BMC Cancer 2010, 10, 501. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. MSCs, macrophages, and cancer: A dangerous menage-a-trois. Cell Stem Cell 2012, 11, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.T.; Gadupudi, G.S. Possible roles of excess tryptophan metabolites in cancer. Environ. Mol. Mutagen. 2011, 52, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.T.; Murdock, C.A.; Stevens, S.E., Jr.; Li, Y.S.; Wei, C.I.; Huang, T.S.; Chou, M.W. Mutagenicity and toxicity studies of p-phenylenediamine and its derivatives. Toxicol. Lett. 1995, 81, 23–32. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, X.; Xu, W.; Cao, Z.; Sun, L.; Li, W.; Li, Q.; Zou, P.; Zhao, Z. The different immunoregulatory functions on dendritic cells between mesenchymal stem cells derived from bone marrow of patients with low-risk or high-risk myelodysplastic syndromes. PLoS ONE 2013, 8, e57470. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.G.; Xu, W.; Yu, H.P.; Fang, B.L.; Wu, S.H.; Li, F.; Li, W.M.; Li, Q.B.; Chen, Z.C.; Zou, P. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett. 2012, 317, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Sun, Z.; Liu, L.; Chen, B.; Cao, Y.; Li, K.; Zhao, R.C. Impairment in immuno-modulatory function of Flk1(+)CD31(-)CD34(-) MSCs from MDS-RA patients. Leuk. Res. 2007, 31, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Aluigi, M.; Pandolfi, S.; Ferri, E.; Isidori, A.; Salvestrini, V.; Durelli, I.; Horenstein, A.L.; Fiore, F.; Massaia, M.; et al. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia 2007, 21, 353–355. [Google Scholar] [CrossRef] [PubMed]
- El Kholy, N.M.; Sallam, M.M.; Ahmed, M.B.; Sallam, R.M.; Asfour, I.A.; Hammouda, J.A.; Habib, H.Z.; bu-Zahra, F. Expression of indoleamine 2,3-dioxygenase in acute myeloid leukemia and the effect of its inhibition on cultured leukemia blast cells. Med. Oncol. 2011, 28, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of. Blood 2007, 109, 2871–2877. [Google Scholar] [PubMed]
- Curti, A.; Trabanelli, S.; Onofri, C.; Aluigi, M.; Salvestrini, V.; Ocadlikova, D.; Evangelisti, C.; Rutella, S.; De Cristofaro, R.; Ottaviani, E.; et al. Indoleamine 2,3-dioxygenase-expressing leukemic dendritic cells impair a leukemia-specific immune response by inducing potent T regulatory cells. Haematologica 2010, 95, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Corm, S.; Berthon, C.; Imbenotte, M.; Biggio, V.; Lhermitte, M.; Dupont, C.; Briche, I.; Quesnel, B. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients’ sera by HPLC and is inducible by IFN-gamma. Leuk. Res. 2009, 33, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Chamuleau, M.E.; van de Loosdrecht, A.A.; Hess, C.J.; Janssen, J.J.; Zevenbergen, A.; Delwel, R.; Valk, P.J.; Löwenberg, B.; Ossenkoppele, G.J. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica 2008, 93, 1894–1898. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Fontenay, M.; Corm, S.; Briche, I.; Allorge, D.; Hennart, B.; Lhermitte, M.; Quesnel, B. Metabolites of tryptophan catabolism are elevated in sera of patients with myelodysplastic syndromes and inhibit hematopoietic progenitor amplification. Leuk. Res. 2013, 37, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Su, J.; Roberts, A.I.; Shou, P.; Rabson, A.B.; Ren, G. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 2012, 33, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Szabo, E.; Fajka-Boja, R.; Kriston-Pal, E.; Hornung, A.; Makra, I.; Kudlik, G.; Uher, F.; Katona, R.L.; Monostori, E.; Czibula, A. Licensing by inflammatory cytokines abolishes heterogeneity of immunosuppressive function of mesenchymal stem cell population. Stem Cells Dev. 2015, 24, 2171–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Zhang, L.; Ren, G.; Yuan, Z.; Zhang, Y.; Zhao, R.C.; Shi, Y. Immunosuppressive properties of cloned bone marrow mesenchymal stem cells. Cell Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Crop, M.J.; Baan, C.C.; Korevaar, S.S.; Ijzermans, J.N.; Pescatori, M.; Stubbs, A.P.; van Ijcken, W.F.; Dahlke, M.H.; Eggenhofer, E.; Weimar, W.; et al. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin. Exp. Immunol. 2010, 162, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Wang, S.K.; Tian, H.; Xin, L.; Zou, Z.G.; Hu, Y.L.; Chang, C.M.; Wang, X.Y.; Yin, Q.S.; Zhang, X.H.; et al. TNF-alpha increases bone marrow mesenchymal stem cell migration to ischemic tissues. Cell Biochem. Biophys. 2012, 62, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M. Mesenchymal stromal cell ‘licensing’: A multistep process. Leukemia 2011, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef] [PubMed]
- Liotta, F.; Angeli, R.; Cosmi, L.; Fili, L.; Manuelli, C.; Frosali, F.; Mazzinghi, B.; Maggi, L.; Pasini, A.; Lisi, V.; et al. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells 2008, 26, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wu, M.; Yuan, Y.; Wang, Z.Z.; Jiang, H.; Chen, T. Priming of Toll-like receptor 4 pathway in mesenchymal stem cells increases expression of B cell activating factor. Biochem. Biophys. Res. Commun. 2014, 448, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cheng, Q.; Li, L.; Wang, J.; Xia, L.; Xu, X.; Sun, Z. Toll-like receptors 2 and 4 mediate the capacity of mesenchymal stromal cells to support the proliferation and differentiation of CD34+ cells. Exp. Cell Res. 2012, 318, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Vural, A.; Kehrl, J.H. Autophagy in macrophages: Impacting inflammation and bacterial infection. Scientifica 2014, 2014, 825463. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, J.; Liu, Y.; Qin, Y.; Luo, Q.; Wang, Q.; Duan, H. TLR4 plays a crucial role in MSC-induced inhibition of NK cell function. Biochem. Biophys. Res. Commun. 2015, 464, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Henkle, S.L.; Betancourt, A.M. Mesenchymal stem cell 1 (MSC1)-based therapy attenuates tumor growth whereas MSC2-treatment promotes tumor growth and metastasis. PLoS ONE 2012, 7, e45590. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, M.T.; Patel, K.; Denecke, B.; Giebel, B.; Widera, D. Paracrine effects of TLR4-polarised mesenchymal stromal cells are mediated by extracellular vesicles. J. Transl. Med. 2016, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Pommey, S.; Eliopoulos, N.; Galipeau, J. Interferon-gamma-stimulated marrow stromal cells: A new type of nonhematopoietic antigen-presenting cell. Blood 2006, 107, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.L.; Tang, K.C.; Patel, A.P.; Bonilla, L.M.; Pierobon, N.; Ponzio, N.M.; Rameshwar, P. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood 2006, 107, 4817–4824. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Romieu-Mourez, R.; Stock-Martineau, S.; Boivin, M.N.; Bramson, J.L.; Galipeau, J. Mesenchymal stromal cells cross-present soluble exogenous antigens as part of their antigen-presenting cell properties. Blood 2009, 114, 2632–2638. [Google Scholar] [CrossRef] [PubMed]
- Tomchuck, S.L.; Zwezdaryk, K.J.; Coffelt, S.B.; Waterman, R.S.; Danka, E.S.; Scandurro, A.B. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells 2008, 26, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Lutz, C.; Lanz, T.V.; Tritschler, I.; Koppel, A.; Tolosa, E.; Hoberg, M.; Anderl, J.; Aicher, W.K.; et al. Toll-like receptor engagement enhances the immunosuppressive properties of human bone marrow-derived mesenchymal stem cells by inducing indoleamine-2,3-dioxygenase-1 via interferon-β and protein kinase R. Stem Cells 2009, 27, 909–919. [Google Scholar] [CrossRef] [PubMed]
- DelaRosa, O.; Lombardo, E. Modulation of adult mesenchymal stem cells activity by toll-like receptors: Implications on therapeutic potential. Mediat. Inflamm. 2010, 2010, 865601. [Google Scholar] [CrossRef] [PubMed]
- DelaRosa, O.; Dalemans, W.; Lombardo, E. Toll-like receptors as modulators of mesenchymal stem cells. Front. Immunol. 2012, 3, 182. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Auletta, J.J.; Deans, R.J.; Bartholomew, A.M. Emerging roles for multipotent, bone marrow-derived stromal cells in host defense. Blood 2012, 119, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Ganan-Gomez, I.; Wei, Y.; Yang, H.; Pierce, S.; Bueso-Ramos, C.; Calin, G.; Boyano-Adanez, M.C.; Garcia-Manero, G. Overexpression of miR-125a in myelodysplastic syndrome CD34+ cells modulates NF-kappaB activation and enhances erythroid differentiation arrest. PLoS ONE 2014, 9, e93404. [Google Scholar]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Megias, J.; Yanez, A.; Moriano, S.; O’Connor, J.E.; Gozalbo, D.; Gil, M.L. Direct Toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells 2012, 30, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Cannova, J.; Breslin, S.J.P.; Zhang, J. Toll-like receptor signaling in hematopoietic homeostasis and the pathogenesis of hematologic diseases. Front. Med. 2015, 9, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, D.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Shu, J.; Hu, Q.; Zhou, S.H.; Qian, Y.M.; Hu, M.H.; Hu, L.Y.; Wang, Y.G.; Zhou, Y.M.; Lu, J.H. Apoptosis in human myelodysplastic syndrome CD34+ cells is modulated by the upregulation of TLRs and histone H4 acetylation via a beta-arrestin 1 dependent mechanism. Exp. Cell Res. 2016, 340, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Li, L.; Giannopoulos, K.; Greiner, J.; Reinhardt, P.; Wiesneth, M.; Schmitt, M. Quantitative expression of Toll-like receptor-2, -4, and -9 in dendritic cells generated from blasts of patients with acute myeloid leukemia. Transfusion 2008, 48, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Dimicoli, S.; Wei, Y.; Bueso-Ramos, C.; Yang, H.; Dinardo, C.; Jia, Y.; Zheng, H.; Fang, Z.; Nguyen, M.; Pierce, S.; et al. Overexpression of the toll-like receptor (TLR) signaling adaptor MYD88, but lack of genetic mutation, in myelodysplastic syndromes. PLoS ONE 2013, 8, e71120. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Driss, V.; Liu, J.; Kuranda, K.; Leleu, X.; Jouy, N.; Hetuin, D.; Quesnel, B. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol. Immunother. 2010, 59, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Dorfel, D.; Lichtenegger, F.S.; Geiger, C.; Lindner, L.; Merk, M.; Schendel, D.J.; Subklewe, M. Effects of TLR agonists on maturation and function of 3-day dendritic cells from AML patients in complete remission. J. Transl. Med. 2011, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.L.; Cools, N.; Lion, E.; Van, C.K.; Ponsaerts, P.; Berneman, Z.N.; Van, T.V. The Toll-like receptor 7/8 agonist resiquimod greatly increases the immunostimulatory capacity of human acute myeloid leukemia cells. Cancer Immunol. Immunother. 2010, 59, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Nourizadeh, M.; Masoumi, F.; Memarian, A.; Alimoghaddam, K.; Moazzeni, S.M.; Hadjati, J. Synergistic effect of Toll-like receptor 4 and 7/8 agonists is necessary to generate potent blast-derived dendritic cells in Acute Myeloid Leukemia. Leuk. Res. 2012, 36, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Nourizadeh, M.; Masoumi, F.; Memarian, A.; Alimoghaddam, K.; Moazzeni, S.M.; Yaghmaie, M.; Hadjati, J. In vitro induction of potent tumor-specific cytotoxic T lymphocytes using TLR agonist-activated AML-DC. Target Oncol. 2014, 9, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Li, H.; Messer, K.; Lane, T.A.; Zhou, J.; Ball, E.D. Augmentation of autologous T cell reactivity with acute myeloid leukemia (AML) blasts by Toll-like receptor (TLR) agonists. Cancer Immunol. Immunother. 2015, 64, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.J.; Cooley, S.; DeFor, T.; Weisdorf, D.J.; Panoskaltsis-Mortari, A.; Chen, W.; Blazar, B.R.; Miller, J.S. Prolonged subcutaneous administration of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced hematologic malignancies. Am. J. Hematol. 2012, 87, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Ignatz-Hoover, J.J.; Wang, H.; Moreton, S.A.; Chakrabarti, A.; Agarwal, M.K.; Sun, K.; Gupta, K.; Wald, D.N. The role of TLR8 signaling in acute myeloid leukemia differentiation. Leukemia 2015, 29, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Nuschke, A.; Rodrigues, M.; Stolz, D.B.; Chu, C.T.; Griffith, L.; Wells, A. Human mesenchymal stem cells/multipotent stromal cells consume accumulated autophagosomes early in differentiation. Stem Cell Res. Ther. 2014, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Oczypok, E.A.; Oury, T.D.; Chu, C.T. It’s a cell-eat-cell world: Autophagy and phagocytosis. Am. J. Pathol. 2013, 182, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Tso, G.H.; Law, H.K.; Tu, W.; Chan, G.C.; Lau, Y.L. Phagocytosis of apoptotic cells modulates mesenchymal stem cells osteogenic differentiation to enhance IL-17 and RANKL expression on CD4+ T cells. Stem Cells 2010, 28, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.S.; Mortensen, M.; Simon, A.K. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle 2011, 10, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.; Romer, W. Considering autophagy, beta-Catenin and E-Cadherin as innovative therapy aspects in AML. Cell Death Dis. 2015, 6, e1950. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Johnstone, R.; Ekert, P.G. Autophagy and AML-food for thought. Cell Death Differ. 2016, 23, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Cui, N.; Wang, H.; Fu, R.; Qu, W.; Ruan, E.; Wang, X.; Wang, G.; Wu, Y.; Liu, H.; et al. Autophagy level of bone marrow mononuclear cells in patients with myelodysplastic syndromes. Zhonghua Xue Ye Xue Za Zhi 2015, 36, 1016–1019. [Google Scholar] [PubMed]
- Houwerzijl, E.J.; Pol, H.W.; Blom, N.R.; van der Want, J.J.; de Wolf, J.T.; Vellenga, E. Erythroid precursors from patients with low-risk myelodysplasia demonstrate ultrastructural features of enhanced autophagy of mitochondria. Leukemia 2009, 23, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Karlic, H.; Herrmann, H.; Varga, F.; Thaler, R.; Reitermaier, R.; Spitzer, S.; Ghanim, V.; Blatt, K.; Sperr, W.R.; Valent, P.; et al. The role of epigenetics in the regulation of apoptosis in myelodysplastic syndromes and acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2014, 90, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.Y.; Zhang, R.; Wang, Y.Y.; Cen, J.N.; Zhou, J.; Yang, Y.; Jiang, F.; Chen, Z.X. Expression of autophagy related gene Beclin1 in myelodysplastic syndrome patients and its significance. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2013, 21, 936–939. [Google Scholar] [PubMed]
- Qian, W.; Liu, J.; Jin, J.; Ni, W.; Xu, W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk. Res. 2007, 31, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Fabre, C.; Carvalho, G.; Tasdemir, E.; Braun, T.; Ades, L.; Grosjean, J.; Boehrer, S.; Metivier, D.; Souquere, S.; Pierron, G.; et al. NF-κB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene 2007, 26, 4071–4083. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood 2012, 120, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.M.; et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Orfali, N.; O’Donovan, T.R.; Nyhan, M.J.; Britschgi, A.; Tschan, M.P.; Cahill, M.R.; Mongan, N.P.; Gudas, L.J.; McKenna, S.L. Induction of autophagy is a key component of all-trans-retinoic acid-induced differentiation in leukemia cells and a potential target for pharmacologic modulation. Exp. Hematol. 2015, 43, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhong, L.; Liu, L.; Fang, Y.; Qi, X.; Cao, J.; Yang, B.; He, Q.; Ying, M. Autophagy contributes to dasatinib-induced myeloid differentiation of human acute myeloid leukemia cells. Biochem. Pharmacol. 2014, 89, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Kastrinaki, M.C.; Pontikoglou, C.; Klaus, M.; Stavroulaki, E.; Pavlaki, K.; Papadaki, H.A. Biologic characteristics of bone marrow mesenchymal stem cells in myelodysplastic syndromes. Curr. Stem Cell Res. Ther. 2011, 6, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Soenen-Cornu, V.; Tourino, C.; Bonnet, M.L.; Guillier, M.; Flamant, S.; Kotb, R.; Bernheim, A.; Bourhis, J.H.; Preudhomme, C.; Fenaux, P.; et al. Mesenchymal cells generated from patients with myelodysplastic syndromes are devoid of chromosomal clonal markers and support short- and long-term hematopoiesis in vitro. Oncogene 2005, 24, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Klaus, M.; Stavroulaki, E.; Kastrinaki, M.C.; Fragioudaki, P.; Giannikou, K.; Psyllaki, M.; Pontikoglou, C.; Tsoukatou, D.; Mamalaki, C.; Papadaki, H.A. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem Cells Dev. 2010, 19, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, H.; Dong, J.; Li, H.; Zhang, H. Defective proliferative potential of MSCs from pediatric myelodysplastic syndrome patients is associated with cell senescence. Int. J. Clin. Exp. Pathol. 2015, 8, 13059–13066. [Google Scholar] [PubMed]
- Zhao, Z.G.; Liang, Y.; Li, K.; Li, W.M.; Li, Q.B.; Chen, Z.C.; Zou, P. Phenotypic and functional comparison of mesenchymal stem cells derived from the bone marrow of normal adults and patients with hematologic malignant diseases. Stem Cells Dev. 2007, 16, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Flores-Figueroa, E.; Arana-Trejo, R.M.; Gutierrez-Espindola, G.; Perez-Cabrera, A.; Mayani, H. Mesenchymal stem cells in myelodysplastic syndromes: Phenotypic and cytogenetic characterization. Leuk. Res. 2005, 29, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Blau, O.; Hofmann, W.K.; Baldus, C.D.; Thiel, G.; Serbent, V.; Schumann, E.; Thiel, E.; Blau, I.W. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp. Hematol. 2007, 35, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Blau, O.; Baldus, C.D.; Hofmann, W.K.; Thiel, G.; Nolte, F.; Burmeister, T.; Turkmen, S.; Benlasfer, O.; Schumann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Flores-Figueroa, E.; Mayani, H. Chromosomal abnormalities in marrow stromal cells from myelodysplastic syndromes (MDS). Blood 2006, 108, 3948–3949. [Google Scholar] [CrossRef] [PubMed]
- Pimenova, M.A.; Parovichnikova, E.N.; Kokhno, A.V.; Domracheva, E.V.; Manakova, T.E.; Mal’tseva, I.S.; Konnova, M.L.; Shishigina, L.A.; Savchenko, V.G. Cytogenetic characteristics of hematopoietic and stromal progenitor cells in myelodysplastic syndrome. Ter. Arkh. 2013, 85, 34–42. [Google Scholar] [PubMed]
- Song, L.X.; Guo, J.; He, Q.; Yang, L.P.; Gu, S.C.; Zhang, Z.; Zhang, X.; Wu, L.Y.; Li, X.; Chang, C.K. Study on phenotypic and cytogenetic characteristics of bone marrow mesenchymal stem cells in myelodysplastic syndromes. Zhonghua Xue Ye Xue Za Zhi 2013, 34, 127–132. [Google Scholar] [PubMed]
- Zhang, Y.Z.; Zhao, D.D.; Han, X.P.; Jin, H.J.; Da, W.M.; Yu, L. In vitro study of biological characteristics of mesenchymal stem cells in patients with low-risk myelodysplastic syndrome. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2008, 16, 813–818. [Google Scholar] [PubMed]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Nguyen, A.N.; Sohal, D.; Ying, M.J.; Pahanish, P.; Gundabolu, K.; Hayman, J.; Chubak, A.; Mo, Y.; Bhagat, T.D.; et al. Inhibition of the TGF-β receptor I kinase promotes hematopoiesis in MDS. Blood 2008, 112, 3434–3443. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Brenner, A.K.; Hagen, K.M.; Liseth, K.; Skrede, S.; Hatfield, K.J.; Bruserud, O. The cytokine-mediated crosstalk between primary human acute myeloid cells and mesenchymal stem cells alters the local cytokine network and the global gene expression profile of the mesenchymal cells. Stem Cell Res. 2015, 15, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Muller, L.; Boyiadzis, M.; Whiteside, T.L. Isolation and characterization of CD34+ blast-derived exosomes in acute myeloid leukemia. PLoS ONE 2014, 9, e103310. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T., Jr.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Goloviznina, N.A.; Kamimae-Lanning, A.N.; David, L.L.; Wilmarth, P.A.; Mori, T.; Chevillet, J.R.; Narla, A.; Roberts, C.T., Jr.; et al. Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia 2015, 29, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Yang, J.; Everett, A.D.; Clevenger, C.V.; Koneru, M.; Mishra, P.J.; Kamen, B.; Banerjee, D.; Glod, J. The isolation of novel mesenchymal stromal cell chemotactic factors from the conditioned medium of tumor cells. Exp. Cell Res. 2008, 314, 3107–3117. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Oyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Bair, E.; Frohling, S.; Schlenk, R.F.; Tibshirani, R.; Dohner, H.; Pollack, J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bhatia, R. Influence of bone marrow microenvironment on leukemic stem cells: Breaking up an intimate relationship. Adv. Cancer Res. 2015, 127, 227–252. [Google Scholar] [PubMed]
- Corselli, M.; Chin, C.J.; Parekh, C.; Sahaghian, A.; Wang, W.; Ge, S.; Evseenko, D.; Wang, X.; Montelatici, E.; Lazzari, L.; et al. Perivascular support of human hematopoietic stem/progenitor cells. Blood 2013, 121, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, X. CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 2013, 330, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Carlesso, N.; Cardoso, A.A. Stem cell regulatory niches and their role in normal and malignant hematopoiesis. Curr. Opin. Hematol. 2010, 17, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M. Advances in understanding the leukaemia microenvironment. Br. J. Haematol. 2014, 164, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Barrett, A.J.; Dutra, A.; Pak, E.; Miner, S.; Keyvanfar, K.; Hensel, N.F.; Rezvani, K.; Muranski, P.; Liu, P.; et al. Long term maintenance of myeloid leukemic stem cells cultured with unrelated human mesenchymal stromal cells. Stem Cell Res. 2015, 14, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Vasold, J.; Wagner, M.; Drolle, H.; Deniffel, C.; Kutt, A.; Oostendorp, R.; Sironi, S.; Rieger, C.; Fiegl, M. The bone marrow microenvironment is a critical player in the NK cell response against acute myeloid leukaemia in vitro. Leuk. Res. 2015, 39, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, T.D.; Bartenstein, M.; Yu, Y.; Marcondes, A.M.Q.; Will, B.; Giricz, O.; Sohal, D.P.; Mantzaris, I.; Wickrema, A.; Mcmahon, C.; et al. Myelodysplastic syndrome marrow stroma shows widespread aberrant hypermethylation that is abrogated by treatment with dnmt inhibitors. Blood 2014, 124, 4379. [Google Scholar]
- Huberle, C.; Wenk, C.; Witham, D.; Garz, A.K.; Pagel, C.; Müller-Thomas, C.; Kaur-Bollinger, P.; Oostendorp, R.; Peschel, C.; Goetze, K.S. Azacitidine directly modulates function of mesenchymal stromal cells to alter bone marrow niche composition and suppress malignant hematopoeitic progenitors in MDS. Leuk. Res. 2015. [Google Scholar] [CrossRef]
- Poloni, A.; Maurizi, G.; Mattiucci, D.; Costantini, B.; Mariani, M.; Mancini, S.; Fanelli, M.; Olivieri, A.; Leoni, P. Azacitidine treatment in high risk myelodysplastic patients in complete haematological remission reverts mesenchymal stem cells to a normal phenotype. Blood 2014, 124, 1904. [Google Scholar]
- Colmone, A.; Sipkins, D.A. Beyond angiogenesis: The role of endothelium in the bone marrow vascular niche. Transl. Res. 2008, 151, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Awaya, N.; Rupert, K.; Bryant, E.; Torok-Storb, B. Failure of adult marrow-derived stem cells to generate marrow stroma after successful hematopoietic stem cell transplantation. Exp. Hematol. 2002, 30, 937–942. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Bourgeois, E.; Lobe, I.; Braun, T.; Visentin, G.; Bourhis, J.H.; Fenaux, P.; Chouaib, S.; Caignard, A. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.J.; Broudy, V.C.; Magenis, R.E.; Olson, S.; Tomar, D.; Barton, S.; Fitchen, J.H.; Bagby, G.C., Jr. Cytogenetic evidence for involvement of B lymphocytes in acquired idiopathic sideroblastic anemias. Blood 1987, 70, 1003–1005. [Google Scholar] [PubMed]
- Ma, L.; Delforge, M.; van Duppen, V.; Verhoef, G.; Emanuel, B.; Boogaerts, M.; Hagemeijer, A.; Vandenberghe, P. Circulating myeloid and lymphoid precursor dendritic cells are clonally involved in myelodysplastic syndromes. Leukemia 2004, 18, 1451–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteo Rigolin, G.; Howard, J.; Buggins, A.; Sneddon, C.; Castoldi, G.; Hirst, W.J.; Mufti, G.J. Phenotypic and functional characteristics of monocyte-derived dendritic cells from patients with myelodysplastic syndromes. Br. J. Haematol. 1999, 107, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Meers, S.; Vandenberghe, P.; Boogaerts, M.; Verhoef, G.; Delforge, M. The clinical significance of activated lymphocytes in patients with myelodysplastic syndromes: A single centre study of 131 patients. Leuk. Res. 2008, 32, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Miura, I.; Kobayashi, Y.; Takahashi, N.; Saitoh, K.; Miura, A.B. Involvement of natural killer cells in patients with myelodysplastic syndrome carrying monosomy 7 revealed by the application of fluorescence in situ hybridization to cells collected by means of fluorescence-activated cell sorting. Br. J. Haematol. 2000, 110, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Koike, K.; Agematsu, K.; Itoh, S.; Hagimoto, R.; Kitazawa, Y.; Higuchi, T.; Sawai, N.; Matsui, H.; Komiyama, A. Cytogenetic clonality analysis in monosomy 7 associated with juvenile myelomonocytic leukemia: Clonality in B and NK cells, but not in T cells. Leuk. Res. 1998, 22, 887–892. [Google Scholar] [CrossRef]
- Nilsson, L.; Astrand-Grundström, I.; Arvidsson, I.; Jacobsson, B.; Hellström-Lindberg, E.; Hast, R.; Jacobsen, S.E. Isolation and characterization of hematopoietic progenitor/stem cells in 5q-deleted myelodysplastic syndromes: Evidence for involvement at the hematopoietic stem cell level. Blood 2000, 96, 2012–2021. [Google Scholar] [PubMed]
- Simmons, P.J.; Przepiorka, D.; Thomas, E.D.; Torok-Storb, B. Host origin of marrow stromal cells following allogeneic bone marrow transplantation. Nature 1987, 328, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Thanopoulou, E.; Cashman, J.; Kakagianne, T.; Eaves, A.; Zoumbos, N.; Eaves, C. Engraftment of NOD/SCID-beta2 microglobulin null mice with multilineage neoplastic cells from patients with myelodysplastic syndrome. Blood 2004, 103, 4285–4293. [Google Scholar] [CrossRef] [PubMed]
- Van Lom, K.; Hagemeijer, A.; Smit, E.; Hahlen, K.; Groeneveld, K.; Lowenberg, B. Cytogenetic clonality analysis in myelodysplastic syndrome: Monosomy 7 can be demonstrated in the myeloid and in the lymphoid lineage. Leukemia 1995, 9, 1818–1821. [Google Scholar] [PubMed]
- White, N.J.; Nacheva, E.; Asimakopoulos, F.A.; Bloxham, D.; Paul, B.; Green, A.R. Deletion of chromosome 20q in myelodysplasia can occur in a multipotent precursor of both myeloid cells and B cells. Blood 1994, 83, 2809–2816. [Google Scholar] [PubMed]
- Houghton, J.; Li, H.; Fan, X.; Liu, Y.; Liu, J.H.; Rao, V.P.; Poutahidis, T.; Taylor, C.L.; Jackson, E.A.; Hewes, C.; et al. Mutations in bone marrow-derived stromal stem cells unmask latent malignancy. Stem Cells Dev. 2010, 19, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.A.; Choi, Q.; Hwang, S.M.; Han, S.H.; Kwon, S.; Oh, I.H.; et al. Asymmetric aneuploidy in mesenchymal stromal cells detected by in situ karyotyping and fluorescence in situ hybridization: Suggestions for reference values for stem cells. Stem Cells Dev. 2015, 24, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Eckert, C.; Seeger, K.; Pfau, M.; Prada, J.; Henze, G.; Blankenstein, T.; Kammertoens, T. Leukemia-associated genetic aberrations in mesenchymal stem cells of children with acute lymphoblastic leukemia. J. Mol. Med. 2010, 88, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Campioni, D.; Lanza, F.; Moretti, S.; Dominici, M.; Punturieri, M.; Pauli, S.; Hofmann, T.; Horwitz, E.; Castoldi, G.L. Functional and immunophenotypic characteristics of isolated CD105+ and fibroblast+ stromal cells from AML: Implications for their plasticity along endothelial lineage. Cytotherapy 2003, 5, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Mandel, K.; Yang, Y.; Schambach, A.; Glage, S.; Otte, A.; Hass, R. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013, 22, 3114–3127. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Mercapide, J.; Lorico, A. Spontaneous formation of tumorigenic hybrids between breast cancer and multipotent stromal cells is a source of tumor heterogeneity. Am. J. Pathol. 2012, 180, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.H.; Gao, X.; Luo, D.; Zhou, X.D.; Xiong, W.; Liu, G.X. EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLoS ONE 2014, 9, e87893. [Google Scholar] [CrossRef] [PubMed]
- Martin-Padura, I.; Marighetti, P.; Gregato, G.; Agliano, A.; Malazzi, O.; Mancuso, P.; Pruneri, G.; Viale, A.; Bertolini, F. Spontaneous cell fusion of acute leukemia cells and macrophages observed in cells with leukemic potential. Neoplasia 2012, 14, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Kurose, K.; Gilley, K.; Matsumoto, S.; Watson, P.H.; Zhou, X.P.; Eng, C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002, 32, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Maffini, M.V.; Soto, A.M.; Calabro, J.M.; Ucci, A.A.; Sonnenschein, C. The stroma as a crucial target in rat mammary gland carcinogenesis. J. Cell Sci. 2004, 117, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Walkley, C.R.; Shea, J.M.; Sims, N.A.; Purton, L.E.; Orkin, S.H. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 2007, 129, 1081–1095. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Kinney, M.C.; Scott, L.M.; Zinkel, S.S.; Rebel, V.I. Revisiting the case for genetically engineered mouse models in human myelodysplastic syndrome research. Blood 2015, 126, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Choudry, F.A.; Frontini, M. Epigenetic control of haematopoietic stem cell aging and its clinical implications. Stem Cells Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Stauder, R.; Burgstaller, S.; Schreder, M.; Tinchon, C.; Pfeilstocker, M.; Steinkirchner, S.; Melchardt, T.; Mitrovic, M.; Girschikofsky, M.; et al. Azacitidine in patients with WHO-defined AML—Results of 155 patients from the Austrian Azacitidine Registry of the AGMT-Study Group. J. Hematol. Oncol. 2013, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Germing, U.; Sperr, W.R.; Linkesch, W.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; Schreder, M.; Pfeilstocker, M.; Lang, A.; et al. Azacitidine in CMML: Matched-pair analyses of daily-life patients reveal modest effects on clinical course and survival. Leuk. Res. 2014, 38, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Kim, J.A.; Oh, I.H. Stem cell niche as a prognostic factor in leukemia. BMB. Rep. 2015, 48, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, A.; DiPersio, J.F. Targeting the leukemia-stroma interaction in acute myeloid leukemia: Rationale and latest evidence. Ther. Adv. Hematol. 2016, 7, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Maryanovich, M.; Arnal-Estape, A.; Frenette, P.S. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 2015, 86, 360–373. [Google Scholar] [CrossRef] [PubMed]
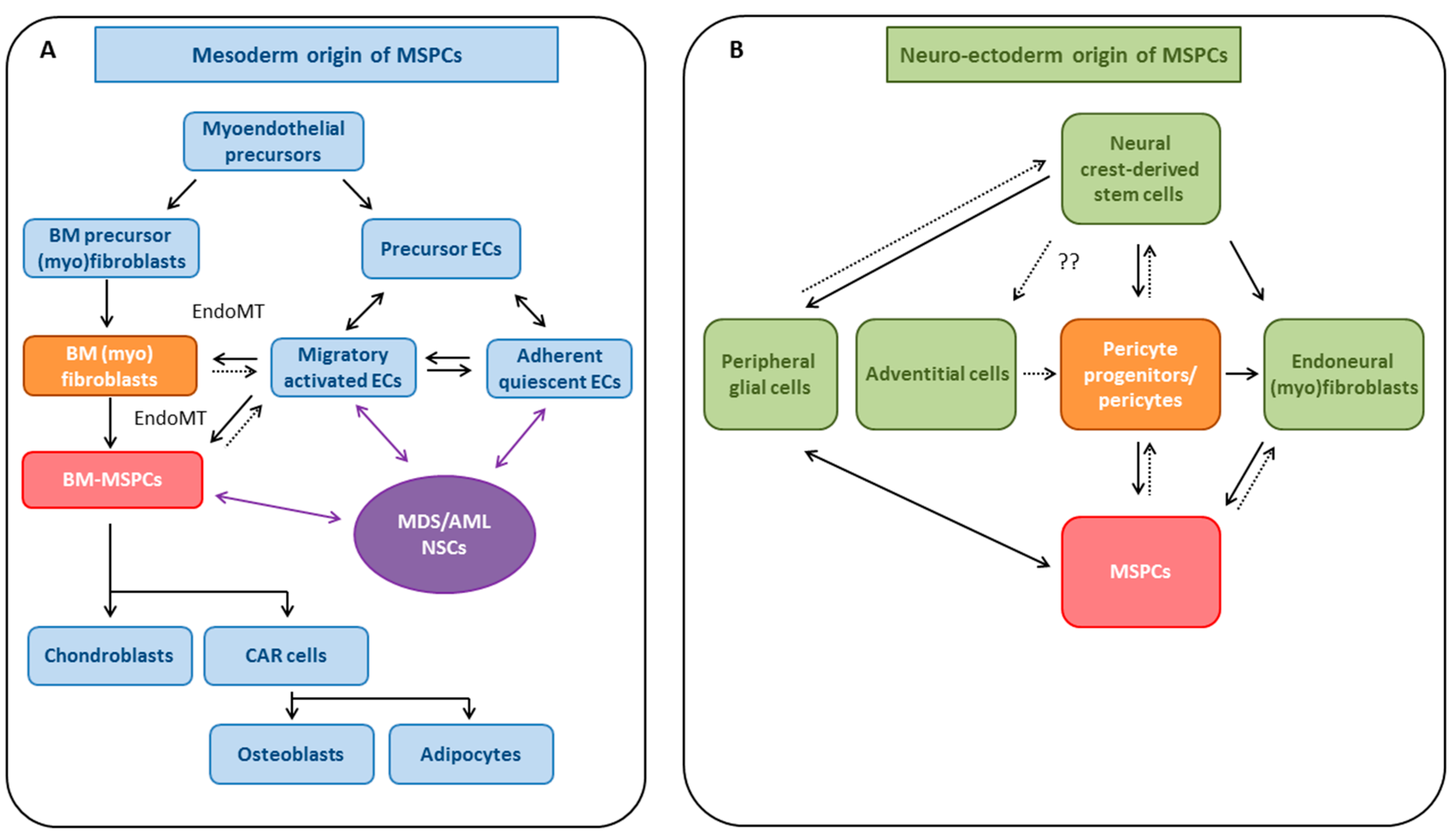
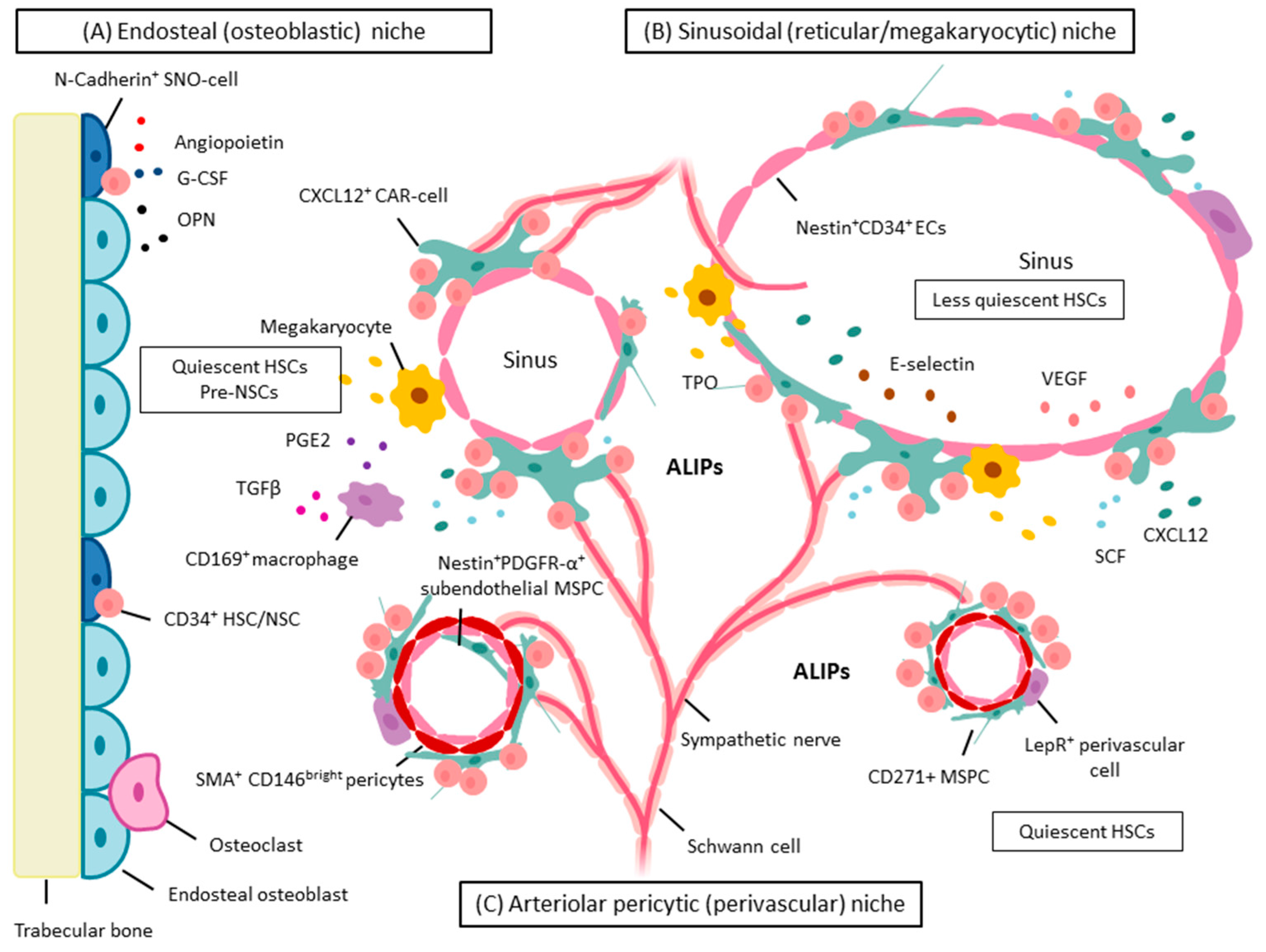

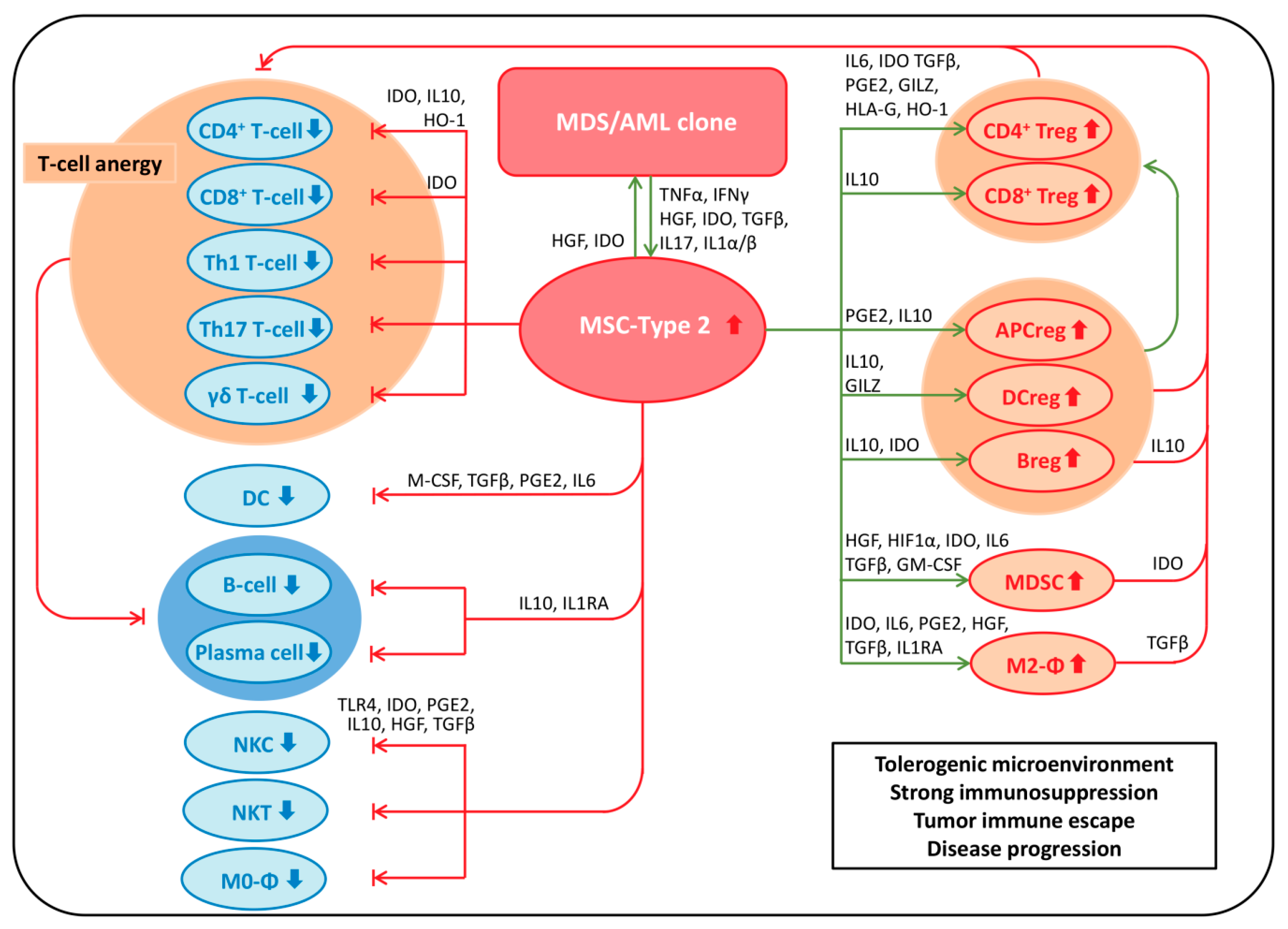

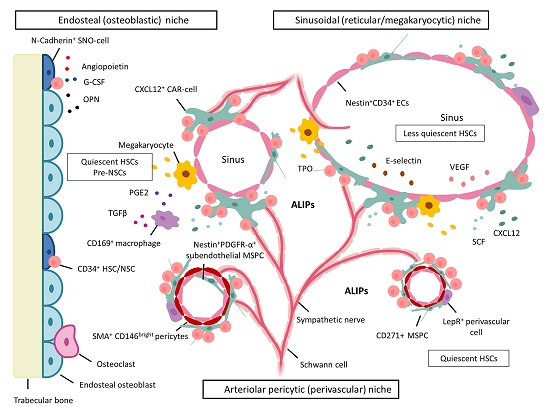{kind=link}
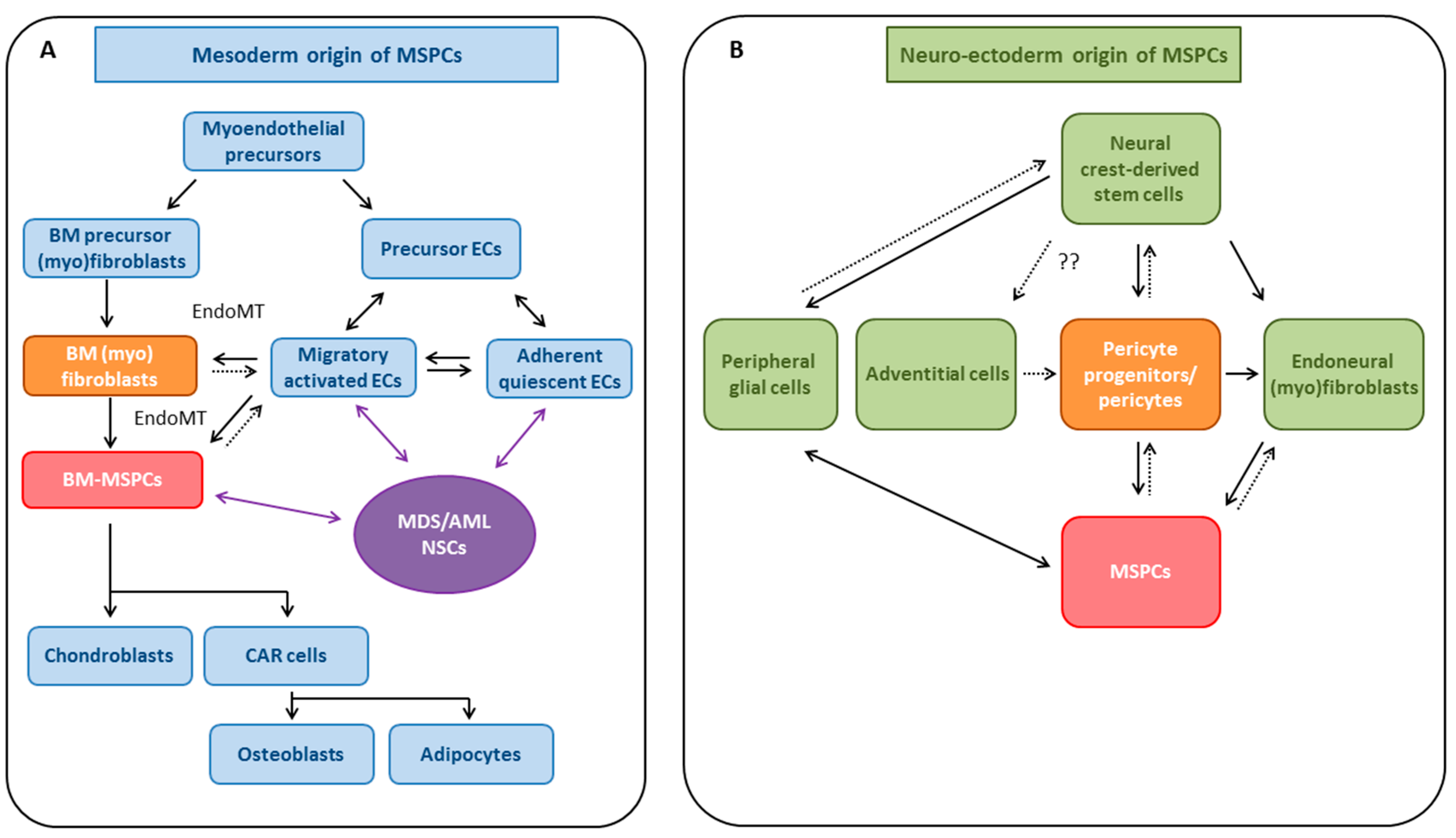{kind=link}
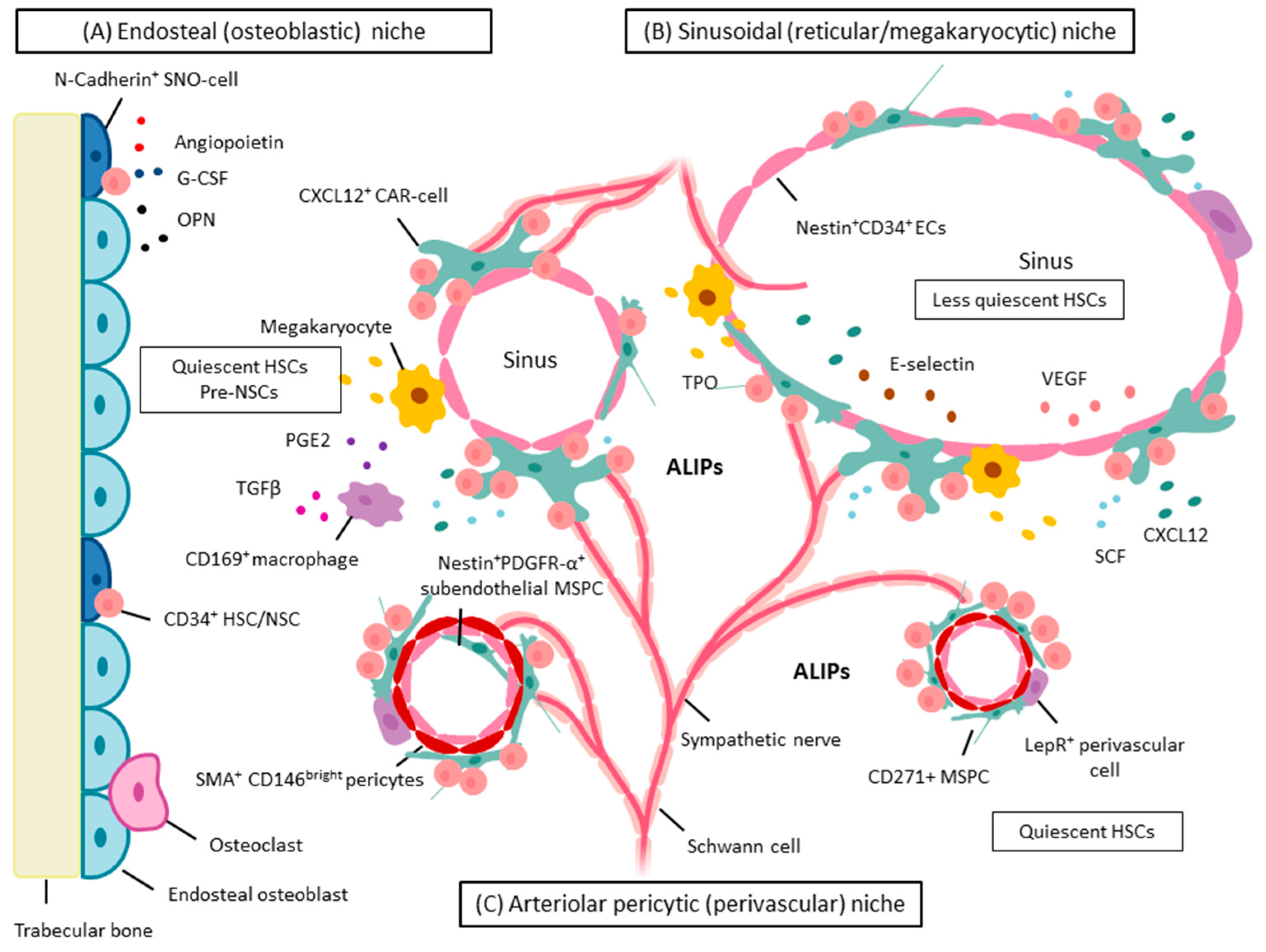{kind=link}
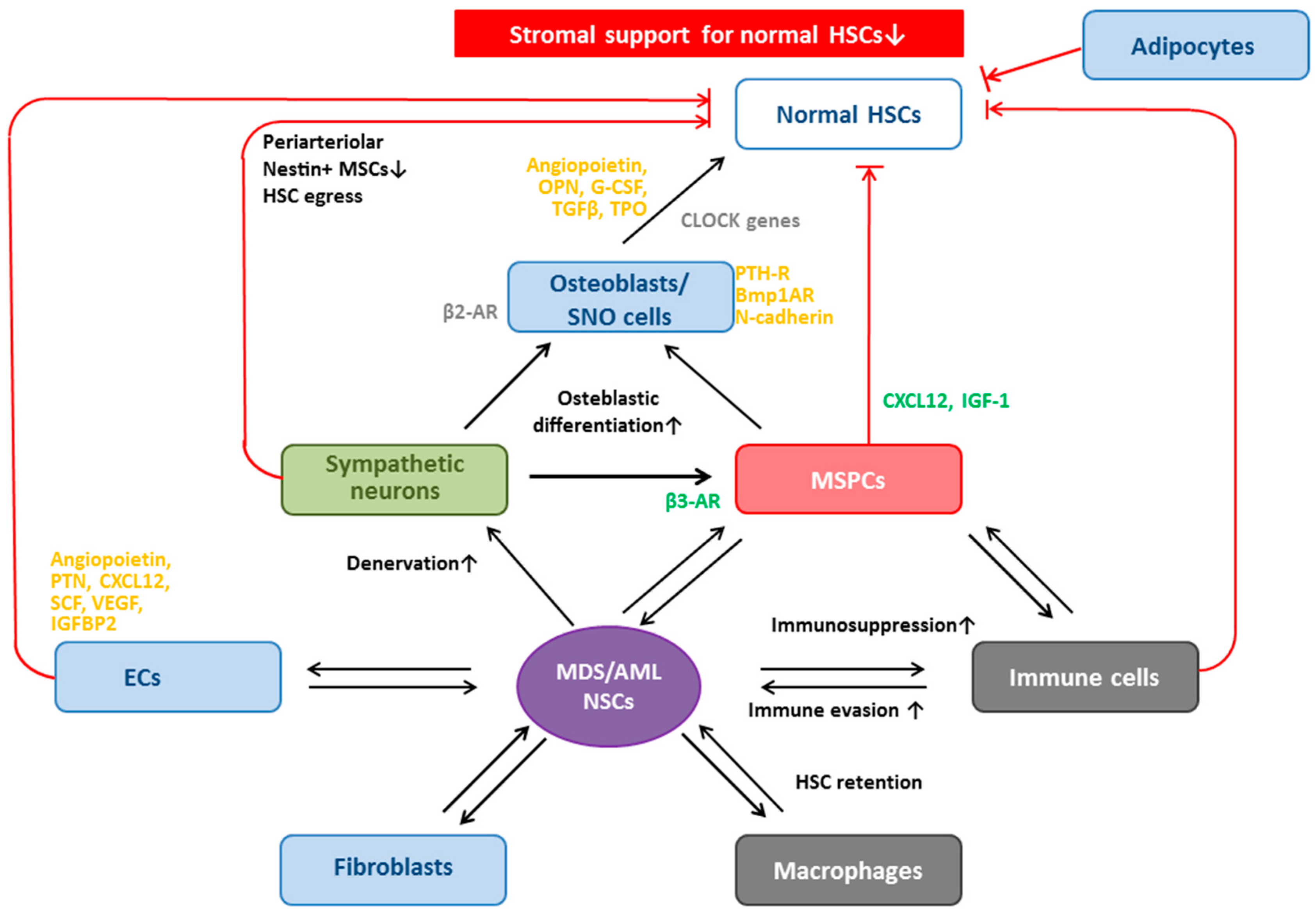{kind=link}
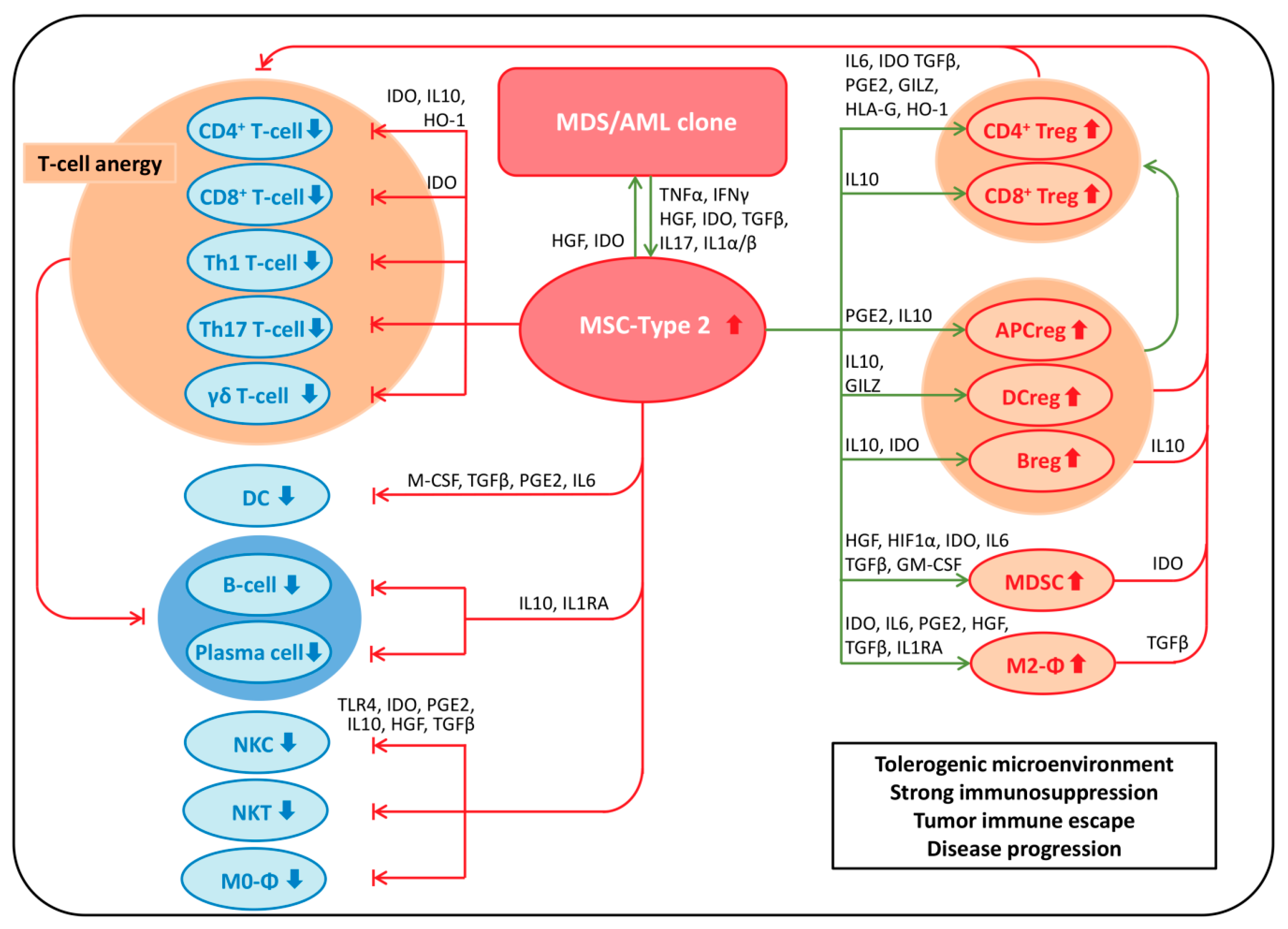{kind=link}
| Grouping A | Grouping B |
|---|---|
| Stromal Cells | Niche Cells |
| Mesenchymal stem cells | Mesenchymal stem cells |
| Fibroblasts | Endothelial cells |
| Endothelial cells | Osteoblastic cells |
| Adipocytes | Sympathetic neurons |
| Tissue macrophages | Non-myelinating Schwann cells |
| Osteoclasts | Perivascular stromal cells |
| Bone marrow macrophages | CXCL12-abundant reticular cells |
| Nestin+ perivascular cells | |
| Leptin-receptor+ perivascular cells | |
| Accessory Cells | Niche Accessory Cells |
| Myeloid regulatory cells | Adipocytes |
| Circulating macrophages | Osteoclastic cells |
| Dendritic cells | Bone marrow macrophages |
| Myeloid-derived suppressor cells | T-regulatory cells |
| Lymphoid regulatory cells | Megakaryocytes |
| T-regulatory cells | |
| Natural killer cells | |
| B-cells |
| MSPCs Suppress Inflammatory Immune Cell Subsets | References |
| CD4+ and CD8+ T-cell proliferation and activation | [403,404,405,406,407,408,409] |
| Th1 and Th17 conversion | [410,411,412,413] |
| γδ T-cell proliferation and possibly cytotoxicity | [344,414,415,416] |
| NKT cell proliferation | [416] |
| NKC proliferation, cytokine production and cytotoxicity, and decreased expression of activating receptors | [408,417,418,419,420] |
| DC maturation, proliferation, differentiation, and pro-inflammatory function, and/or antigen-presenting capacity, with ensuing inability to prime T-cells | [421,422,423,424,425,426,427,428] |
| B-cell proliferation, differentiation, and function (in part through the modulation of T-cell help by MSPCs) | [408,413,429,430,431,432,433,434] |
| Plasma cell formation | [413,433,435] |
| Inflammatory potential of activated neutrophils | [436] |
| MSPCs Favor Immunosuppressive Regulatory Cell Subsets | |
| CD4+ Tregs | [410,418,437,438,439,440,441,442,443,444,445] |
| CD8+ Tregs | [444,446] |
| Invariant NK regulatory cells (NKregs) | [180,416] |
| Regulatory DCs (DCregs) with T-cell suppressive properties, induction of T-cell anergy, and the capacity to induce Tregs | [388,447,448,449] |
| Polarization of macrophages towards the anti-inflammatory M2-phenotype with T-cell-suppressive properties | [413,422,450,451,452,453,454] |
| Regulatory antigen-presenting cells (APCregs) with T-cell-suppressive properties | [455,456] |
| MDSCs | [457,458] |
| B regulatory cells | [433,435,459,460] |
| Human MSPC/Stromal Cell Alterations | Disease | References |
|---|---|---|
| Impaired proliferative and clonogenic potential in cell passages, growth and differentiation defects, altered morphology, disrupted clonal architecture (less CFU, less cobble-stone area formation) | MDS, AML | [20,25,26,30,33,66,68,274,279,592,593,594,595] |
| Higher apoptotic index | MDS | [32] |
| Increased senescence | MDS | [468] |
| Increased density of primitive MSPCs (CD271+ or Nestin+) | MDS, AML | [81,182] |
| Reduced support for HSCs and/or hematopoiesis | MDS, AML | [25,33,34,35,80,274,592] |
| Chromosomal abnormalities | MDS, AML | [66,324,593,596,597,598,599,600,601,602] |
| Epigenetic changes (altered methylation profile) | MDS | [33] |
| Altered adhesion molecule profiles | MDS | [68,274] |
| Altered levels of cytokine or chemokine production | MDS, AML | [32,33,274,324,592,603] |
| Deregulated signaling (Wnt/β-catenin, Notch/Jagged1, KIT/SCF, senescence-associated CDKN1A/2A/2B) | MDS, AML | [26,30,31,33,332] |
| DNA methylation changes | MDS, AML | [33,592] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pleyer, L.; Valent, P.; Greil, R. Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis—Masters of Survival and Clonality? Int. J. Mol. Sci. 2016, 17, 1009. https://doi.org/10.3390/ijms17071009
Pleyer L, Valent P, Greil R. Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis—Masters of Survival and Clonality? International Journal of Molecular Sciences. 2016; 17(7):1009. https://doi.org/10.3390/ijms17071009
Chicago/Turabian StylePleyer, Lisa, Peter Valent, and Richard Greil. 2016. "Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis—Masters of Survival and Clonality?" International Journal of Molecular Sciences 17, no. 7: 1009. https://doi.org/10.3390/ijms17071009