
Echinacoside Inhibits Glutamate Release by Suppressing Voltage-Dependent Ca2+ Entry and Protein Kinase C in Rat Cerebrocortical Nerve Terminals


Abstract
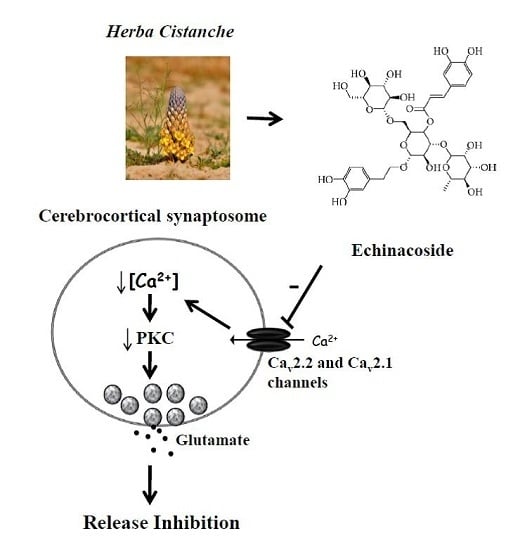
:
1. Introduction
2. Results
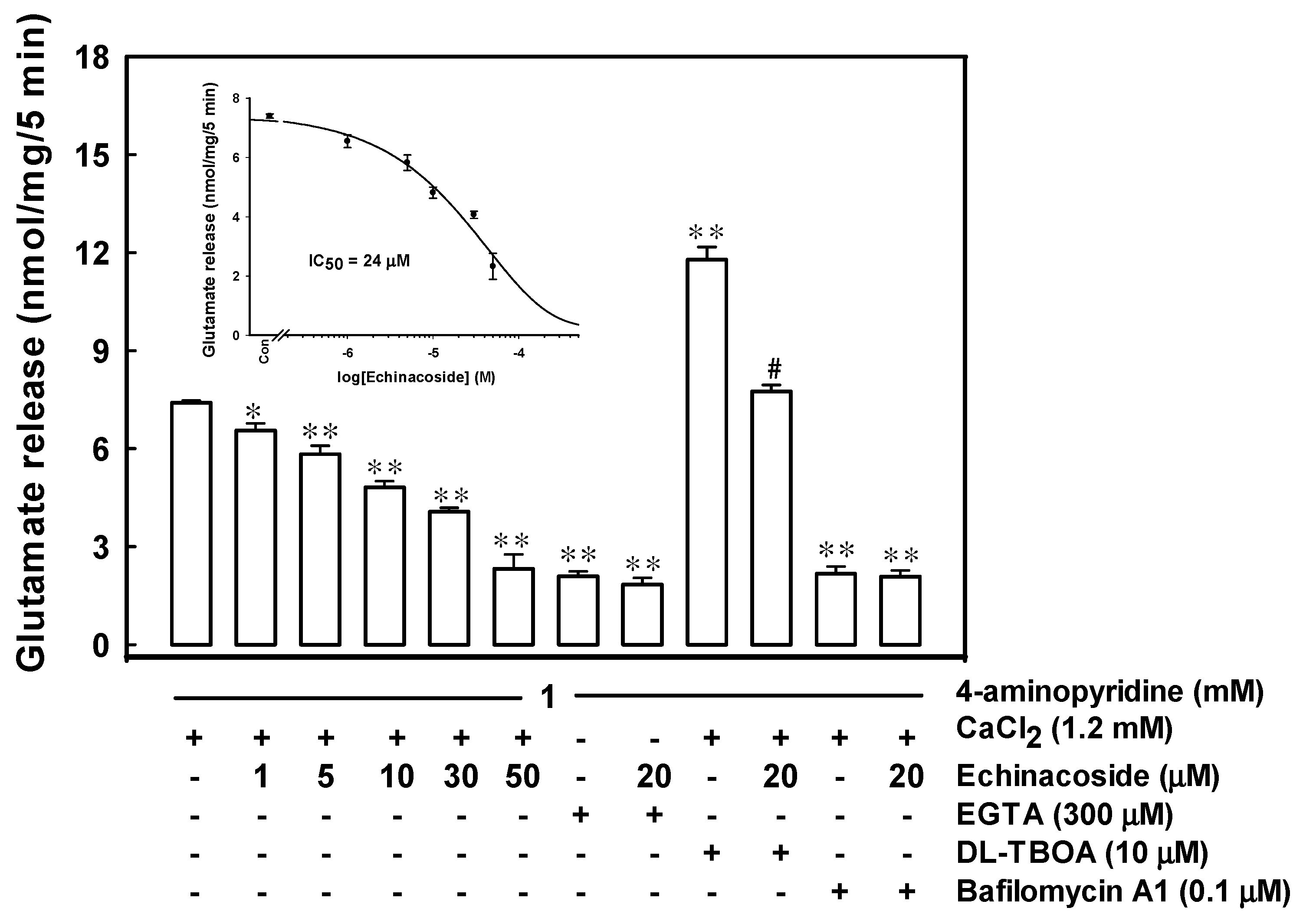
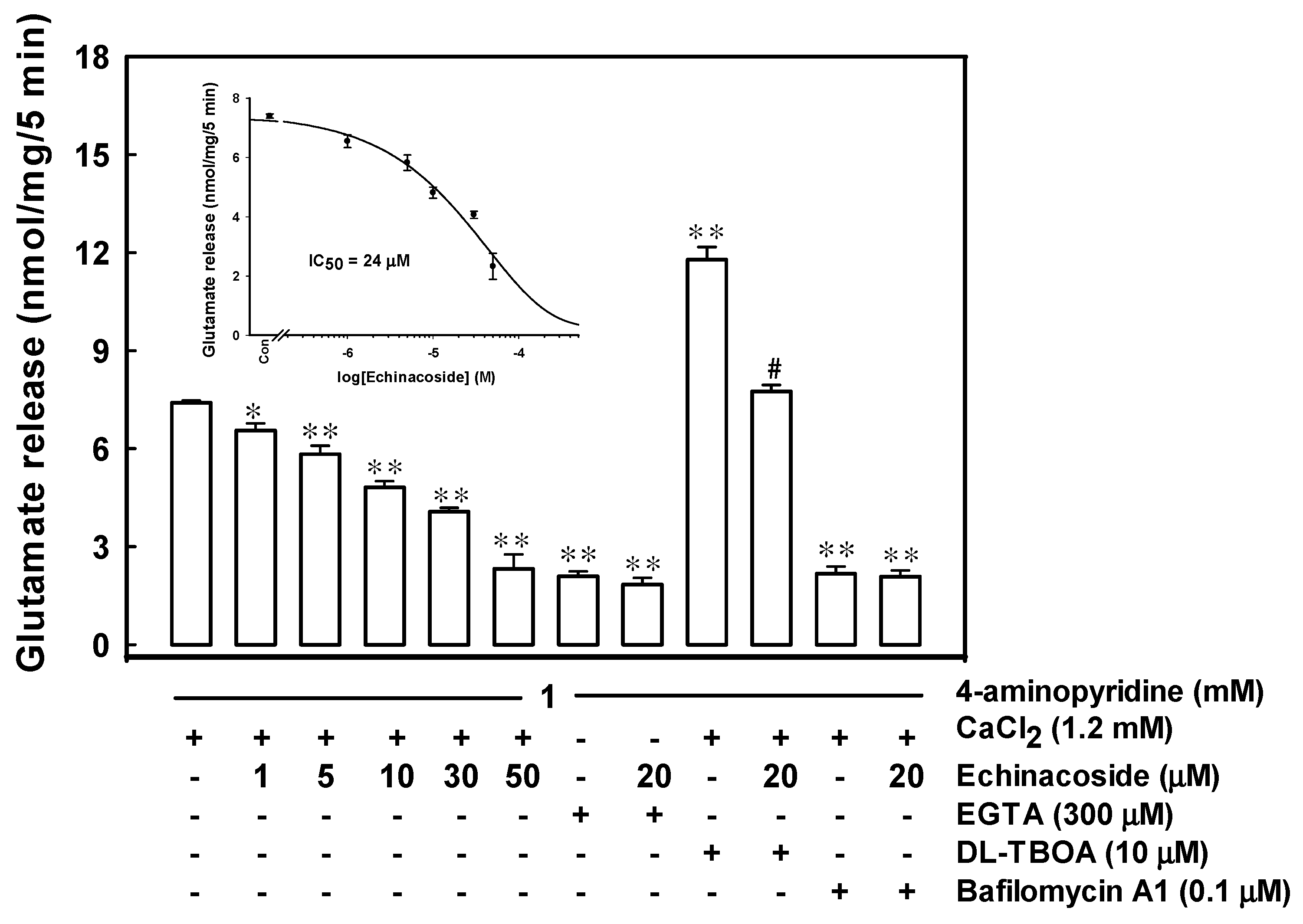
2.1. Echinacoside Inhibits 4-Aminopyridine-Evoked Glutamate Release from Rat Cerebrocortical Nerve Terminals by Reducing Vesicular Exocytosis
2.2. Echinacoside Reduces Cytosolic Ca2+ Concentration but Does Not Alter the Synaptosomal Membrane Potential
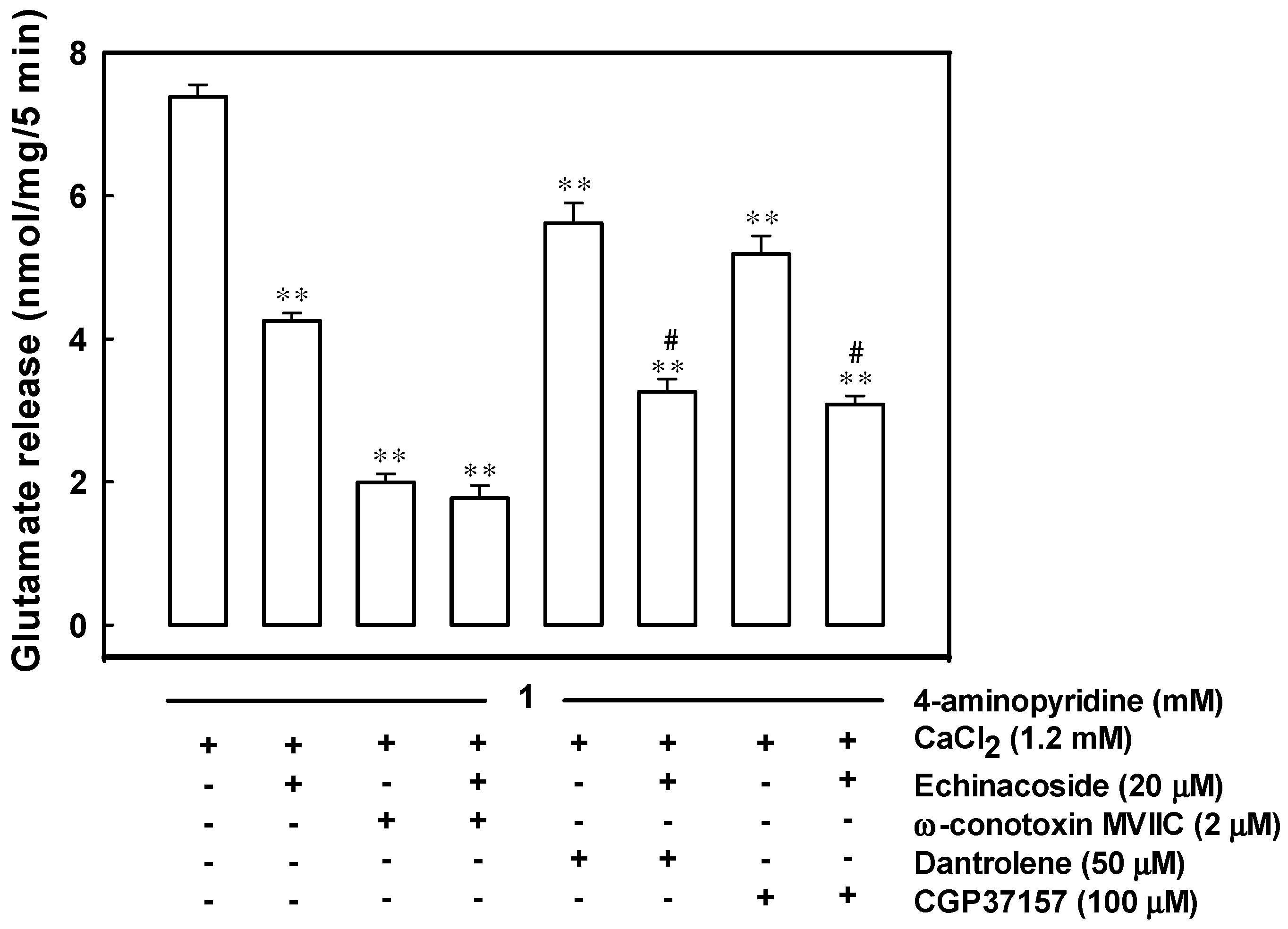
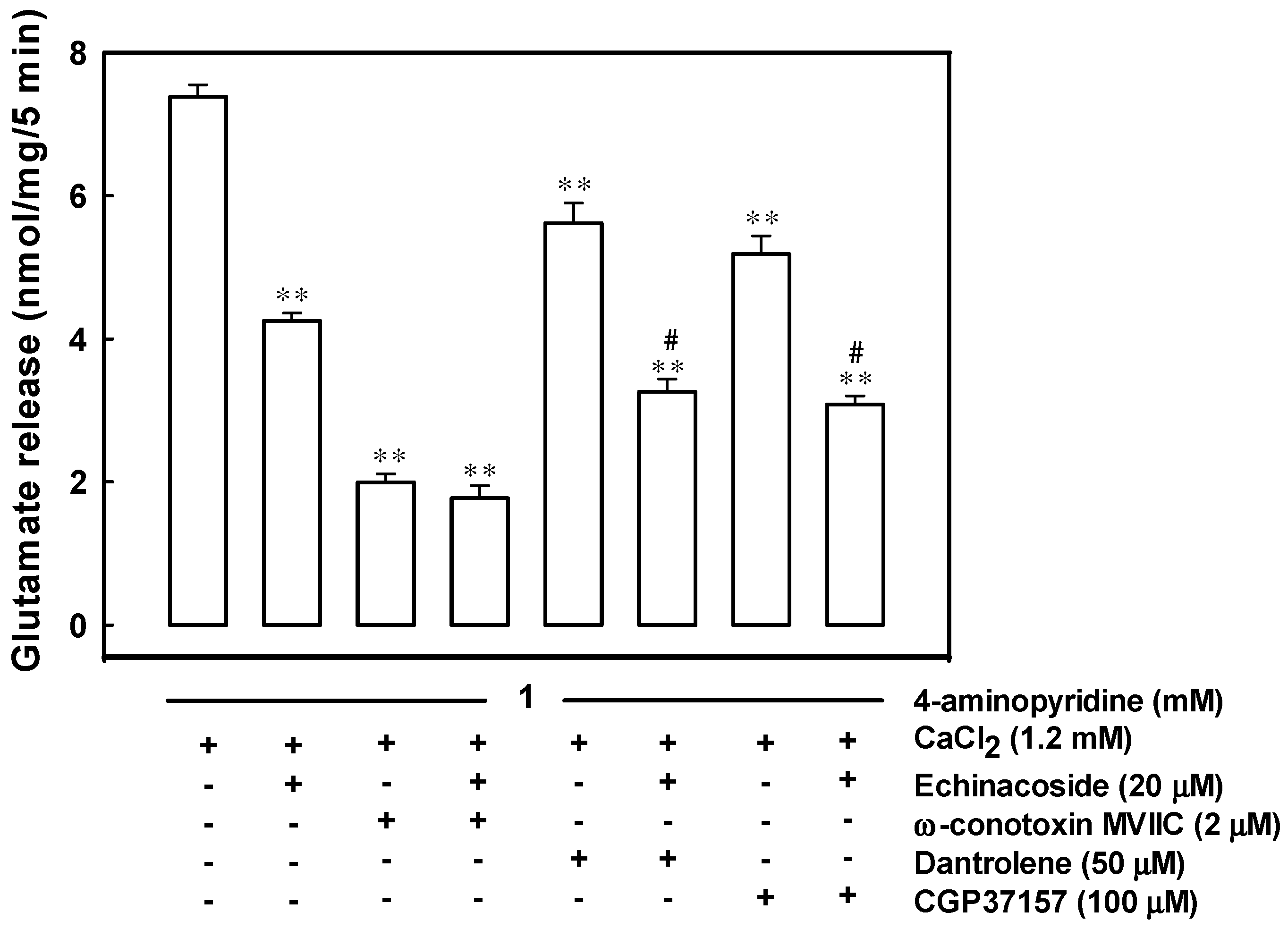
2.3. Reduced Ca2+ Influx through the Cav2.2 (N-Type) and Cav2.1 (P/Q-Type) Channels May Be Associated with the Inhibition of 4-Aminopyridine-Evoked Glutamate Release by Echinacoside
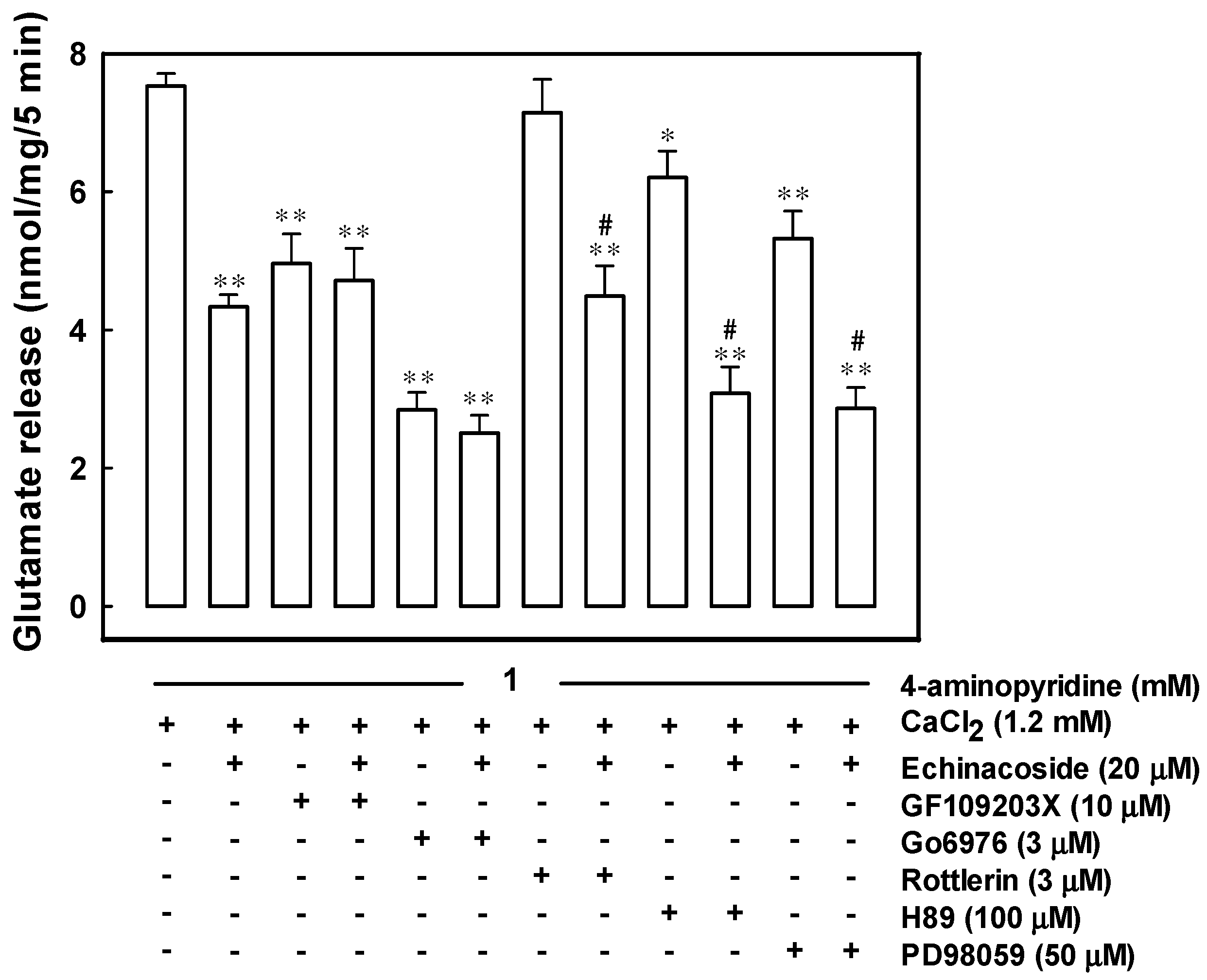
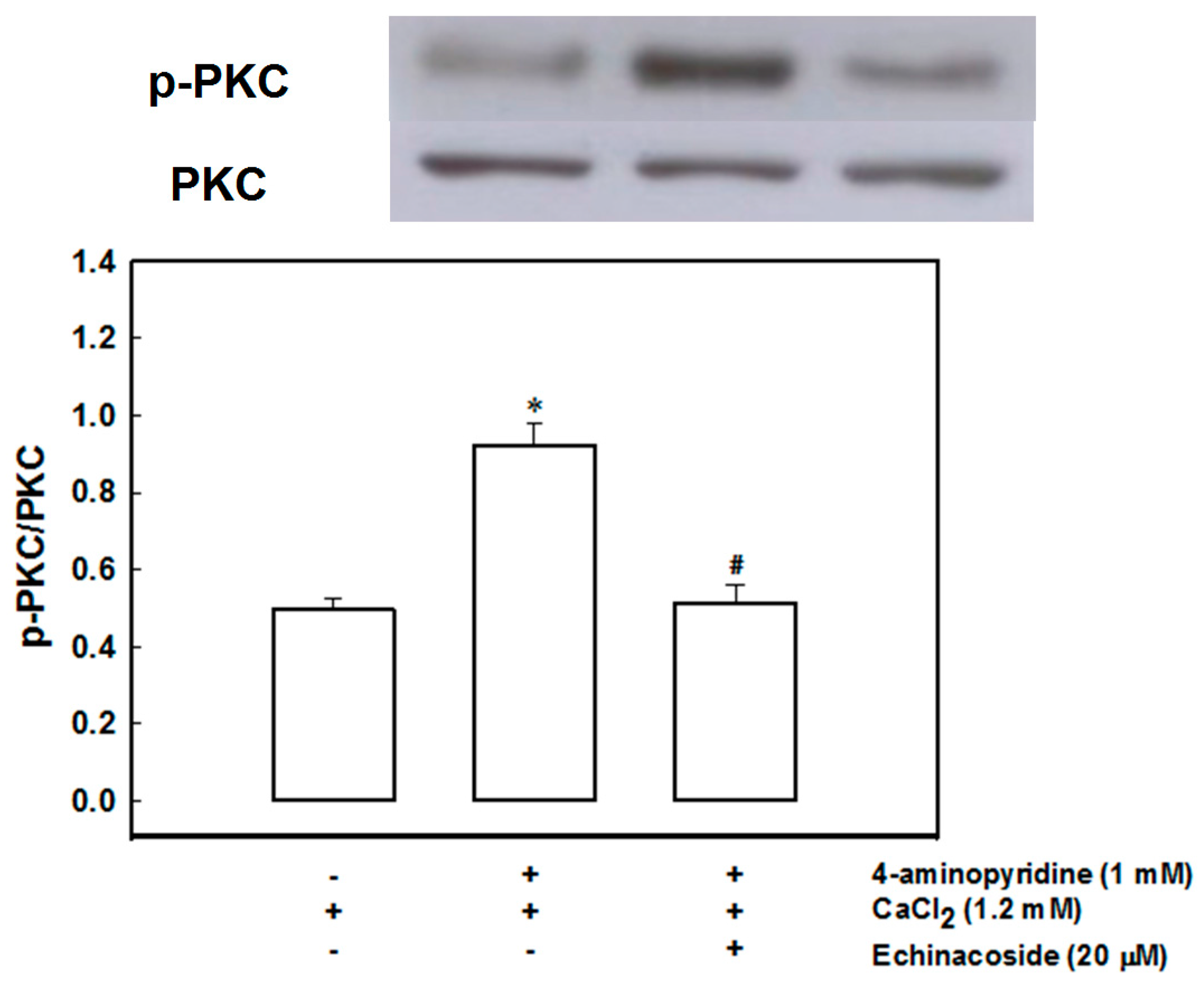
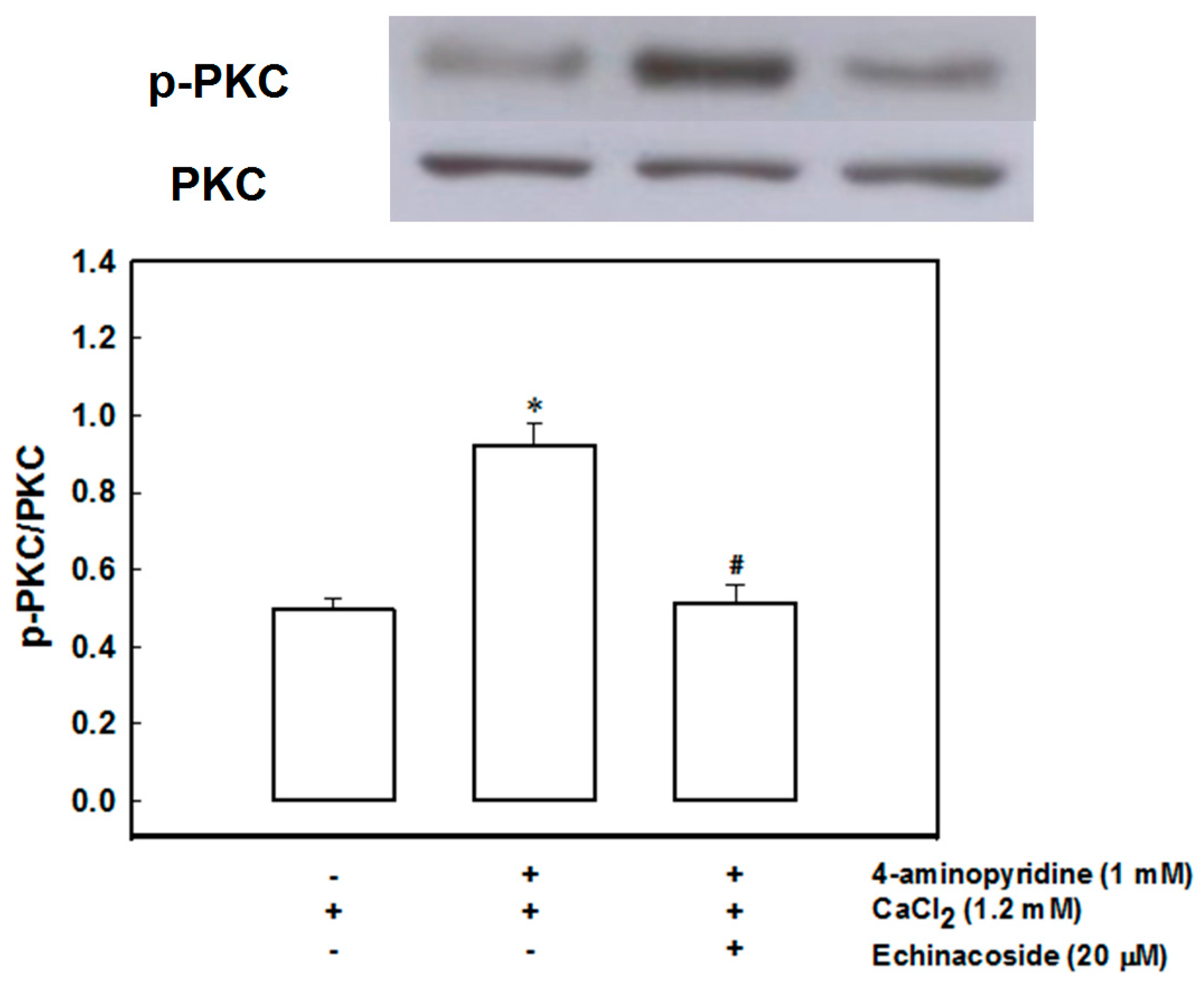
2.4. Echinacoside Inhibits 4-Aminopyridine-Evoked Glutamate Release by Signaling through Protein Kinase C
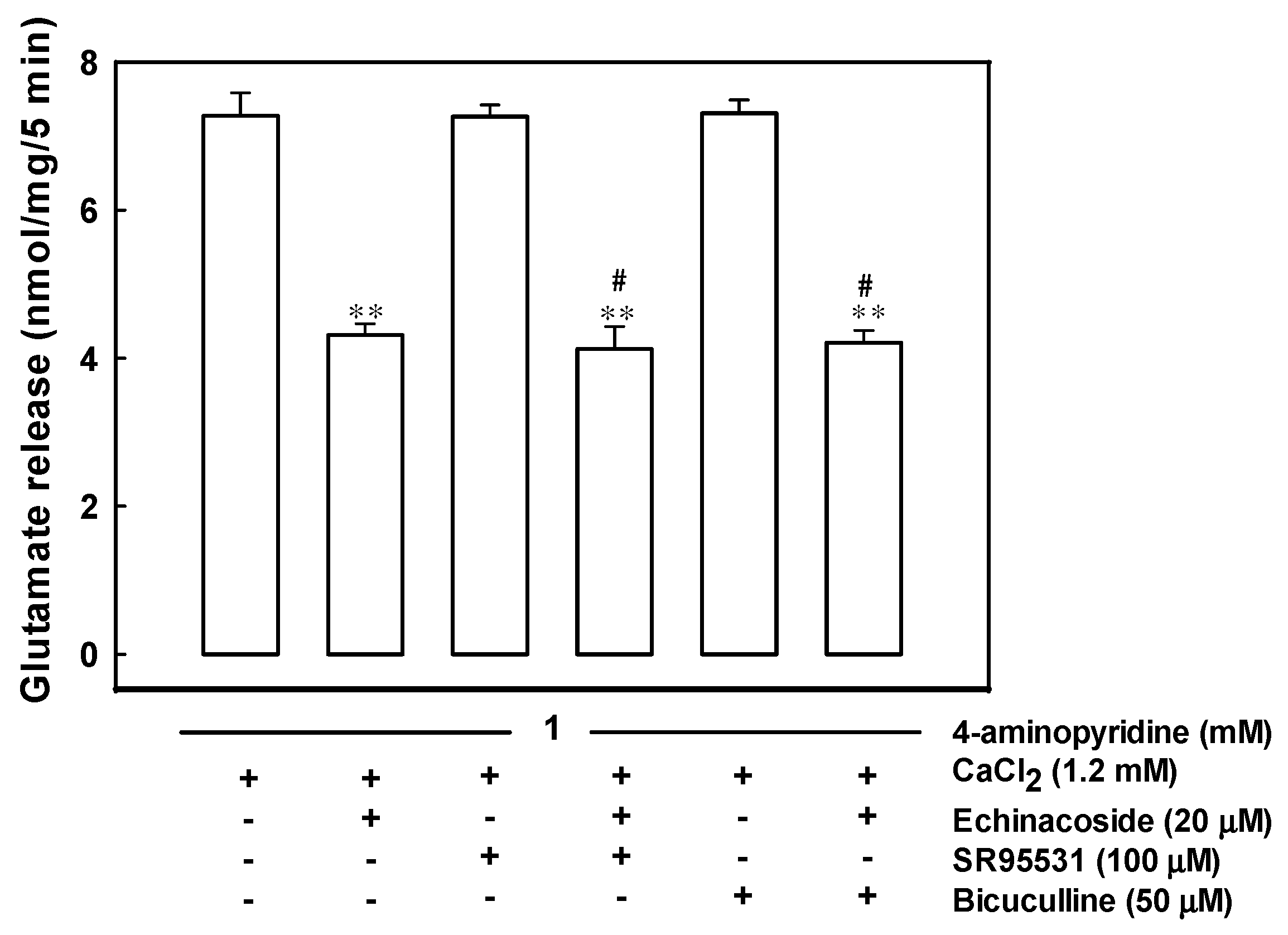
2.5. Echinacoside-Mediated Inhibition of Glutamate Release Does Not Involve a Gamma-Aminobutyric Acid Type A (GABAA) Receptor
3. Discussion
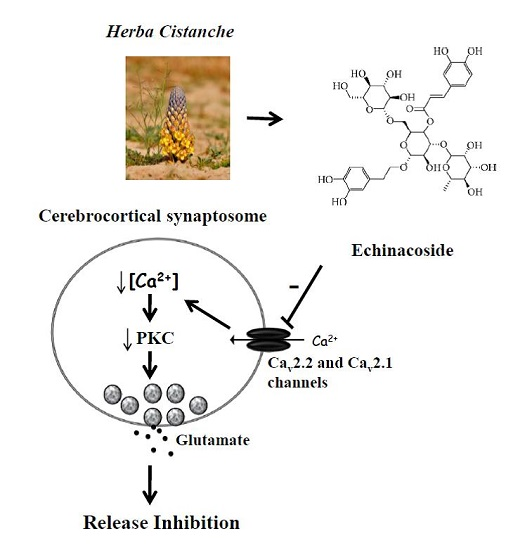
3.1. Mechanisms Underlying Echinacoside-Mediated Inhibition of Glutamate Release
3.2. Therapeutic Implications
4. Materials and Methods
4.1. Chemicals
4.2. Animals
4.3. Synaptosomal Preparations
4.4. Glutamate Release
4.5. Plasma Membrane Potential
4.6. Cytosolic Ca2+ Concentration ([Ca2+]C)
4.7. Western Blotting
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tu, P.F.; Wang, B.; Deyama, T.; Zhang, Z.G.; Lou, Z.C. Analysis of phenylethanoid glycosides of Herba cistanchis by RP-HPLC. Acta Pharm. Sin. 1997, 32, 294–300. [Google Scholar]
- Dalby-Brown, L.; Barsett, H.; Landbo, A.K.; Meyer, A.S.; Molgaard, P. Synergistic antioxidative effects of alkamides, caffeic acid derivatives, and polysaccharide fractions from Echinacea purpurea on in vitro oxidation of human low-density lipoproteins. J. Agric. Food Chem. 2005, 53, 9413–9423. [Google Scholar] [CrossRef] [PubMed]
- Dapas, B.; Dall’Acqua, S.; Bulla, R.; Agostinis, C.; Perissutti, B.; Invernizzi, S.; Grassi, G.; Voinovich, D. Immunomodulation mediated by a herbal syrup containing a standardized Echinacea root extract: A pilot study in healthy human subjects on cytokine gene expression. Phytomedicine 2014, 21, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- He, W.J.; Fang, T.H.; Tu, P.F. Research progress on pharmacological activities of echinacoside. China J. Chin. Mater. Med. 2009, 34, 476–479. [Google Scholar]
- Deng, M.; Zhao, J.Y.; Tu, P.F.; Jiang, Y.; Li, Z.B.; Wang, Y.H. Echinacoside rescues the SHSY5Y neuronal cells from TNFalpha-induced apoptosis. Eur. J. Pharmacol. 2004, 505, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Koo, K.A.; Sung, S.H.; Park, J.H.; Kim, S.H.; Lee, K.Y.; Kim, Y.C. In vitro neuroprotective activities of phenylethanoid glycosides from Callicarpa dichotoma. Planta Med. 2005, 71, 778–780. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Xuan, Z.H.; Tian, S.; Du, G.H. Echinacoside Protects against 6-Hydroxydopamine-Induced Mitochondrial Dysfunction and Inflammatory Responses in PC12 Cells via Reducing ROS Production. Evid. Based Complement. Altern. Med. 2015, 2015, 189239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Lu, C.; Li, W. Transient exposure to echinacoside is sufficient to activate Trk signaling and protect neuronal cells from rotenone. J. Neurochem. 2013, 124, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Tian, X.; Tu, P.; Pu, X. Neuroprotective effects of echinacoside in the mouse MPTP model of Parkinson's disease. Eur. J. Pharmacol. 2007, 564, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.L.; Chen, H.; Jiang, Y.; Tu, P.F.; Zhong, M.; Du, J.; Liu, F.; Wang, L.; Liu, C.Y. Effects of echinacoside on histio-central levels of active mass in middle cerebral artery occlusion rats. Biomed. Environ. Sci. 2012, 25, 238–244. [Google Scholar] [PubMed]
- Wu, C.R.; Lin, H.C.; Su, M.H. Reversal by aqueous extracts of Cistanche tubulosa from behavioral deficits in Alzheimer’s disease-like rat model: Relevance for amyloid deposition and central neurotransmitter function. BMC Complement. Altern. Med. 2014, 14, 202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Gao, J.; Li, W.; Cai, D. Neurotrophic and neurorescue effects of Echinacoside in the subacute MPTP mouse model of Parkinson’s disease. Brain Res. 2010, 1346, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [PubMed]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Calcium and excitotoxic neuronal injury. Ann. N. Y. Acad. Sci. 1994, 747, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers. Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol. Neurobiol. 2001, 24, 107–129. [Google Scholar] [CrossRef]
- Schauwecker, P.E. Neuroprotection by glutamate receptor antagonists against seizure-induced excitotoxic cell death in the aging brain. Exp. Neurol. 2010, 224, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, F.; Nikbakht, F.; Bahmanpour, S.; Rastegar, K.; Namavar, R. Neuroprotective effects of NMDA and group I metabotropic glutamate receptor antagonists against neurodegeneration induced by homocysteine in rat hippocampus: In vivo study. J. Mol. Neurosci. 2013, 50, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Muir, K.W. Glutamate-based therapeutic approaches: Clinical trials with NMDA antagonists. Curr. Opin. Pharmacol. 2006, 6, 53–60. [Google Scholar] [CrossRef] [PubMed]
- González, J.C.; Egea, J.; del Carmen Godino, M.; Fernandez-Gomez, F.J.; Sánchez-Prieto, J.; Gandía, L.; García, A.G.; Jordán, J.; Hernández-Guijo, J.M. Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca2+ signalling in hippocampal neurons. Eur. J. Neurosci. 2007, 26, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, T.Y.; Wang, S.J. Memantine depresses glutamate release through inhibition of voltage-dependent Ca2+ entry and protein kinase C in rat cerebral cortex nerve terminals: An NMDA receptor-independent mechanism. Neurochem. Int. 2010, 57, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Sihra, T.S. Noncompetitive metabotropic glutamate 5 receptor antagonist (E)-2-methyl-6-styryl-pyridine (SIB1893) depresses glutamate release through inhibition of voltage-dependent Ca2+ entry in rat cerebrocortical nerve terminals (synaptosomes). J. Pharmacol. Exp. Ther. 2004, 309, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, P.R.; Jarvie, P.E.; Heath, J.W.; Kidd, G.J.; Rostas, J.A. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res. 1986, 372, 115–129. [Google Scholar] [CrossRef]
- Araque, A.; Li, N.; Doyle, R.T.; Haydon, P.G. SNARE protein-dependent glutamate release from astrocytes. J. Neurosci. 2000, 20, 666–673. [Google Scholar] [PubMed]
- Dunlop, J. Glutamate-based therapeutic approaches: Targeting the glutamate transport system. Curr. Opin. Pharmacol. 2006, 6, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R.; Ronca-Testoni, S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: Modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 1997, 49, 1–51. [Google Scholar] [PubMed]
- Fan, Y.; Li, J.; Zhang, Y.Q.; Jiang, L.H.; Zhang, Y.N.; Yan, C.Q. Protein kinase C delta mediated cytotoxicity of 6-Hydroxydopamine via sustained extracellular signal-regulated kinase 1/2 activation in PC12 cells. Neurol. Res. 2014, 36, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Gschwendt, M.; Müller, H.J.; Kielbassa, K.; Zang, R.; Kittstein, W.; Rincke, G.; Marks, F. Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun. 1994, 199, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Sihra, T.S.; Sanchez-Prieto, J. Calcium-dependent and-independent release of glutamate from synaptosomes monitored by continuous fluorometry. J. Neurochem. 1987, 49, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.A. The coupling of neurotransmitter receptors to ion channels in the brain. Science 1988, 241, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997, 20, 204–212. [Google Scholar] [CrossRef]
- Turner, T.J.; Dunlap, K. Pharmacological characterization of presynaptic calcium channels using subsecond biochemical measurements of synaptosomal neurosecretion. Neuropharmacology 1995, 34, 1469–1478. [Google Scholar] [CrossRef]
- Millan, C.; Sanchez-Prieto, J. Differential coupling of N- and P/Q-type calcium channels to glutamate exocytosis in the rat cerebral cortex. Neurosci. Lett. 2002, 330, 29–32. [Google Scholar] [CrossRef]
- Vazquez, E.; Sanchez-Prieto, J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur. J. Neurosci. 1997, 9, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Long, P.; Mercer, A.; Begum, R.; Stephens, G.J.; Sihra, T.S.; Jovanovic, J.N. Nerve Terminal GABAA Receptors Activate Ca2+/Calmodulin-dependent Signaling to Inhibit Voltage-gated Ca2+ Influx and Glutamate Release. J. Biol. Chem. 2009, 284, 8726–8737. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R.; Angle, J.M.; Black, E.D.; Joyal, J.L.; Sacks, D.B.; Madara, J.L. PKC-dependent regulation of transepithelial resistance: Roles of MLC and MLC kinase. Am. J. Physiol. 1999, 277, C554–C562. [Google Scholar] [PubMed]
- Vaughan, P.F.; Walker, J.H.; Peers, C. The regulation of neurotransmitter secretion by protein kinase C. Mol. Neurobiol. 1998, 18, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T.; Sihra, T.S.; Nicholls, D.G.; Pocock, J.M. Phosphorylation of synapsin I and MARCKS in nerve terminals is mediated by Ca2+ entry via an Aga-GI sensitive Ca2+ channel which is coupled to glutamate exocytosis. FEBS Lett. 1994, 353, 264–268. [Google Scholar] [CrossRef]
- Obrenovitch, T.P.; Urenjak, J. Altered glutamatergic transmission in neurological disorders: From high extracellular glutamate to excessive synaptic efficacy. Prog. Neurobiol. 1997, 51, 39–87. [Google Scholar] [CrossRef]
- Zhang, D.; Li, H.; Wang, J.B. Echinacoside inhibits amyloid fibrillization of HEWL and protects against Aβ-induced neurotoxicity. Int. J. Biol. Macromol. 2015, 72, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Lu, C.W.; Wang, C.C.; Lu, J.F.; Wang, S.J. Hispidulin inhibits the release of glutamate in rat cerebrocortical nerve terminals. Toxicol. Appl. Pharmacol. 2012, 263, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Lin, T.Y.; Lu, C.W.; Huang, S.K.; Wang, Y.C.; Chou, S.S.; Wang, S.J. Hesperidin inhibits glutamate release and exerts neuroprotection against excitotoxicity induced by kainic acid in the hippocampus of rats. Neurotoxicology 2015, 2015, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Sihra, T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986, 321, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Kuo, J.R.; Wang, S.J. Dimebon, an antihistamine drug, inhibits glutamate release in rat cerebrocortical nerve terminals. Eur. J. Pharmacol. 2014, 734, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Tibbs, G.R.; Barrie, A.P.; van Mieghem, F.J.; Mc-Mahon, H.T.; Nicholls, D.G. Repetitive action potentials in isolated nerve terminals in the presence of 4-aminopyridine: Effects on cytosolic free Ca2+ and glutamate release. J. Neurochem. 1989, 53, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Akerman, K.E.; Scott, L.G.; Heikkila, J.E.; Heinonen, E. Ionic dependence of membrane potential and glutamate receptor-linked responses in synaptoneurosomes as measured with a cyanine dye, DiSC2(5). J. Neurochem. 1987, 48, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Sihra, T.S.; Bogonez, E.; Nicholls, D.G. Localized Ca2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release. J. Biol. Chem. 1992, 267, 1983–1989. [Google Scholar] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane Potential (Fluorescence Units) | Cytosolic [Ca2+] (nM) | |||
|---|---|---|---|---|
| Basal | 4-Aminopyridine (1 mM) | Basal | 4-Aminopyridine (1 mM) | |
| Control | 5.6 ± 0.3 | 14.8 ± 0.4 | 144.6 ± 3.1 | 219.3 ± 6.9 |
| Echinacoside | 5.2 ± 0.3 | 14.8 ± 0.4 | 143.7 ± 3.5 | 176.5 ± 6.7 ** |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Echinacoside Inhibits Glutamate Release by Suppressing Voltage-Dependent Ca2+ Entry and Protein Kinase C in Rat Cerebrocortical Nerve Terminals. Int. J. Mol. Sci. 2016, 17, 1006. https://doi.org/10.3390/ijms17071006
Lu CW, Lin TY, Huang SK, Wang SJ. Echinacoside Inhibits Glutamate Release by Suppressing Voltage-Dependent Ca2+ Entry and Protein Kinase C in Rat Cerebrocortical Nerve Terminals. International Journal of Molecular Sciences. 2016; 17(7):1006. https://doi.org/10.3390/ijms17071006
Chicago/Turabian StyleLu, Cheng Wei, Tzu Yu Lin, Shu Kuei Huang, and Su Jane Wang. 2016. "Echinacoside Inhibits Glutamate Release by Suppressing Voltage-Dependent Ca2+ Entry and Protein Kinase C in Rat Cerebrocortical Nerve Terminals" International Journal of Molecular Sciences 17, no. 7: 1006. https://doi.org/10.3390/ijms17071006