
A Theoretical Study of the Hydration of Methane, from the Aqueous Solution to the sI Hydrate-Liquid Water-Gas Coexistence


Abstract
:1. Introduction
2. Results and Discussion
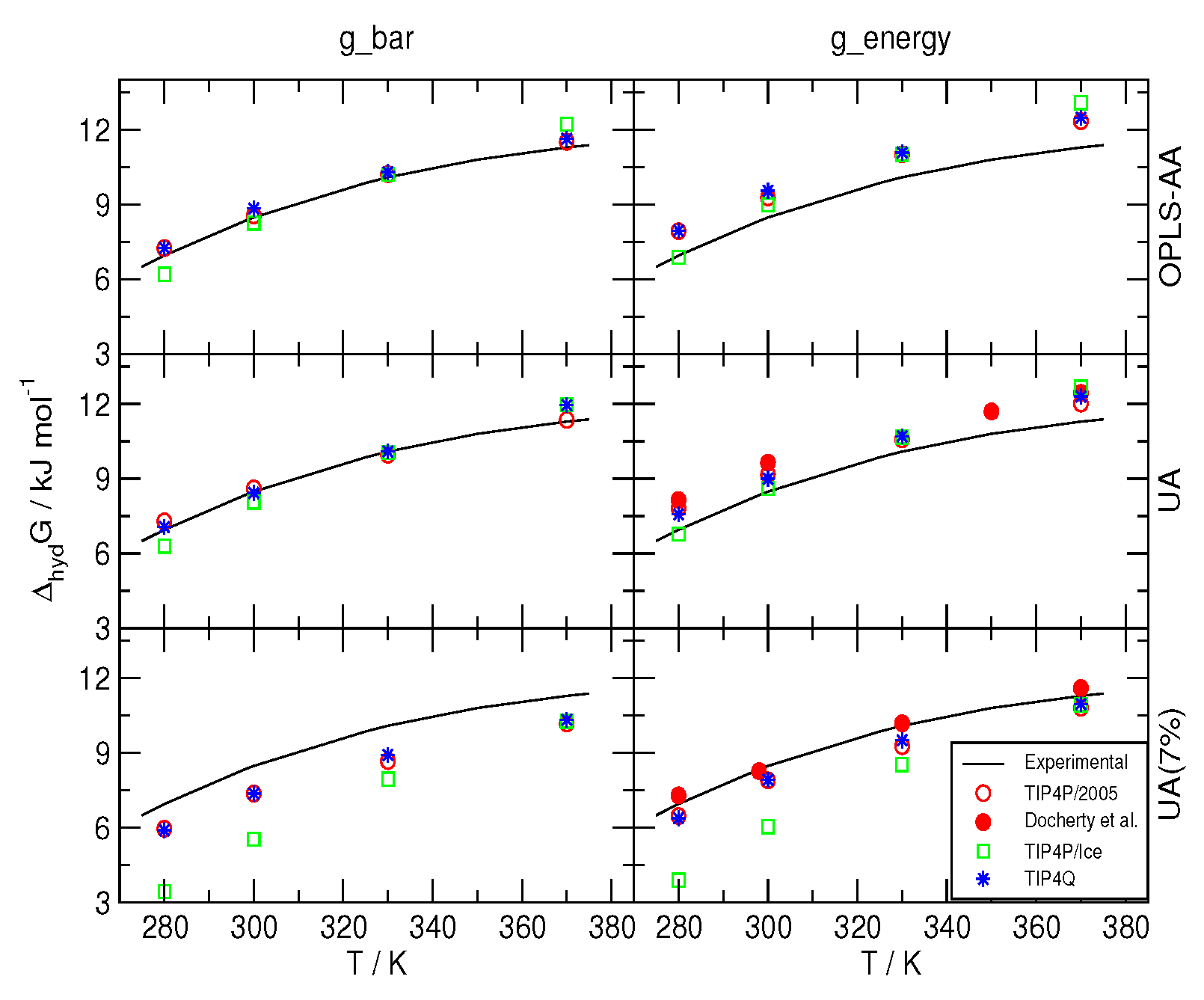
2.1. Free Energies of Hydration
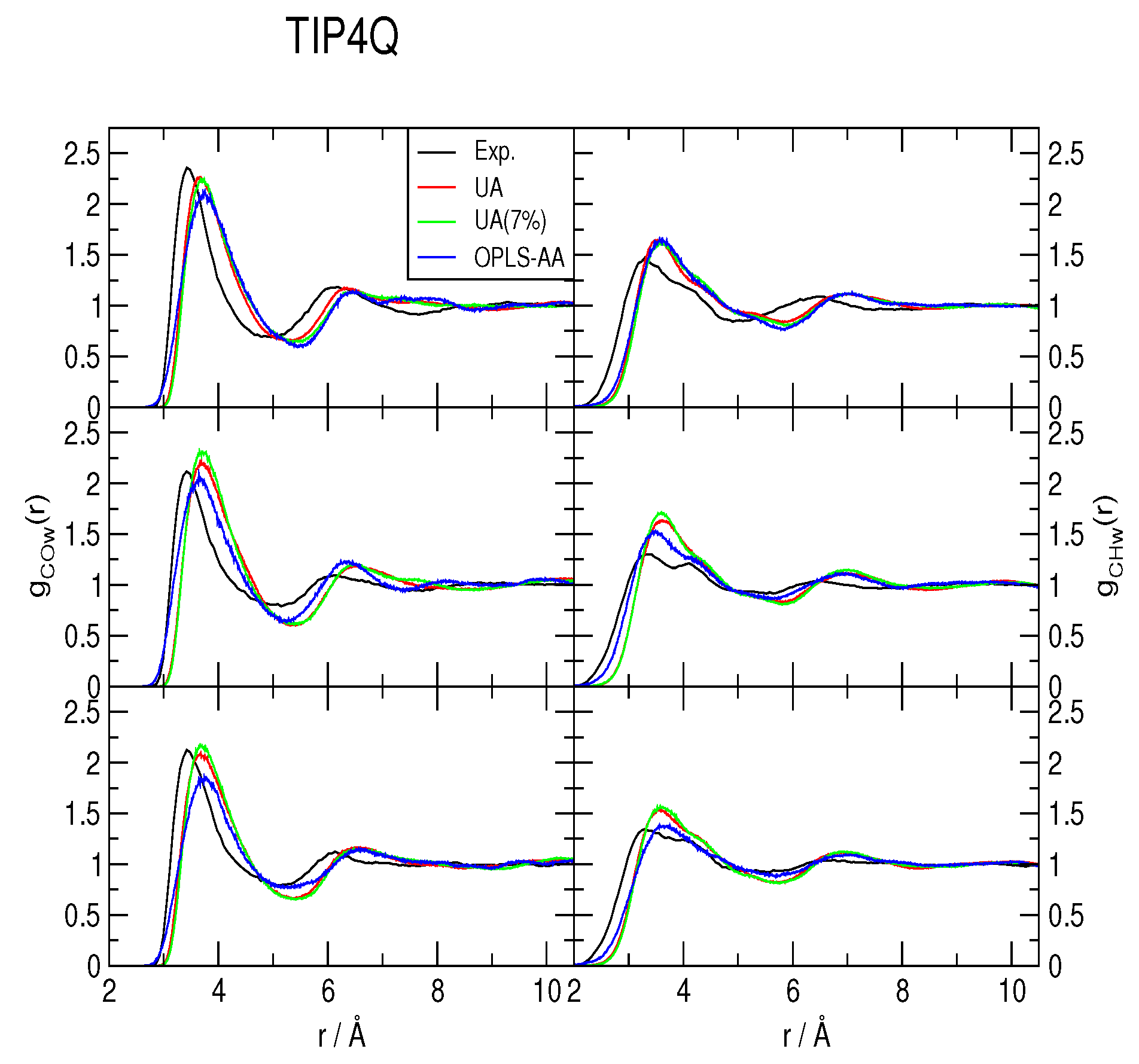
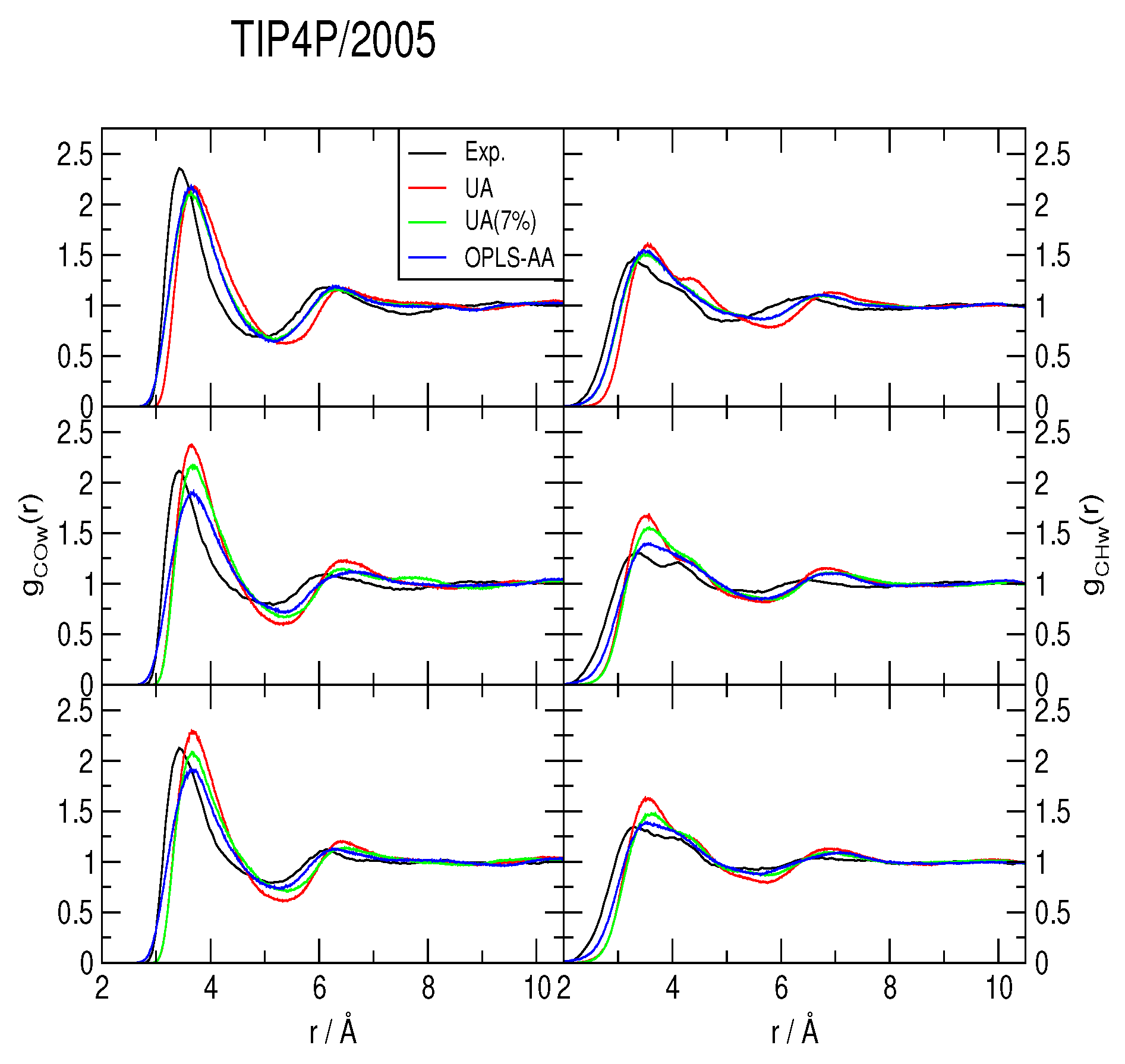
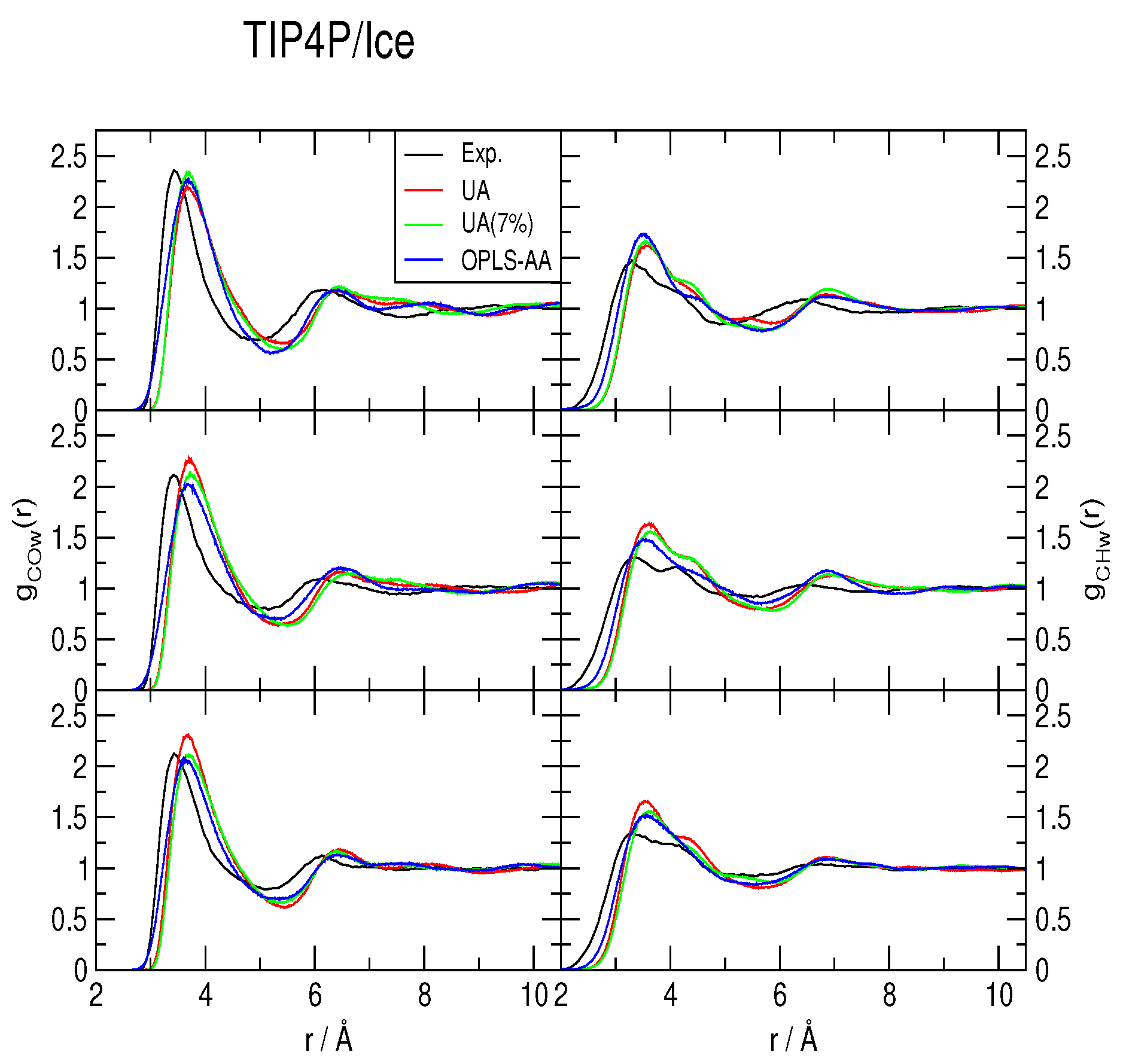
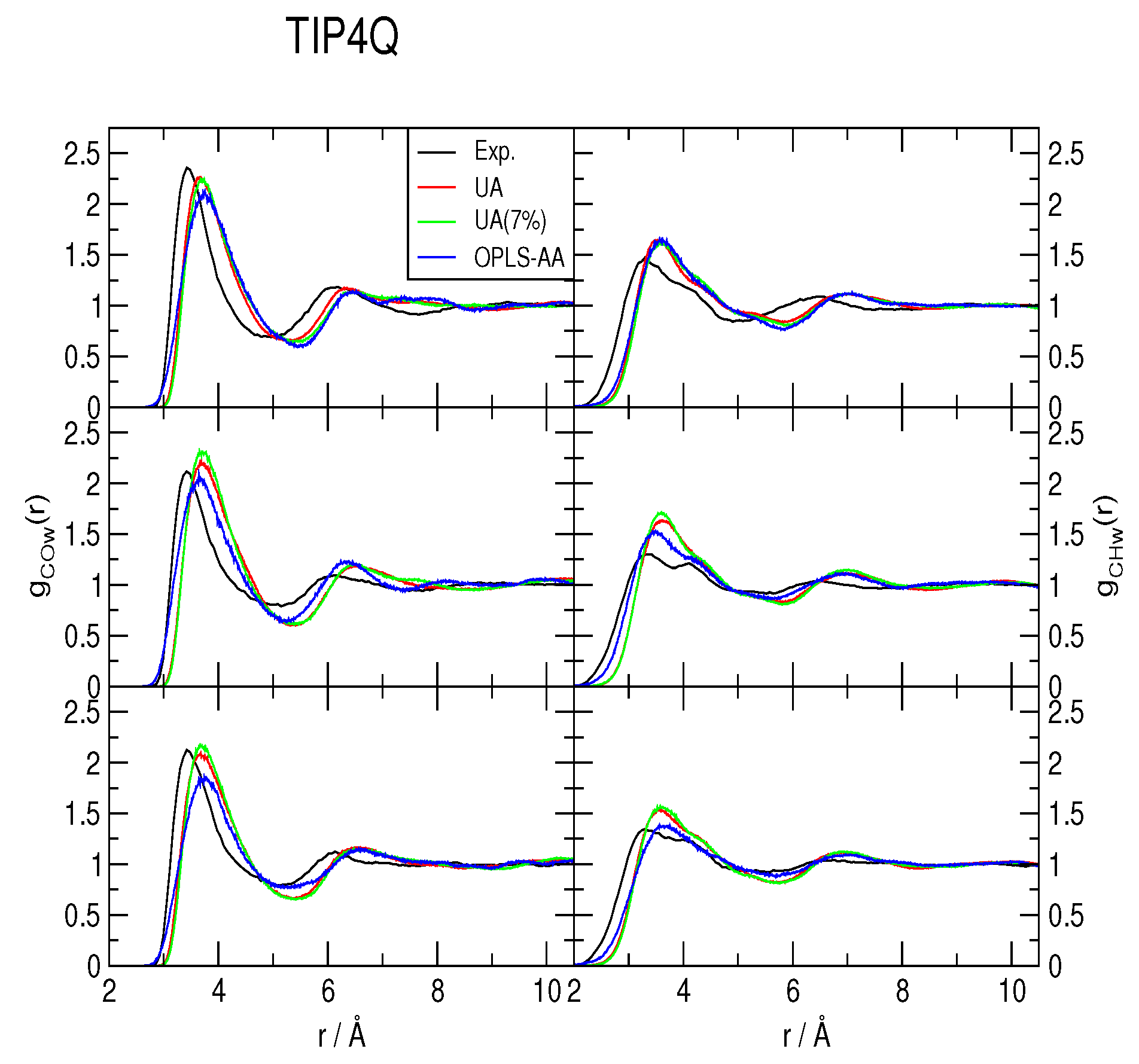
2.2. Radial Distribution Functions and Coordination Numbers nH
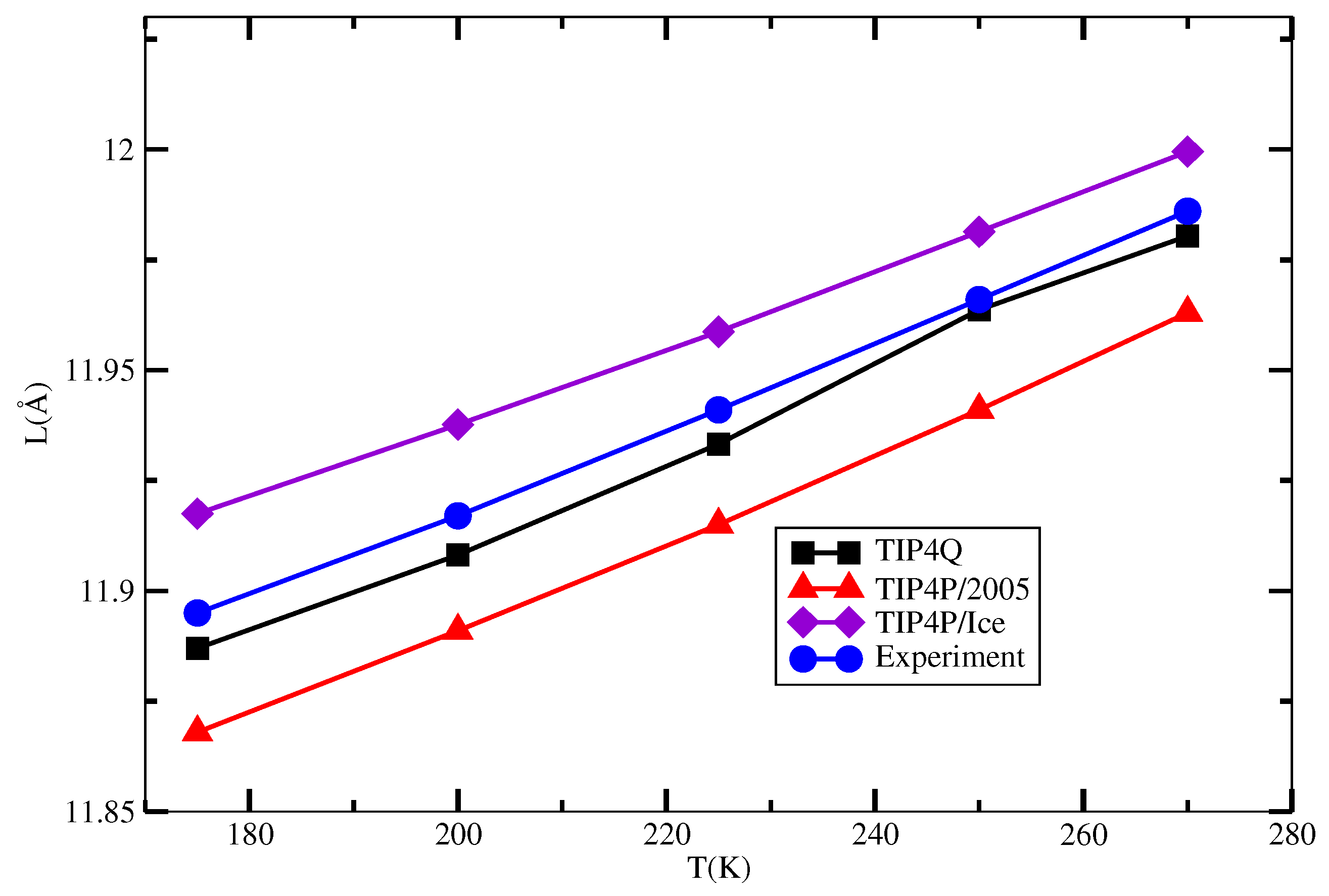
2.3. The sI Hydrate
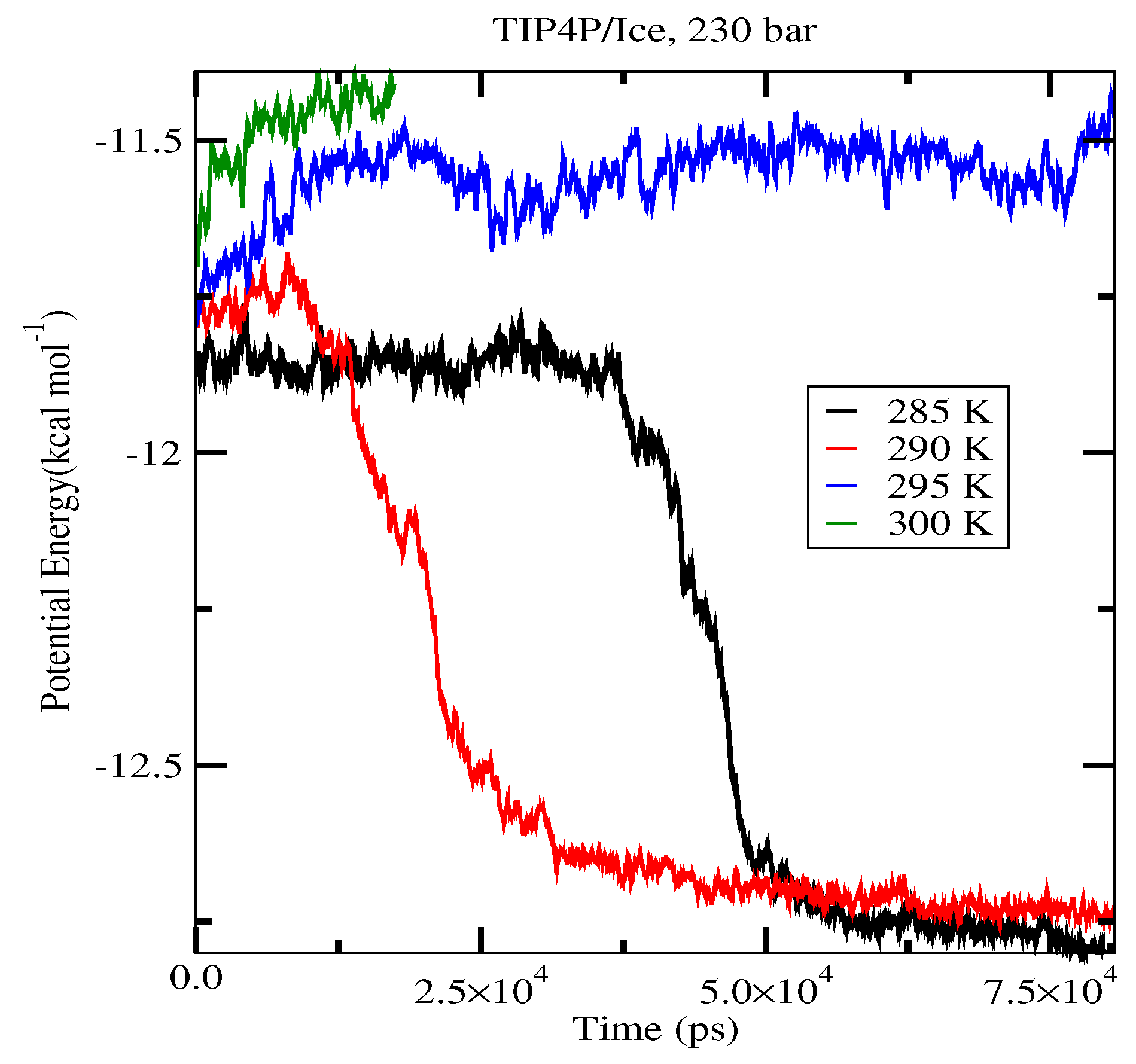
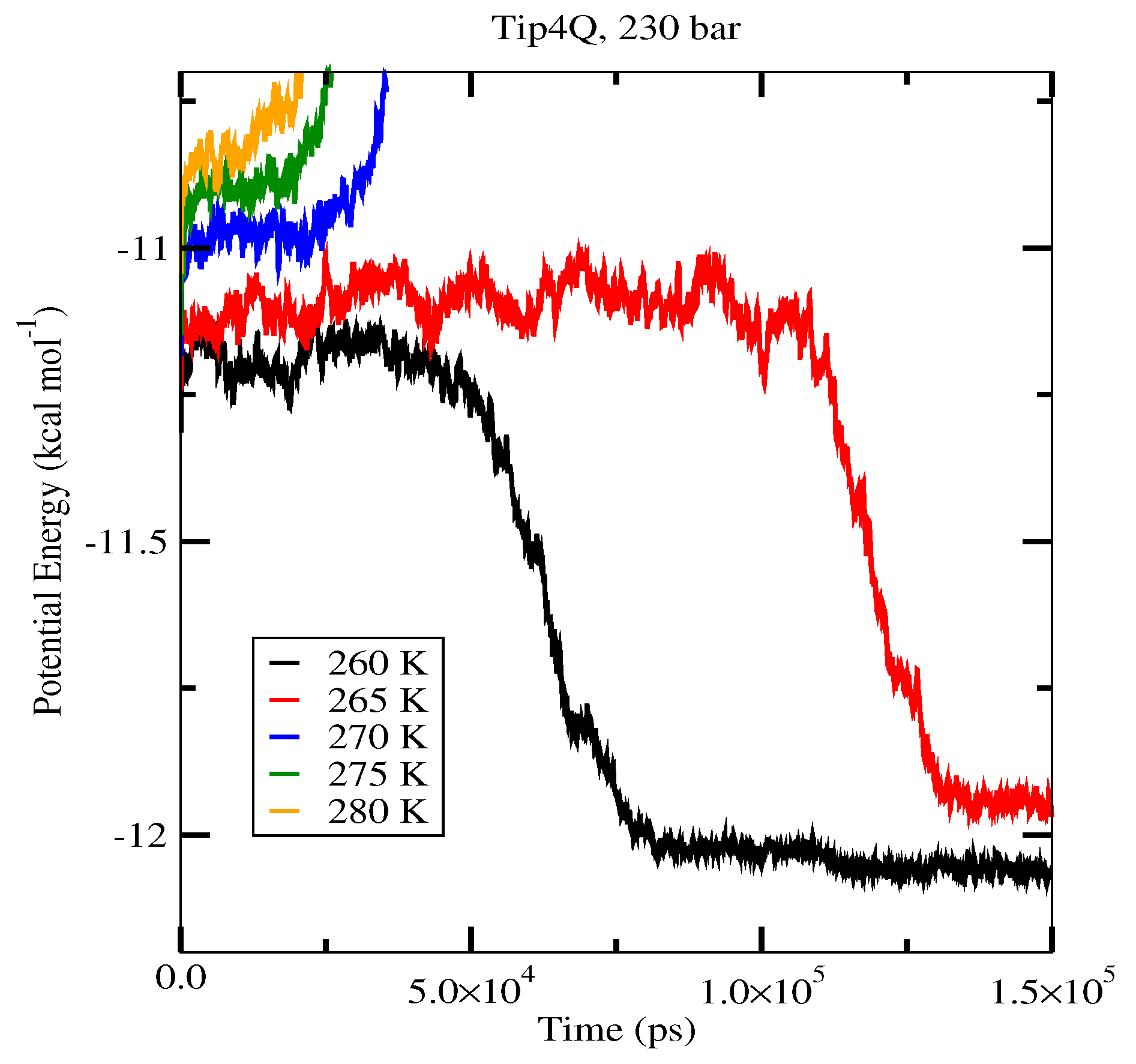
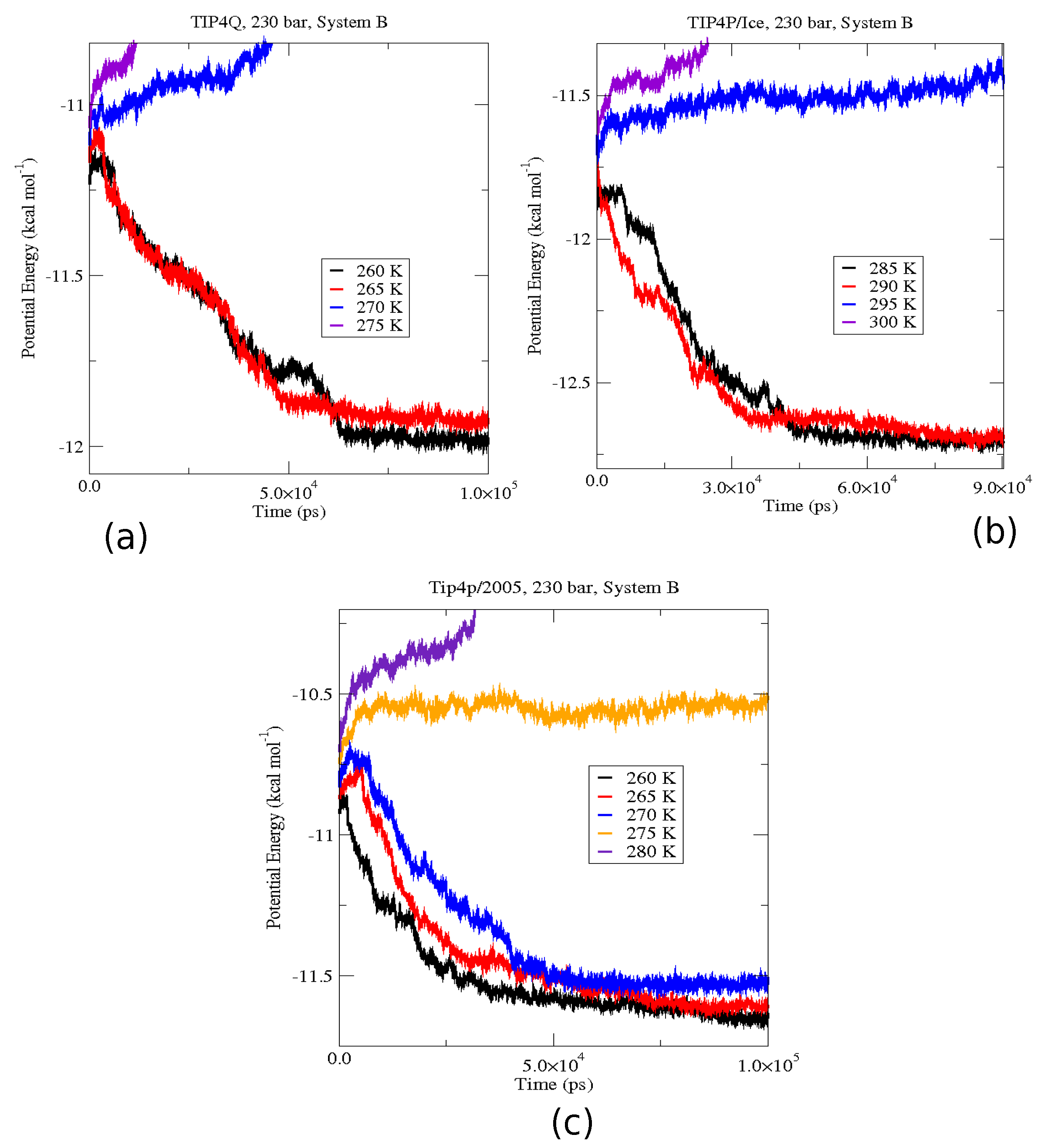
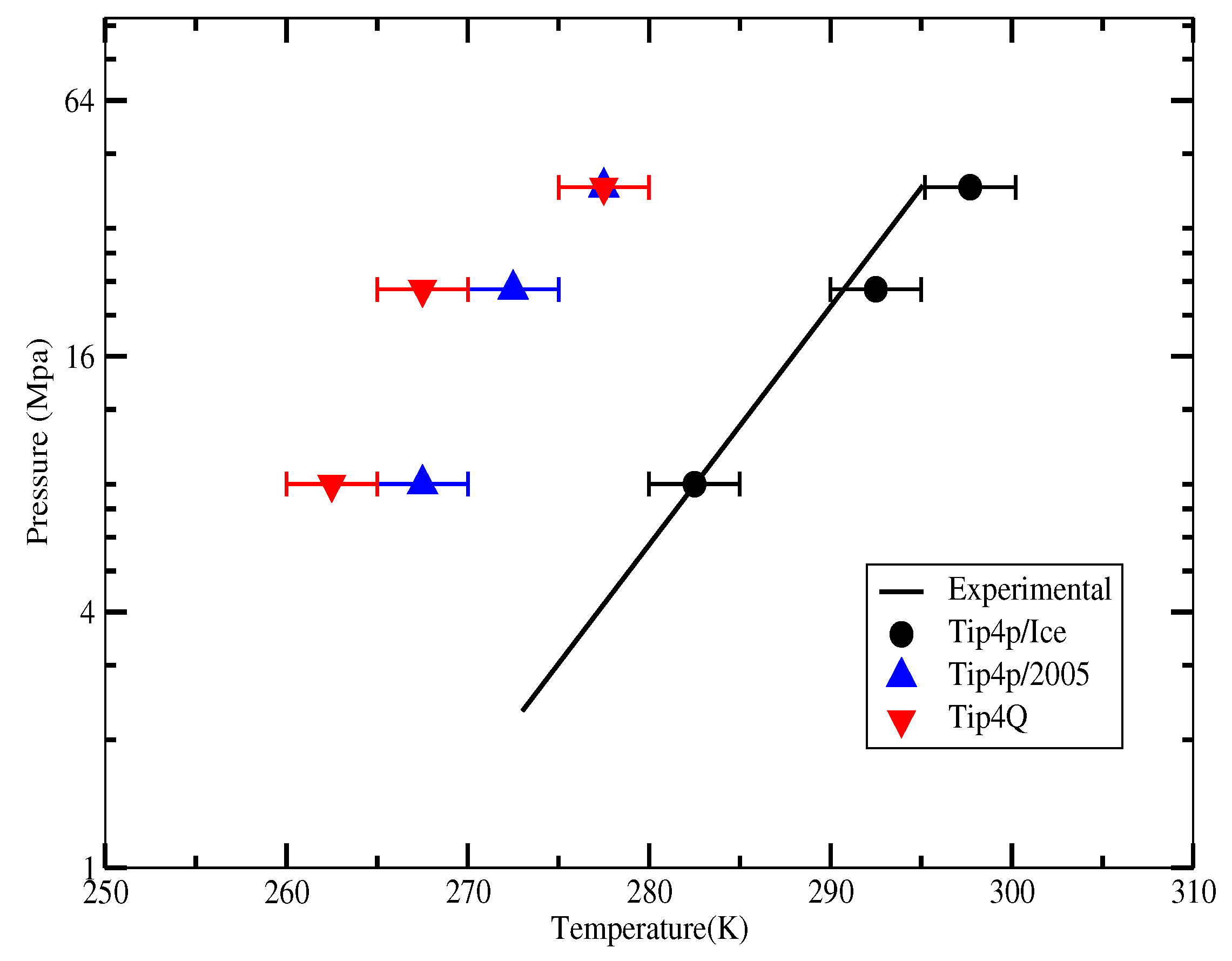
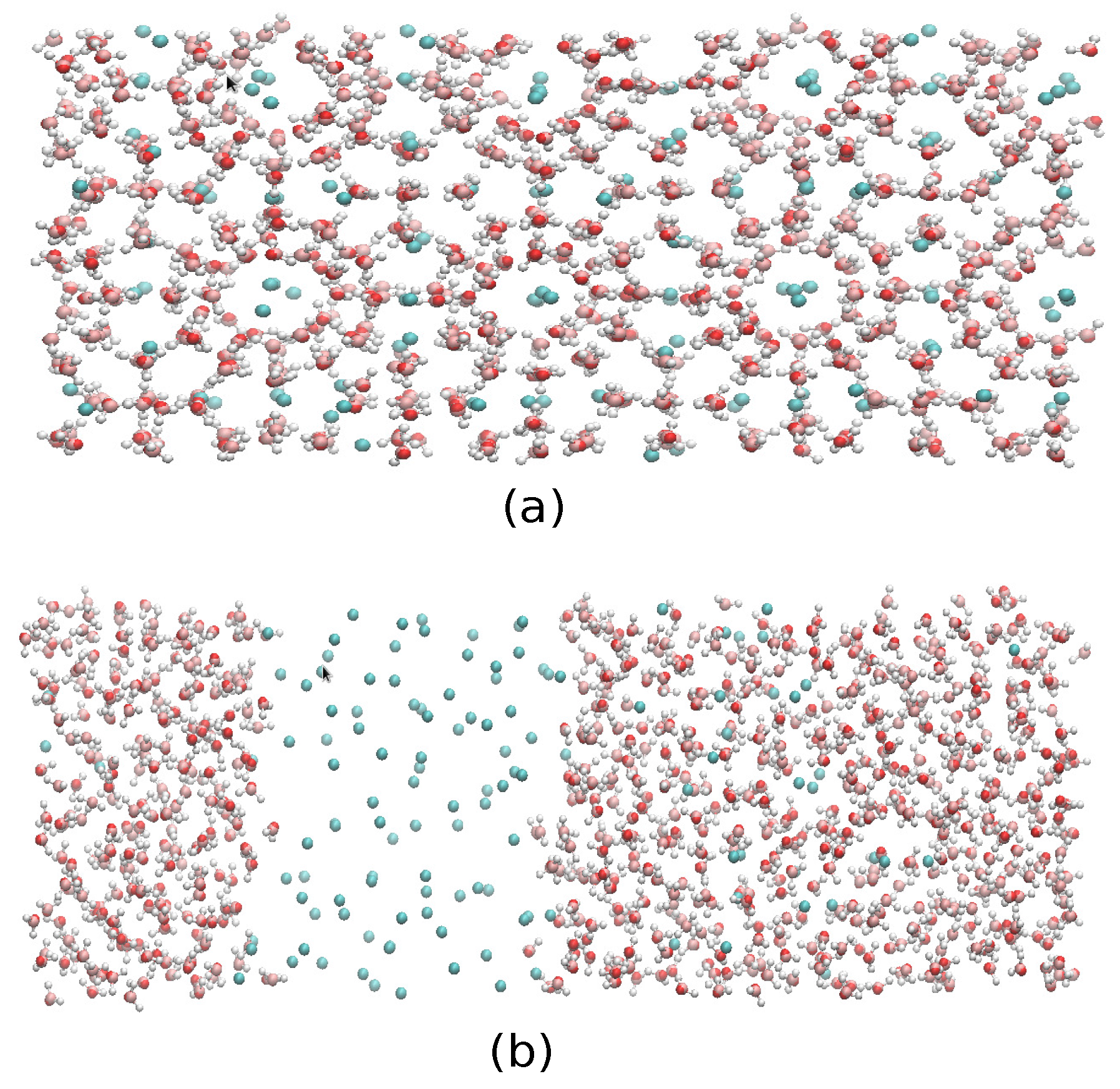
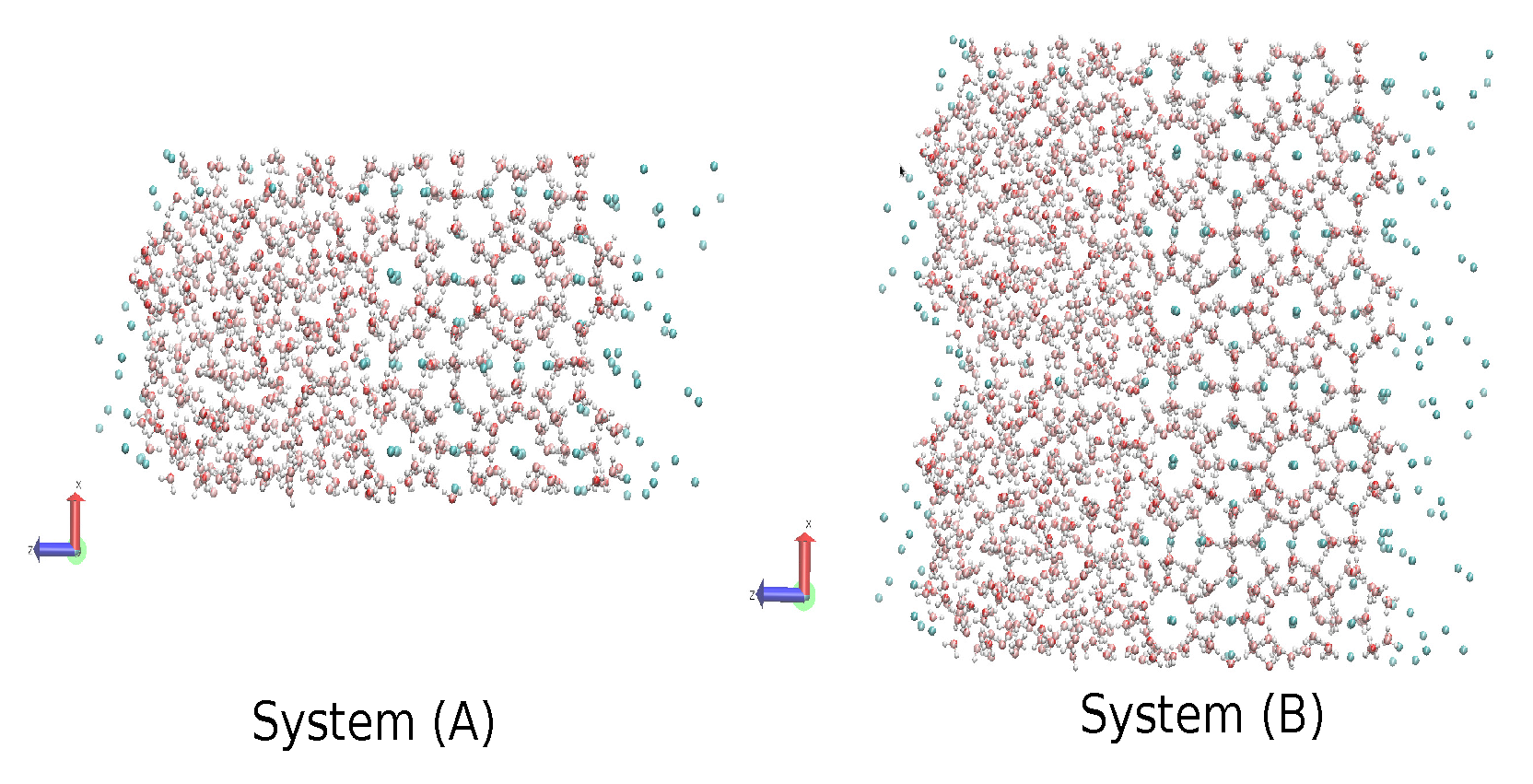
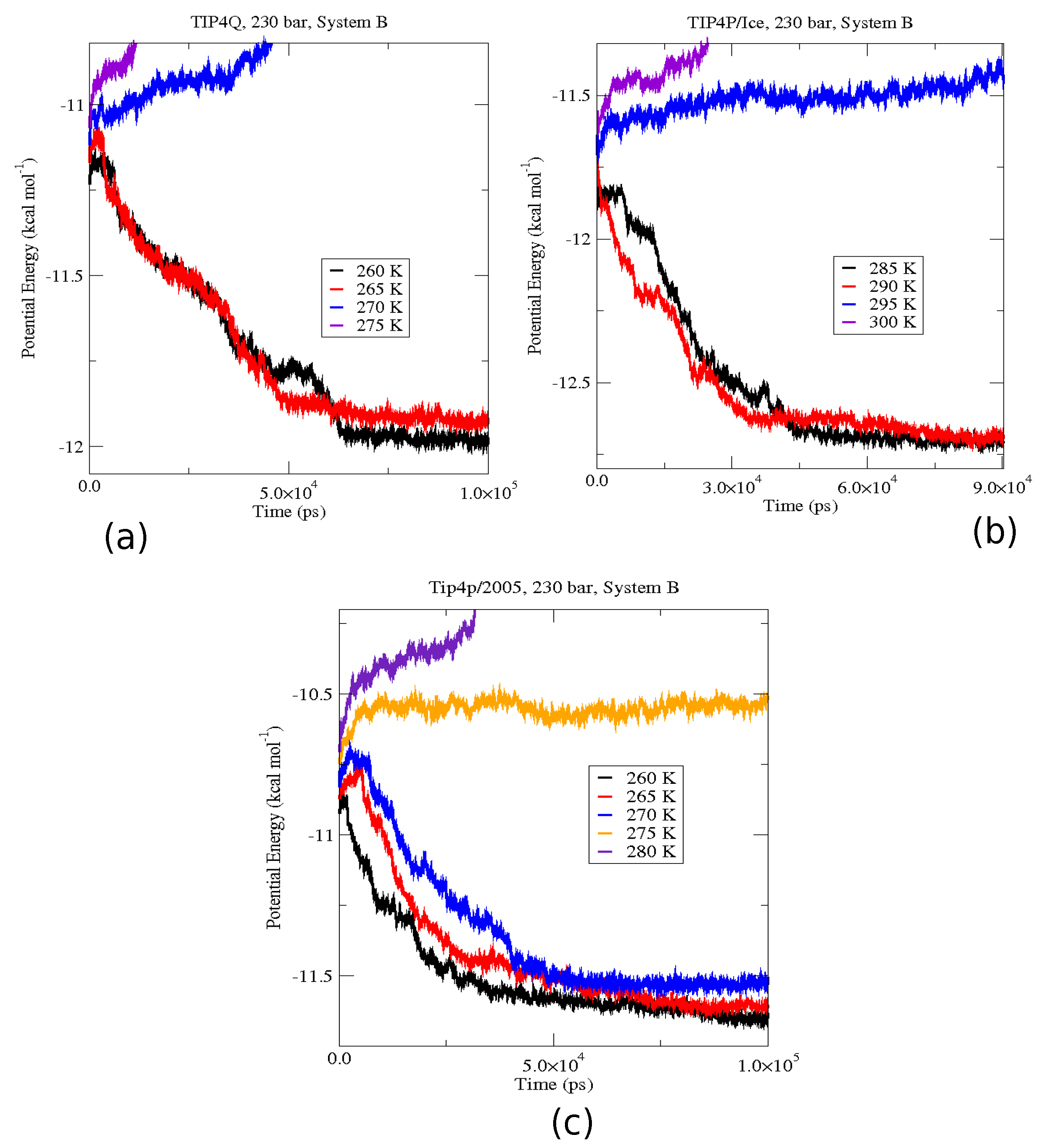
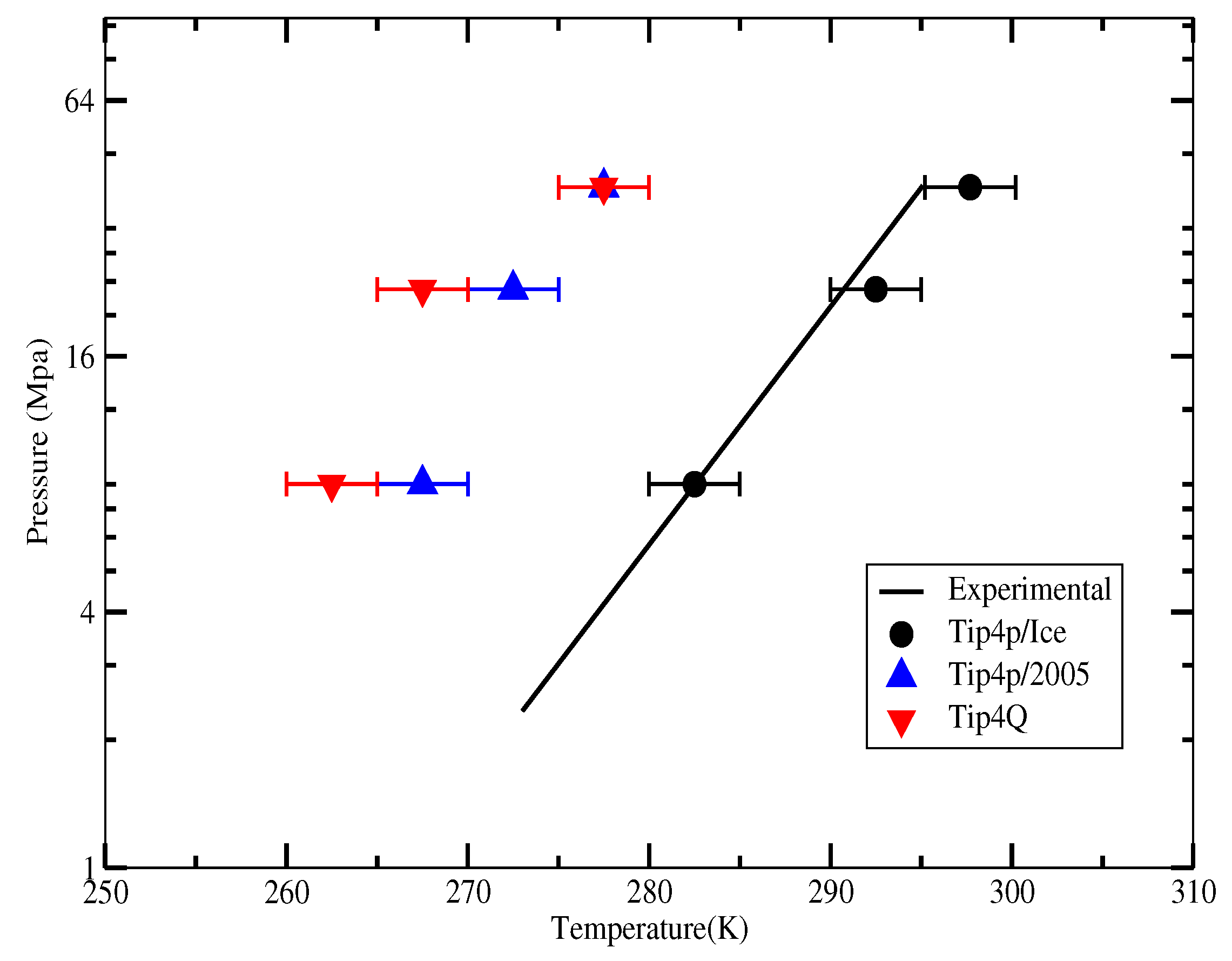
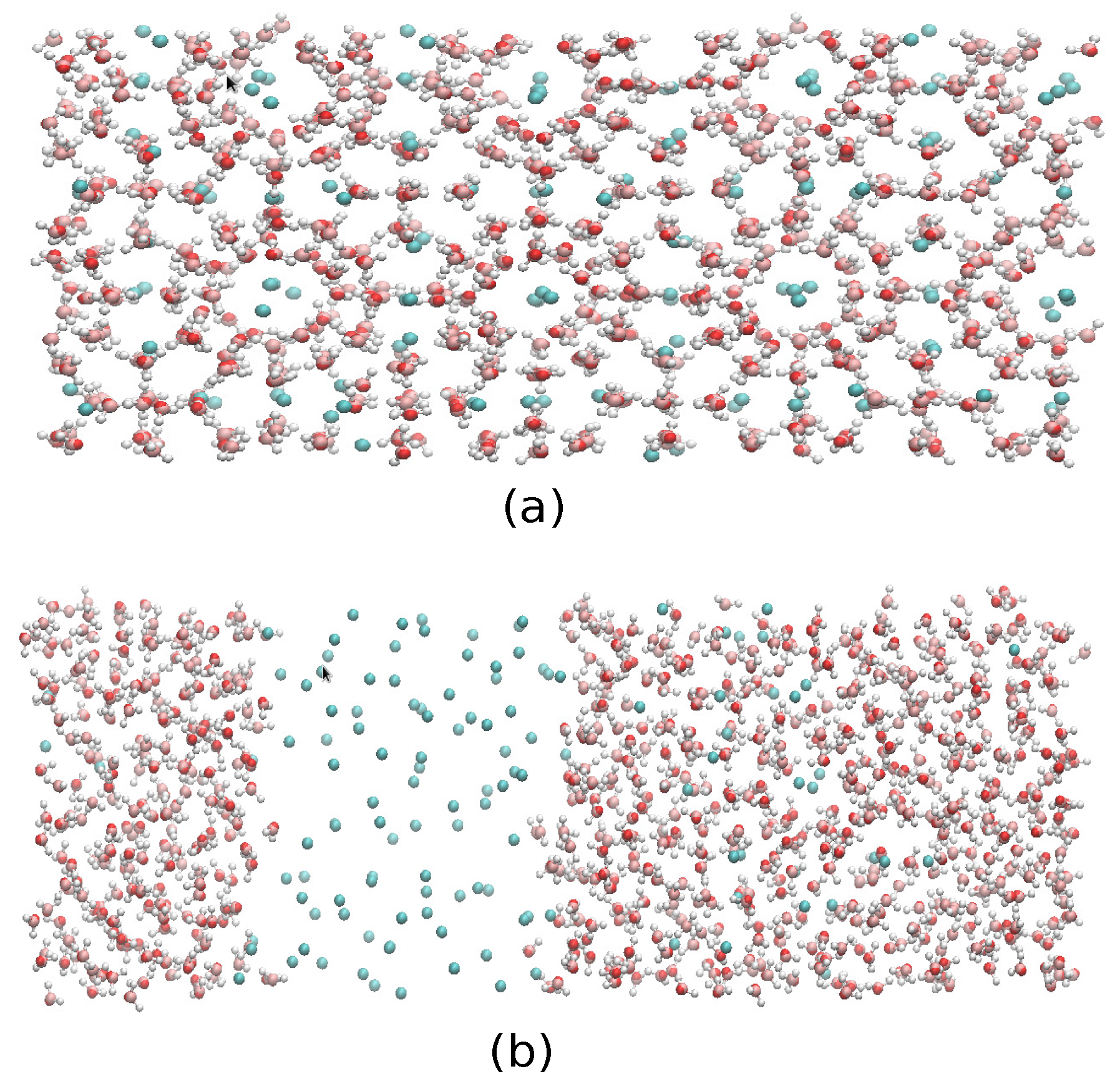
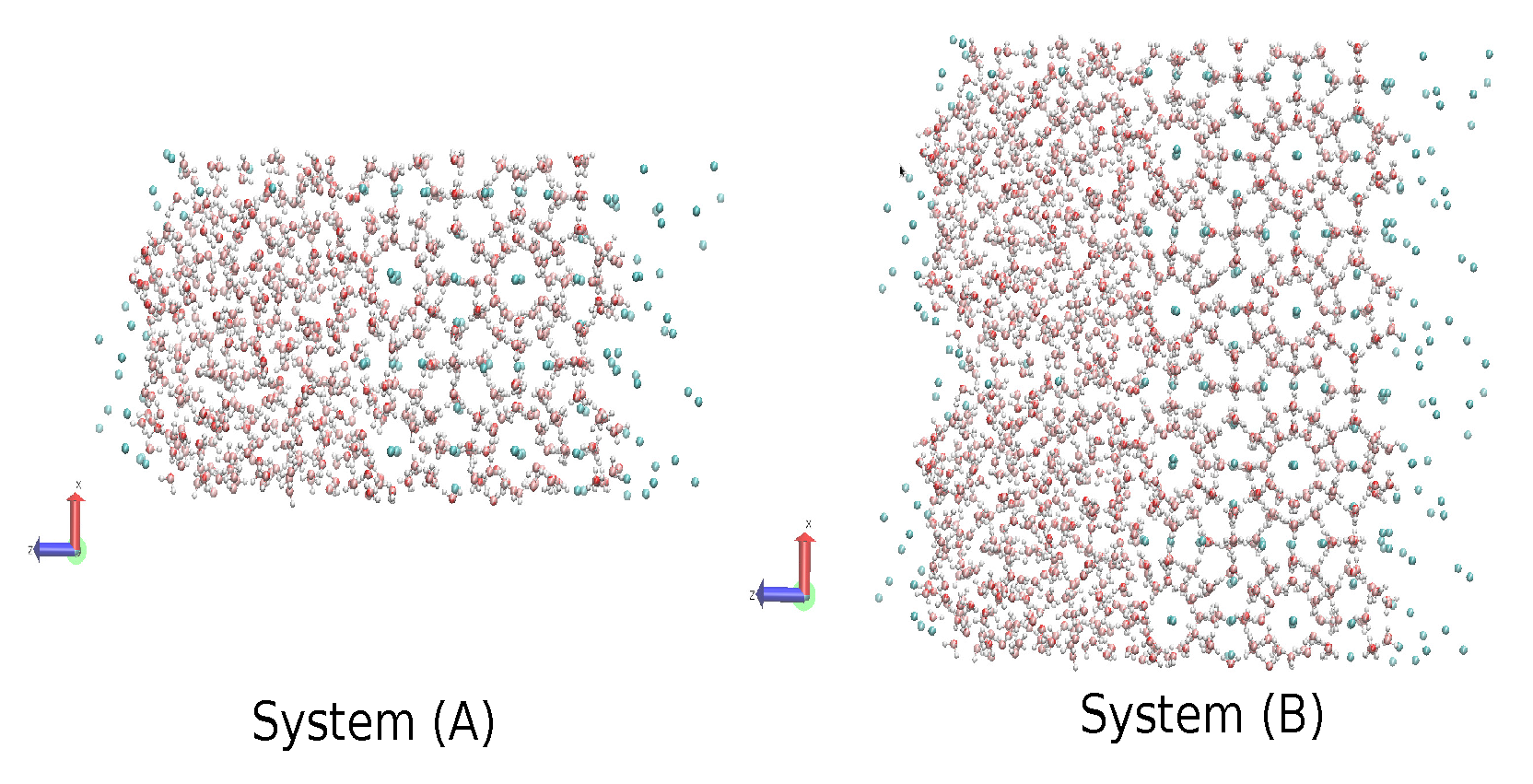
2.4. The Gas-Liquid-Hydrate Coexistence
3. Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sloan, E.D., Jr.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Koh, C.A. Towards a fundamental understanding of natural gas hydrates. Chem. Soc. Rev. 2002, 31, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Claussen, W.F. Suggested structures of water in inert gas hydrates. J. Chem. Phys. 1951, 19, 259–260. [Google Scholar] [CrossRef]
- Stackelberg, M.V.; Müller, H.R. Zur Struktur der Gashydrate. Naturwissenschaften 1951, 38, 456. [Google Scholar] [CrossRef]
- Gutt, C.; Asmussen, B.; Press, W.; Johnson, M.R.; Handa, Y.P.; Tse, J.S. The structure of deuterated methane-hydrate. J. Chem. Phys. 2000, 113, 4713–4721. [Google Scholar] [CrossRef]
- Kirchner, M.T.; Boese, R.; Billups, W.E.; Norman, L.R. Gas hydrate single-crystal structure analyses. J. Am. Chem. Soc. 2004, 126, 9407–9412. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.J.; Drew, M.G.B.; Rodger, P. Inhibition of crystal growth in methane hydrate. J. Chem. Soc. Faraday Trans. 1995, 91, 3449–3460. [Google Scholar] [CrossRef]
- Zangi, R.; Mark, A. Electrofreezing of confined water. J. Chem. Phys. 2004, 120, 7123–7130. [Google Scholar] [CrossRef] [PubMed]
- Girardi, M.; Figueiredo, W. Three-dimensional square water in the presence of an external electric field. J. Chem. Phys. 2006, 125, 094508. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Xiaobin, X.; Hong, Z.; Chuanxiang, X. Effects of dipole polarization of water molecules on ice formation under an electrostatic field. Cryobiology 2008, 56, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Orlowska, M.; Havet, M.; Le-Bail, A. Controlled ice nucleation under high voltage DC electrostatic field conditions. Food Res. Int. 2009, 42, 879–884. [Google Scholar] [CrossRef]
- Luis, D.P.; López-Lemus, J.; Mayorga, M.; Romero-Salazar, L. Performance of rigid water models in the phase transition of clathrates. Mol. Simul. 2010, 36, 35–40. [Google Scholar] [CrossRef]
- Luis, D.P.; López-Lemus, J.; Mayorga, M. Electrodissociation of clathrate-like structures. Mol. Simul. 2010, 36, 461–467. [Google Scholar] [CrossRef]
- Aragones, J.L.; MacDowell, L.G.; Siepmann, J.I.; Vega, C. Phase diagram of water under an applied electric field. Phys. Rev. Lett. 2011, 107, 155702. [Google Scholar] [CrossRef] [PubMed]
- Luis, D.P.; Herrera-Hernández, E.C.; Saint-Martin, H. A theoretical study of the dissociation of the sI methane hydrate induced by an external electric field. J. Chem. Phys. 2015, 143, 204503. [Google Scholar] [CrossRef] [PubMed]
- English, N.J.; MacElroy, J.M.D. Theoretical studies of the kinetics of methane hydrate crystallization in external electromagnetic fields. J. Chem. Phys. 2004, 120, 10247–10256. [Google Scholar] [CrossRef] [PubMed]
- Myshakin, E.M.; Jiag, H.; Warzinski, R.P.; Jordan, K.D. Molecular dynamics simulations of methane hydrate decomposition. J. Phys. Chem. A 2009, 113, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- English, N.J.; Johnson, J.K.; Taylor, C.E. Molecular-dynamics simulations of methane hydrate dissociation. J. Chem. Phys. 2005, 123, 244503. [Google Scholar] [CrossRef] [PubMed]
- Yagasaki, T.; Matsumoto, M.; Andoh, Y.; Okazaki, S.; Tanaka, H. Dissociation of methane hydrate in aqueous NaCl solutions. J. Phys. Chem. B 2014, 118, 11797–11804. [Google Scholar] [CrossRef] [PubMed]
- Báez, L.A.; Clancy, P. Computer-simulation of the crystal-growth and dissolution of natural-gas hydrates. Ann. N. Y. Acad. Sci. 1994, 715, 177–186. [Google Scholar] [CrossRef]
- Walsh, M.R.; Koh, C.A.; Sloan, E.D., Jr.; Sum, A.K.; Wu, D.T. Microsecond simulations of spontaneous methane hydrate nucleation and growth. Science 2009, 326, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Abascal, J.L.F.; Sanz, E.; García Fernández, R.; Vega, C. A potential model for the study of ices and amorphous water: TIP4P/Ice. J. Chem. Phys. 2005, 122, 234511. [Google Scholar] [CrossRef] [PubMed]
- Mastny, E.A.; Miller, C.A.; Pablo, J.J. The effect of the water/methane interface on methane hydrate cages: The potential of mean force and cage lifetimes. J. Chem. Phys. 2008, 129, 034701. [Google Scholar] [CrossRef] [PubMed]
- English, N.J.; Clarke, E.T. Molecular dynamics study of CO2 hydrate dissociation: Fluctuation-dissipation and non-equilibrium analysis. J. Chem. Phys. 2013, 139, 094701. [Google Scholar] [CrossRef] [PubMed]
- Susilo, R.; Alavi, S.; Ripmeester, J.; Englezos, P. Tuning methane content in gas hydrates via thermodynamic modeling and molecular dynamics simulation. Fluid Phase Equilib. 2008, 263, 6–17. [Google Scholar] [CrossRef]
- Lauricella, M.; Meloni, S.; English, N.J.; Peters, B.; Ciccotti, G. Methane clathrate hydrate nucleation mechanism by advanced molecular simulations. J. Phys. Chem. C 2014, 118, 22847–22857. [Google Scholar] [CrossRef]
- Sarupria, S.; Debenedetti, P.G. Homogeneous nucleation of methane hydrate in microsecond molecular dynamics simulation. Phys. Chem. Lett. 2012, 3, 2942–2947. [Google Scholar] [CrossRef] [PubMed]
- Frank, H.S.; Evans, M.W. Free volume and entropy in condensed systems III. entropy in binary liquid mixtures; partial molal entropy in dilute solutions; structure and thermodynamics in aqueous electrolytes. J. Chem. Phys. 1945, 13, 507–532. [Google Scholar] [CrossRef]
- Silverstein, T.P. Hydrophobic solvation NOT via clathrate water cages. J. Chem. Educ. 2008, 85, 917–918. [Google Scholar] [CrossRef]
- Guillot, B.; Guissani, Y.; Bratos, S. A computer-simulation study of hydrophobic hydration of rare gases and of methane. I. Thermodynamic and structural properties. J. Chem. Phys. 1991, 95, 3643–3648. [Google Scholar] [CrossRef]
- Laaksonen, A.; Stilbs, P. Molecular dynamics and NMR study of methane-water. Mol. Phys. 1991, 74, 747–764. [Google Scholar] [CrossRef]
- Griffith, J.H.; Scheraga, H.A. Statistical thermodynamics of aqueous solutions. I. Water structure, solutions with non-polar solutes, and hydrophobic interactions. J. Mol. Struct. 2004, 682, 97–113. [Google Scholar] [CrossRef]
- Guo, G.J.; Zhang, Y.G.; Li, M.; Wu, C.H. Can the dodecahedral water cluster naturally form in methane aqueous solutions? A molecular dynamics study on the hydrate nucleation mechanisms. J. Chem. Phys. 2008, 128, 194504. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.A.; Wisbey, R.P.; Wu, X.; Westacott, R.E. Water ordering around methane during hydrate formation. J. Chem. Phys. 2000, 113, 6390–6397. [Google Scholar] [CrossRef]
- Buchanan, P.; Aldiwan, N.; Soper, A.K.; Creek, J.L.; Koh, C.A. Decreased structure on dissolving methane in water. Chem. Phys. Lett. 2005, 415, 89–93. [Google Scholar] [CrossRef]
- Dec, S.F.; Bowler, K.E.; Stadterman, L.L.; Koh, C.A.; Sloan, E.D., Jr. Direct Measure of the hydration number of aqueous methane. J. Am. Chem. Soc. 2006, 128, 414–415. [Google Scholar] [CrossRef] [PubMed]
- Abascal, J.L.F.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. [Google Scholar] [CrossRef] [PubMed]
- Alejandre, J.; Chapela, G.A.; Saint-Martin, H.; Mendoza, N. A non-polarizable model of water that yields the dielectric constant and the density anomalies of the liquid: TIP4Q. Phys. Chem. Chem. Phys. 2011, 13, 19728–19740. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Economou, I.G.; Stubos, A.K. Influence of combining rules on the cavity occupancy of clathrate hydrates by Monte Carlo simulations. Mol. Phys. 2014, 112, 2258–2274. [Google Scholar]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Docherty, H.; Galindo, A.; Vega, C.; Sanz, E. A potential model for methane in water describing correctly the solubility of the gas and the properties of the methane hydrate. J. Chem. Phys. 2006, 125, 074510. [Google Scholar] [CrossRef] [PubMed]
- Conde, M.M.; Vega, C. Determining the three-phase coexistence line in methane hydrates using computer simulations. J. Chem. Phys. 2010, 133, 064507. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.; Thomsen, K.; von Solms, N.; Wierzchowski, S.; Walsh, M.R.; Koh, C.A.; Sloan, E.D.; Wu, D.T.; Sum, A.K. Calculation of liquid water-hydrate-methane vapor phase equilibria from molecular simulations. J. Phys. Chem. B 2010, 114, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Michalis, V.K.; Costandy, J.; Tsimpanogiannis, I.N.; Economou, I.G. Prediction of the phase equilibria of methane hydrates using the direct phase coexistence methodology. J. Chem. Phys. 2015, 142, 044501. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Cho, S.; Lee, H.; Lee, C. Measurement and prediction of phase equilibria for water + methane in hydrate forming conditions. Fluid Phase Equilibr. 2001, 185, 53–63. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Widom, B. Some Topics in the Theory of Fluids. J. Chem. Phys. 1963, 39, 2808–2812. [Google Scholar] [CrossRef]
- Tung, Y.T.; Chen, L.J.; Chen, Y.P.; Lin, S.T. The growth of structure I methane hydrate from molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 10804–10813. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.R.; Rainey, J.D.; Lafond, P.G.; Park, D.H.; Beckham, G.T.; Jones, M.D.; Lee, K.H.; Koh, C.A.; Sloan, E.D., Jr.; Wu, D.T.; et al. The cages, dynamics, and structuring of incipient methane clathrate hydrates. Phys. Chem. Chem. Phys. 2011, 13, 19951–19959. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, G.S.; Stegailov, V.V. Melting and superheating of sI methane hydrate: Molecular dynamics study. J. Chem. Phys. 2012, 136, 044523. [Google Scholar] [CrossRef] [PubMed]
- Flyvbjerg, H.; Petersen, H.G. Error estimates on averages of correlated data. J. Chem. Phys. 1989, 91, 461–466. [Google Scholar] [CrossRef]
- English, N.J.; Phelan, G.M. Molecular dynamics study of thermal-driven methane hydrate dissociation. J. Chem. Phys. 2009, 131, 074704. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.R.; Sanz, E.; Valeriani, C.; Vega, C. On fluid-solid direct coexistence simulations: The pseudo-hard sphere model. J. Chem. Phys. 2013, 139, 144502. [Google Scholar] [CrossRef] [PubMed]
- Alavi, S.; Ripmeester, J.A. Nonequilibrium adiabatic molecular dynamics simulations of methane clathrate hydrate decomposition. J. Chem. Phys. 2010, 132, 144703. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunnen, R. Gromacs: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Mark, A. Calculation of the free energy of solvation for neutral analogs of amino acid side chains. J. Comput. Chem. 2002, 23, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Pitera, J.W.; Swope, W.C.; Pande, V.S. Extremely precise free energy calculations of amino acid side chain analogs: Comparison of common molecular mechanics force fields for proteins. J. Chem. Phys. 2003, 119, 5740. [Google Scholar] [CrossRef]
- Pohorille, A.; Jarzynski, C.; Chipot, C. Good practices in free-energy calculations. J. Phys. Chem. B 2010, 114, 10235–10253. [Google Scholar] [CrossRef] [PubMed]
- Christ, C.D.; Mark, A.E.; van Gunsteren, W.F. Basic ingredients of free energy calculations: A review. J. Comput. Chem. 2010, 31, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Apol, E.; Apostolov, R.; Berendsen, H.J.; van Buuren, A.; Bjelkmar, P.; van Drunen, R.; Feenstra, A.; Groenhof, G.; Kasson, P.; Larsson, P.; et al. GROMACS User Manual. Available online: http://www.gromacs.org (accessed on 15 September 2015).
- McMullan, R.K.; Jeffrey, G.A. Polyhedral clathrate hydrates. IX. Structure of ethylene oxide hydrate. J. Chem. Phys. Chem. Phys. 1965, 42, 2725–2732. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics method. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. 1985, A31, 1696–1697. [Google Scholar] [CrossRef]
- Fernández, R.G.; Abascal, J.L.; Vega, C. The melting point of ice Ih for common water models calculated from direct coexistence of the solid-liquid interface. J. Chem. Phys. 2006, 124, 144506. [Google Scholar] [CrossRef] [PubMed]
- English, N.J.; MacElroy, J. Perspectives on molecular simulation of clathrate hydrates: Progress, prospects and challenges. Chem. Eng. Sci. 2015, 121, 133–156. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Water Model | OPLS-AA | UA | Corrected UA |
|---|---|---|---|
| TIP4P/2005 | 2005-1 | 2005-2 | 2005-3 |
| TIP4P/Ice | Ice-1 | Ice-2 | Ice-3 |
| TIP4Q | Q-1 | Q-2 | Q-3 |
| T/K | TIP4Q | TIP4P/2005 | TIP4P/Ice | Expt. |
|---|---|---|---|---|
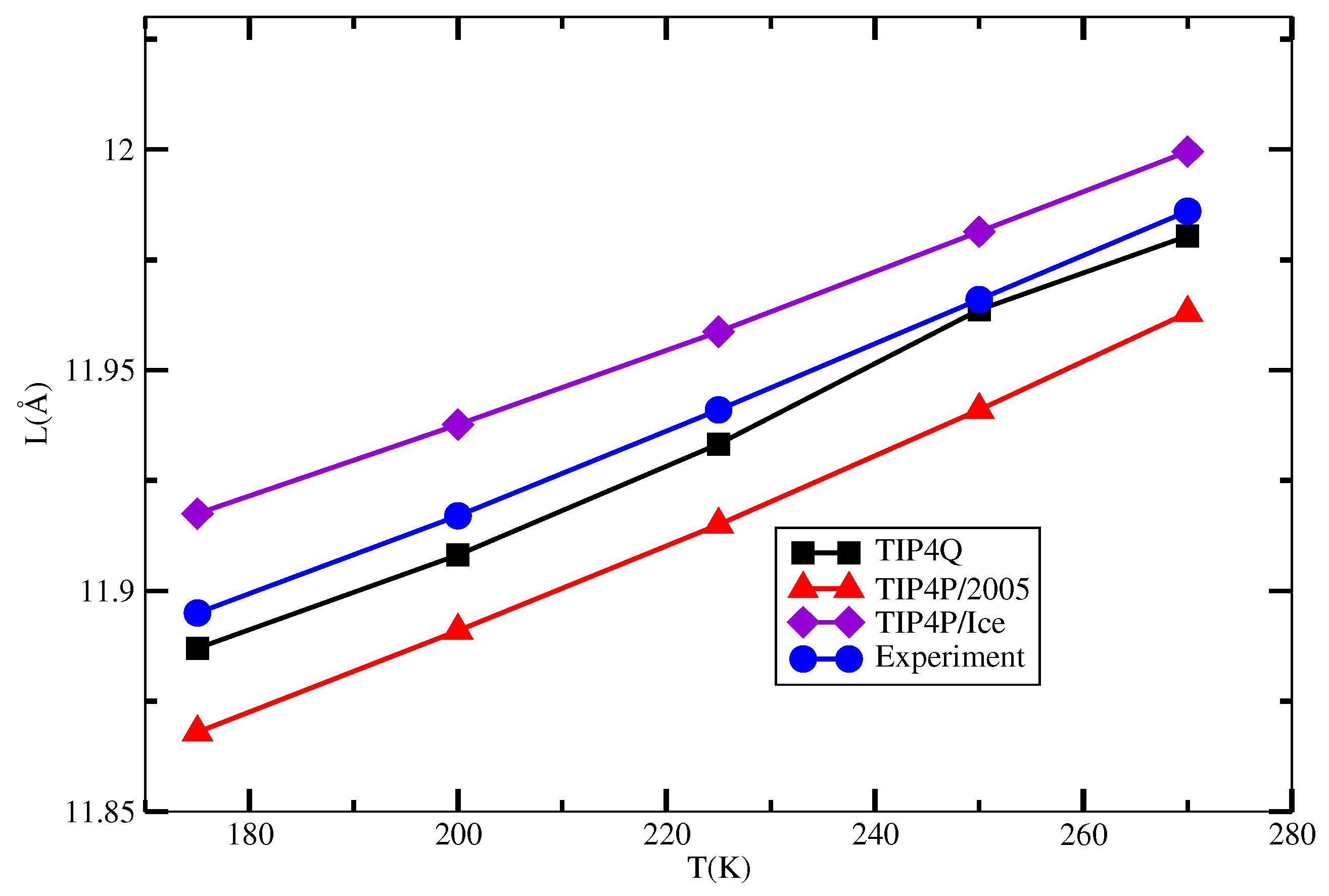
| 175 | 11.887 | 11.868 | 11.917 | 11.895 |
| 200 | 11.908 | 11.891 | 11.937 | 11.917 |
| 225 | 11.933 | 11.915 | 11.958 | 11.941 |
| 250 | 11.964 | 11.941 | 11.981 | 11.966 |
| 270 | 11.980 | 11.963 | 11.999 | 11.986 |
| Pressure/Bar. | TIP4Q | TIP4P/2005 | TIP4P/Ice |
|---|---|---|---|
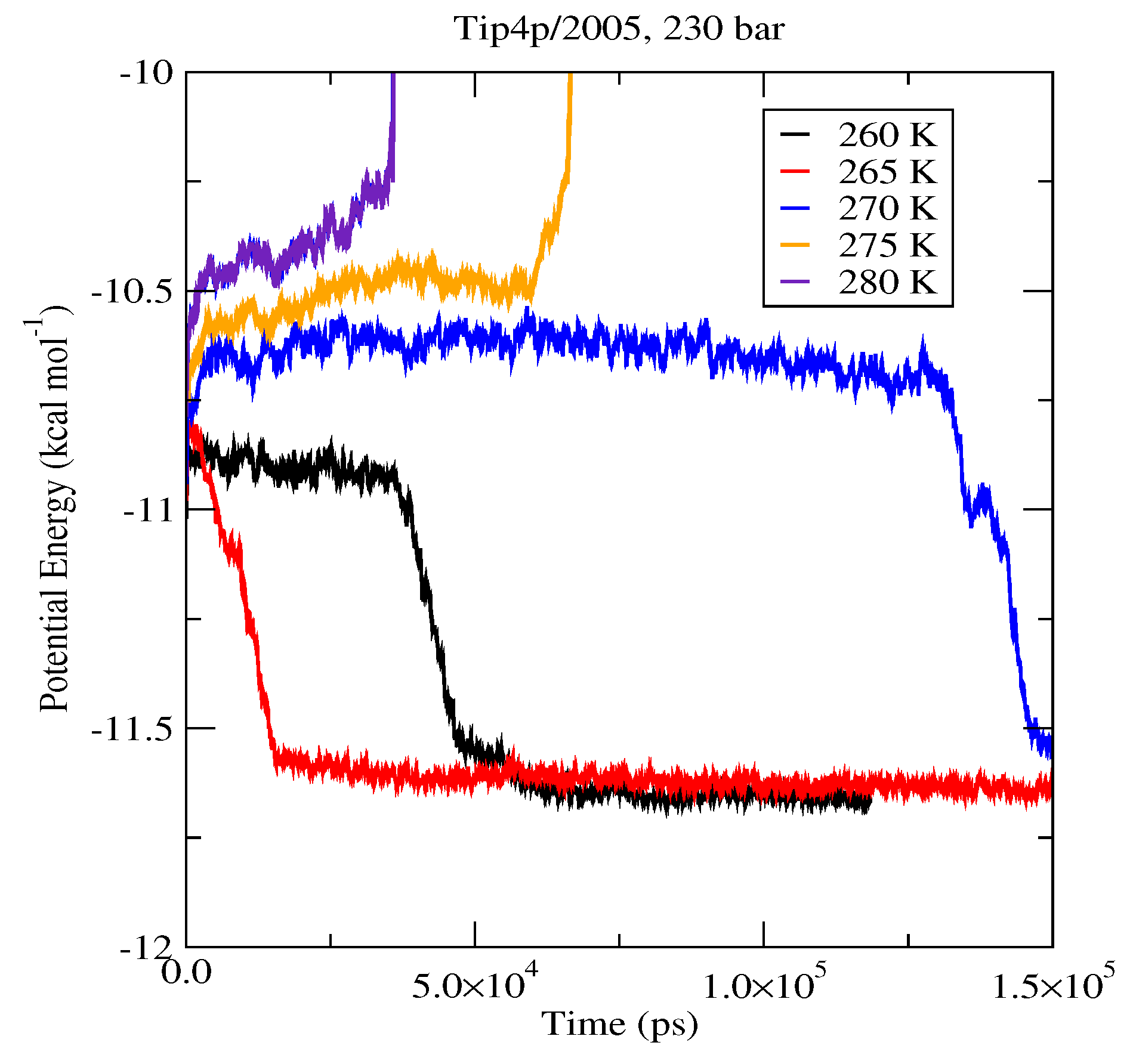
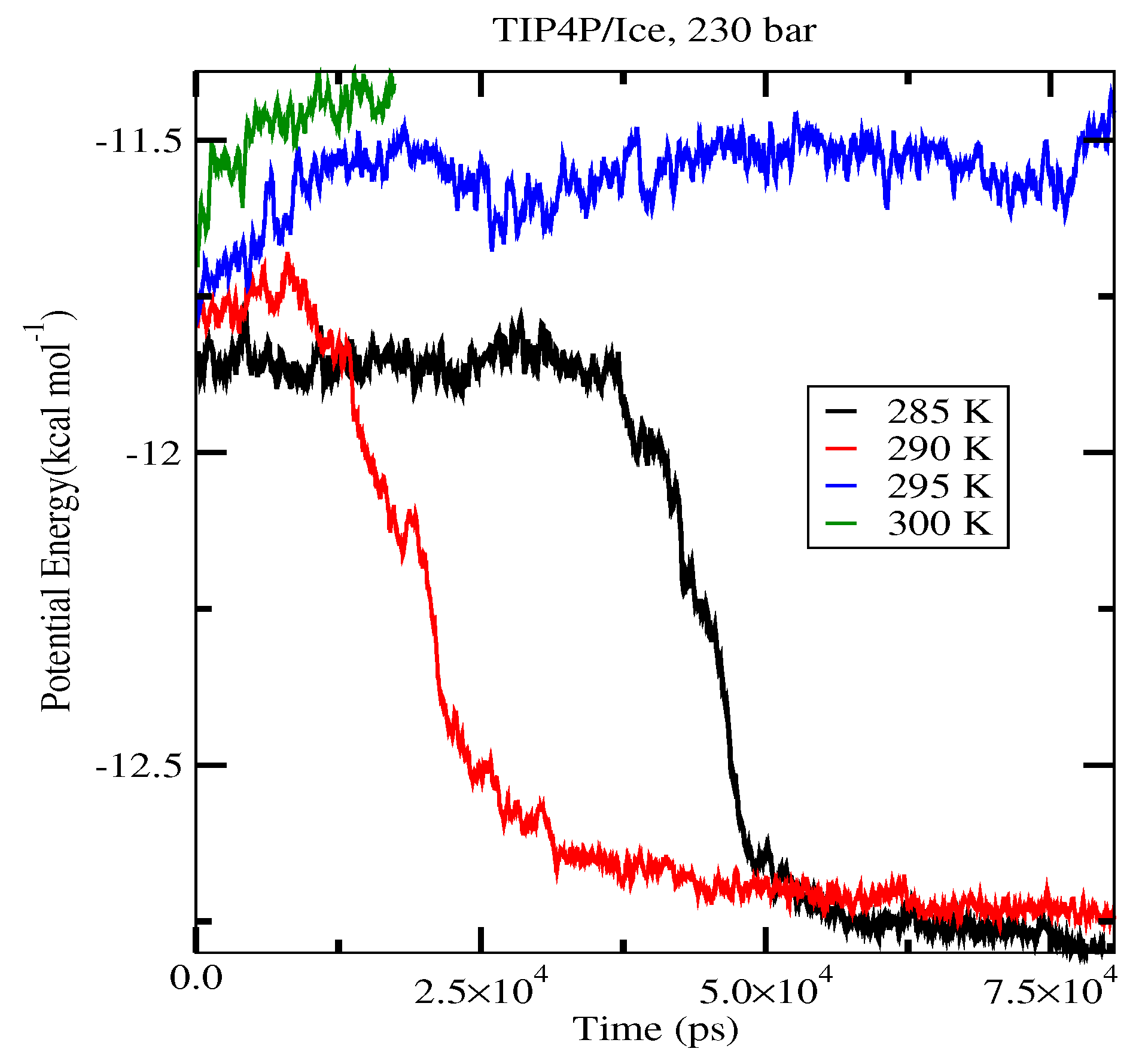
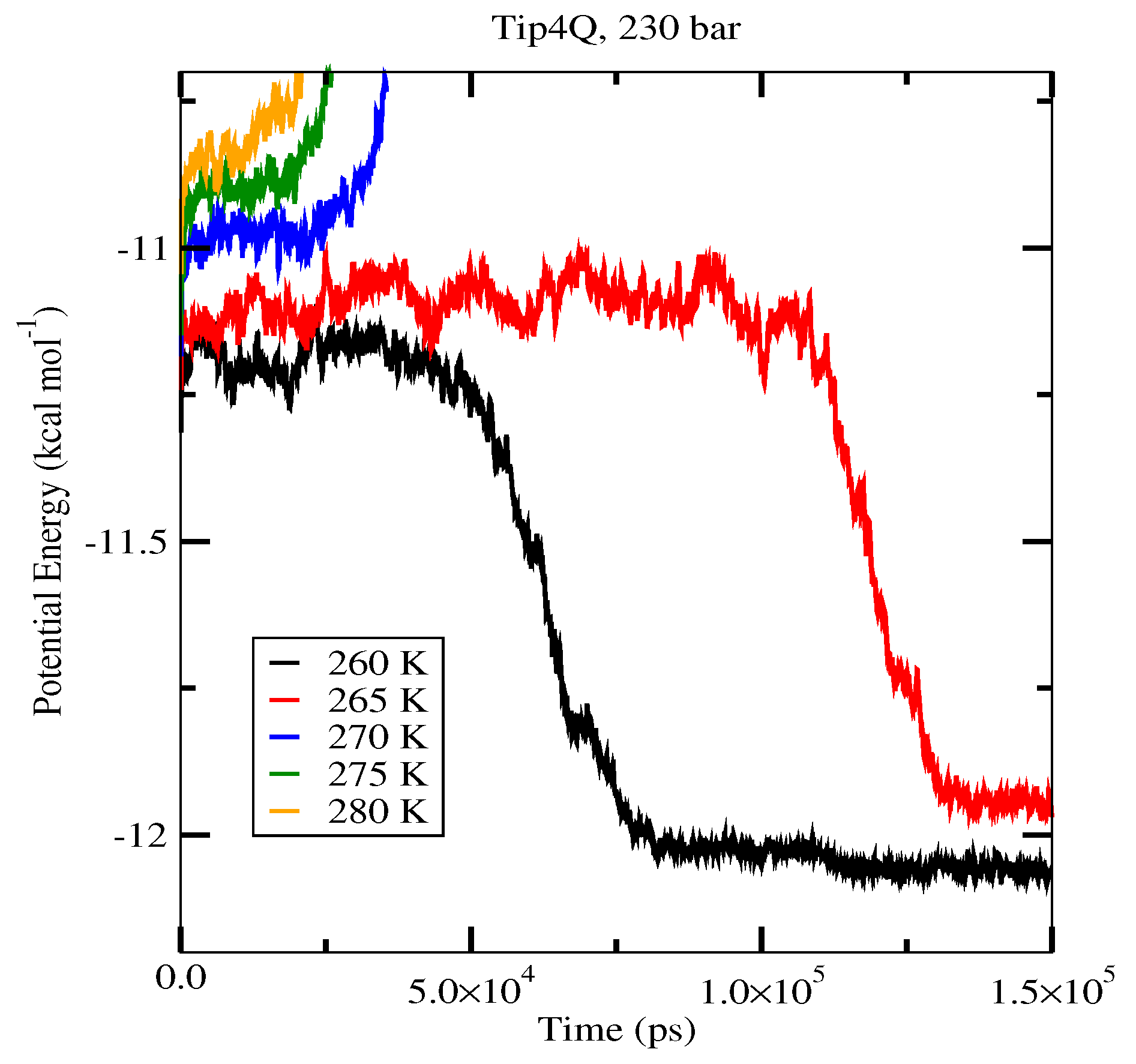
| 80 (System A) | 262.5 K | 267.5 K | 282.5 K |
| 230 (System A) | 267.5 K | 272.5 K | 292.5 K |
| 230 (System B) | 267.5 K | 272.5 K | 292.5 K |
| 400 (System A) | 277.5 K | 277.5 K | 297.5 K |
| Model | σ/Å | (ϵ/R)/K | qH/e | qO/e | lOM/Å |
|---|---|---|---|---|---|
| TIP4P/2005 | 3.1589 | 93.2 | 0.5564 | 0.0 | 0.1546 |
| TIP4P/Ice | 3.1668 | 106.1 | 0.5897 | 0.0 | 0.1577 |
| TIP4Q | 3.1666 | 93.2 | 0.5250 | 0.5 | 0.0690 |
| Model | σ/Å | (ϵ/R)/K | q/e | rCH/Å |
|---|---|---|---|---|
| OPLS-AA C | 3.50 | 33.2123 | −0.240 | 1.094760 |
| OPLS-AA H | 2.50 | 15.0965 | 0.060 | |
| UA CH4 | 3.73 | 147.5 | 0.0 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luis, D.P.; García-González, A.; Saint-Martin, H. A Theoretical Study of the Hydration of Methane, from the Aqueous Solution to the sI Hydrate-Liquid Water-Gas Coexistence. Int. J. Mol. Sci. 2016, 17, 378. https://doi.org/10.3390/ijms17060378
Luis DP, García-González A, Saint-Martin H. A Theoretical Study of the Hydration of Methane, from the Aqueous Solution to the sI Hydrate-Liquid Water-Gas Coexistence. International Journal of Molecular Sciences. 2016; 17(6):378. https://doi.org/10.3390/ijms17060378
Chicago/Turabian StyleLuis, Daniel Porfirio, Alcione García-González, and Humberto Saint-Martin. 2016. "A Theoretical Study of the Hydration of Methane, from the Aqueous Solution to the sI Hydrate-Liquid Water-Gas Coexistence" International Journal of Molecular Sciences 17, no. 6: 378. https://doi.org/10.3390/ijms17060378