
Pathogenetic and Therapeutic Applications of Tumor Necrosis Factor-α (TNF-α) in Major Depressive Disorder: A Systematic Review


Abstract
:1. Introduction
2. Tumor Necrosis Factor (TNF)-α and the Central Nervous System (CNS)
2.1. TNF-α and Signaling Pathways
2.2. Production of TNF-α within CNS
2.3. Brain Signaling by Peripherally Produced TNF-α
3. Evidence from Animal and Human Studies
3.1. Animal Models
3.2. Clinical Studies
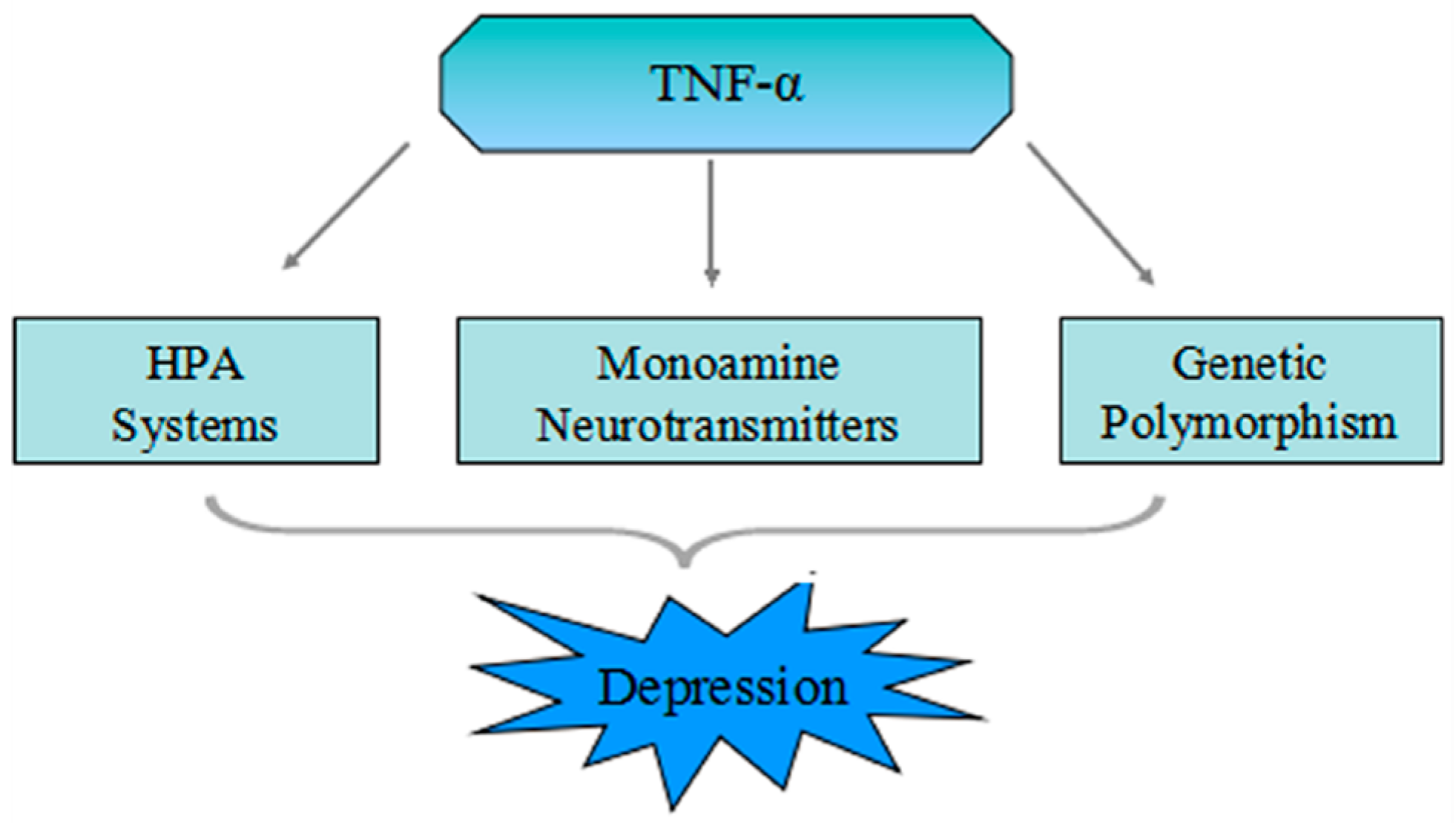
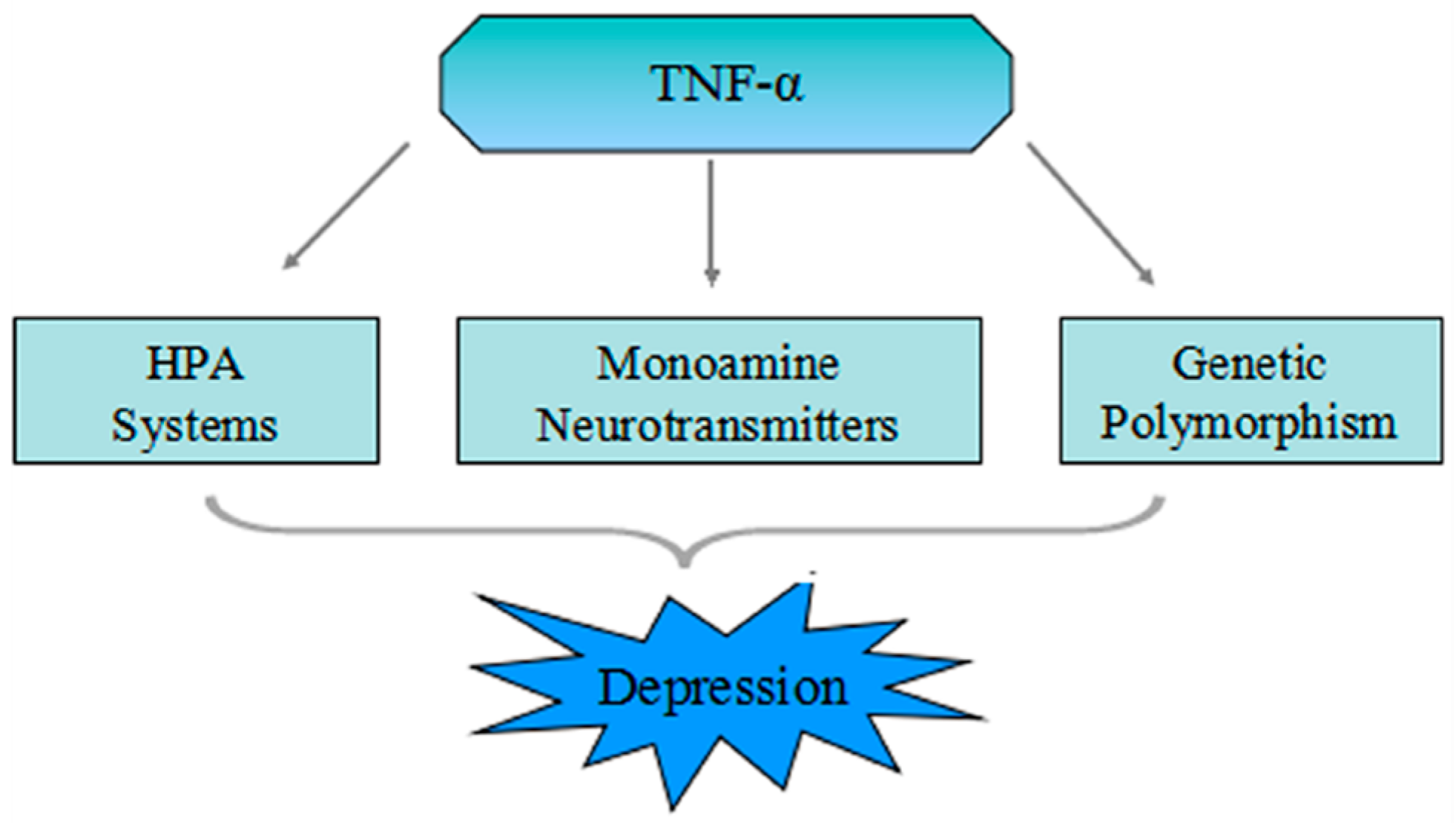
4. The Pathophysiologic Role of TNF-α in Depression
4.1. Mutual Influence of the TNF-α and HPA System
4.2. TNF-α Increases Activation of the Neurotransmitter Transporter
4.3. TNF-α Stimulates the Indoleamine 2,3-Dioxygenase (IDO)
5. The Genetic Polymorphism of TNF-α in MDD
5.1. The Genetic Polymorphism of TNF-α
5.2. Association of TNF-α Genetic Polymorphism in Major Depressive Disorder (MDD)
6. The Therapeutic Implications of TNF-α in MDD
6.1. The Potential Benefits of TNF-α Antagonists on Depression
6.2. The Effect of Antidepressant Medication Treatment on the Level of TNF-α
7. TNF-α and MDD with Autoimmune Diseases
8. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roiser, J.P.; Elliott, R.; Sahakian, B.J. Cognitive mechanisms of treatment in depression. Neuropsychopharmacology 2012, 37, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Waraich, P.; Goldner, E.M.; Somers, J.M.; Hsu, L. Prevalence and incidence studies of mood disorders: A systematic review of the literature. Can. J. Psychiatry. Revue Canadienne de Psychiatrie 2004, 49, 124–138. [Google Scholar] [PubMed]
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25. [Google Scholar] [CrossRef]
- Moller, H.J. Suicide, suicidality and suicide prevention in affective disorders. Acta Psychiatr. Scand. 2003, 108 (Suppl. S418), 73–80. [Google Scholar] [CrossRef]
- Labermaier, C.; Masana, M.; Muller, M.B. Biomarkers predicting antidepressant treatment response: How can we advance the field? Dis. Markers 2013, 35, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [PubMed]
- Postal, M.; Appenzeller, S. The importance of cytokines and autoantibodies in depression. Autoimmun. Rev. 2015, 14, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Consortium, C. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015, 523, 588–591. [Google Scholar]
- Smith, R.S. The macrophage theory of depression. Med. Hypotheses 1991, 35, 298–306. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Hepgul, N.; Pariante, C.M. Inflammation and depression. Curr. Top. Behav. Neurosci. 2013, 14, 135–151. [Google Scholar] [PubMed]
- Liu, Y.; Ho, R.C.; Mak, A. Interleukin (IL)-6, tumour necrosis factor α (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: A meta-analysis and meta-regression. J. Affect. Disord. 2012, 139, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Miller, A.H. Cytokines and psychopathology: Lessons from interferon-α. Biol. Psychiatry 2004, 56, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Pollak, Y.; Yirmiya, R. Cytokine-induced changes in mood and behaviour: Implications for “depression due to a general medical condition”, immunotherapy and antidepressive treatment. Int. J. Neuropsychopharmacol. 2002, 5, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Na, K.S.; Shin, K.H.; Jung, H.Y.; Choi, S.H.; Kim, J.B. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Oehadian, A.; Koide, N.; Mu, M.M.; Hassan, F.; Islam, S.; Yoshida, T.; Yokochi, T. Interferon (IFN)-β induces apoptotic cell death in DHL-4 diffuse large B cell lymphoma cells through tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Cancer Lett. 2005, 225, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Ohnishi, M.; Pawankar, R. IL-1 and TNF-α-mediated regulation of IL-6, IL-8, and GM-CSF release from cultured nasal epithelial cells. Nihon Jibiinkoka Gakkai Kaiho 2000, 103, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Ragab, S.M.; Safan, M.A.; Obeid, O.M.; Sherief, A.S. Lipoprotein-associated phospholipase A2 (Lp-PLA2) and tumor necrosis factor-α (TNF-α) and their relation to premature atherosclerosis in β-thalassemia children. Hematology 2015, 20, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Brynskov, J.; Foegh, P.; Pedersen, G.; Ellervik, C.; Kirkegaard, T.; Bingham, A.; Saermark, T. Tumour necrosis factor α converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut 2002, 51, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.I.; Willemsen, G.; Sullivan, P.F.; Heutink, P.; Meijer, P.; Sondervan, D.; Kluft, C.; Smit, G.; Nolen, W.A.; Zitman, F.G.; et al. Genome-wide association of major depression: Description of samples for the GAIN major depressive disorder study: NTR and NESDA biobank projects. Eur. J. Hum. Genet. EJHG 2008, 16, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaster, M.P.; Gadotti, V.M.; Calixto, J.B.; Santos, A.R.; Rodrigues, A.L. Depressive-like behavior induced by tumor necrosis factor-α in mice. Neuropharmacology 2012, 62, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Ertenli, I.; Ozer, S.; Kiraz, S.; Apras, S.B.; Akdogan, A.; Karadag, O.; Calguneri, M.; Kalyoncu, U. Infliximab, a TNF-α antagonist treatment in patients with ankylosing spondylitis: The impact on depression, anxiety and quality of life level. Rheumatol. Int. 2012, 32, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Tyring, S.; Gottlieb, A.; Papp, K.; Gordon, K.; Leonardi, C.; Wang, A.; Lalla, D.; Woolley, M.; Jahreis, A.; Zitnik, R.; et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: Double-blind placebo-controlled randomised phase III trial. Lancet 2006, 367, 29–35. [Google Scholar] [CrossRef]
- Kapadia, S.; Lee, J.; Torre-Amione, G.; Birdsall, H.H.; Ma, T.S.; Mann, D.L. Tumor necrosis factor-α gene and protein expression in adult feline myocardium after endotoxin administration. J. Clin. Investig. 1995, 96, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.J.; Sprang, S.R. The structure of tumor necrosis factor-α at 2.6 A resolution. Implications for receptor binding. J. Biol. Chem. 1989, 264, 17595–17605. [Google Scholar] [PubMed]
- Theiss, A.L.; Simmons, J.G.; Jobin, C.; Lund, P.K. Tumor necrosis factor (TNF) α increases collagen accumulation and proliferation in intestinal myofibroblasts via tnf receptor 2. J. Biol. Chem. 2005, 280, 36099–36109. [Google Scholar] [CrossRef] [PubMed]
- Ferreccio, C.; Prado, R.B.; Luzoro, A.V.; Ampuero, S.L.; Snijders, P.J.; Meijer, C.J.; Vaccarella, S.V.; Jara, A.T.; Puschel, K.I.; Robles, S.C.; et al. Population-based prevalence and age distribution of human papillomavirus among women in Santiago, Chile. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2271–2276. [Google Scholar]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Reus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, S.; Rivest, S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: A view from the blood-brain barrier. Neuroscience 1999, 93, 1449–1464. [Google Scholar] [CrossRef]
- Khairova, R.A.; Machado-Vieira, R.; Du, J.; Manji, H.K. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int. J. Neuropsychopharmacol. 2009, 12, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Kronfol, Z.; Remick, D.G. Cytokines and the brain: Implications for clinical psychiatry. Am. J. Psychiatry 2000, 157, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Tchelingerian, J.L.; Vignais, L.; Jacque, C. TNF α gene expression is induced in neurones after a hippocampal lesion. Neuroreport 1994, 5, 585–588. [Google Scholar] [CrossRef] [PubMed]
- McNally, L.; Bhagwagar, Z.; Hannestad, J. Inflammation, glutamate, and glia in depression: A literature review. CNS Spectr. 2008, 13, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Kavelaars, A.; Heijnen, C.J.; Dantzer, R. Neuroinflammation and comorbidity of pain and depression. Pharmacol. Rev. 2014, 66, 80–101. [Google Scholar] [CrossRef] [PubMed]
- Lichtblau, N.; Schmidt, F.M.; Schumann, R.; Kirkby, K.C.; Himmerich, H. Cytokines as biomarkers in depressive disorder: Current standing and prospects. Int. Rev. Psychiatry 2013, 25, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Manosso, L.M.; Neis, V.B.; Moretti, M.; Daufenbach, J.F.; Freitas, A.E.; Colla, A.R.; Rodrigues, A.L. Antidepressant-like effect of α-tocopherol in a mouse model of depressive-like behavior induced by TNF-α. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Neis, V.B.; Manosso, L.M.; Moretti, M.; Freitas, A.E.; Daufenbach, J.; Rodrigues, A.L. Depressive-like behavior induced by tumor necrosis factor-α is abolished by agmatine administration. Behav. Brain Res. 2014, 261, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Bruning, C.A.; Martini, F.; Soares, S.M.; Savegnago, L.; Sampaio, T.B.; Nogueira, C.W. Depressive-like behavior induced by tumor necrosis factor-α is attenuated by m-trifluoromethyl-diphenyl diselenide in mice. J. Psychiatr. Res. 2015, 66–67, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Budni, J.; Freitas, A.E.; Neis, V.B.; Ribeiro, C.M.; de Oliveira Balen, G.; Rieger, D.K.; Leal, R.B.; Rodrigues, A.L. TNF-α-induced depressive-like phenotype and p38(MAPK) activation are abolished by ascorbic acid treatment. Eur. Neuropsychopharmacol. 2015, 25, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Grippo, A.J.; Francis, J.; Beltz, T.G.; Felder, R.B.; Johnson, A.K. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol. Behav. 2005, 84, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Kubera, M.; Basta-Kaim, A.; Papp, M. The effect of chronic treatment with imipramine on the immunoreactivity of animals subjected to a chronic mild stress model of depression. Immunopharmacology 1995, 30, 225–230. [Google Scholar] [CrossRef]
- Simen, B.B.; Duman, C.H.; Simen, A.A.; Duman, R.S. TNF-α signaling in depression and anxiety: Behavioral consequences of individual receptor targeting. Biol. Psychiatry 2006, 59, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Himmerich, H.; Fulda, S.; Linseisen, J.; Seiler, H.; Wolfram, G.; Himmerich, S.; Gedrich, K.; Kloiber, S.; Lucae, S.; Ising, M.; et al. Depression, comorbidities and the TNF-α system. Eur. Psychiatry 2008, 23, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Mattei, D.; Westrin, A.; Traskman-Bendz, L.; Brundin, L. Cytokine levels in the blood may distinguish suicide attempters from depressed patients. Brain Behav. Immun. 2011, 25, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.N.; Rizavi, H.S.; Ren, X.; Fareed, J.; Hoppensteadt, D.A.; Roberts, R.C.; Conley, R.R.; Dwivedi, Y. Proinflammatory cytokines in the prefrontal cortex of teenage suicide victims. J. Psychiatr. Res. 2012, 46, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Cytokine, sickness behavior, and depression. Immunol. Allergy Clin. N. Am. 2009, 29, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: Leads for future research and new drug developments in depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar] [PubMed]
- Schiepers, O.J.; Wichers, M.C.; Maes, M. Cytokines and major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Krishnadas, R.; Cavanagh, J. Depression: An inflammatory illness? J. Neurol. Neurosurgery Psychiatry 2012, 83, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Koehl, M.; Darnaudery, M.; Dulluc, J.; van Reeth, O.; Le Moal, M.; Maccari, S. Prenatal stress alters circadian activity of hypothalamo-pituitary-adrenal axis and hippocampal corticosteroid receptors in adult rats of both gender. J. Neurobiol. 1999, 40, 302–315. [Google Scholar] [CrossRef]
- Pariante, C.M. Depression, stress and the adrenal axis. J. Neuroendocrinol. 2003, 15, 811–812. [Google Scholar] [PubMed]
- Du, X.; Pang, T.Y. Is dysregulation of the HPA-axis a core pathophysiology mediating co-morbid depression in neurodegenerative diseases? Front. Psychiatry 2015, 6, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Bodenlos, J.S.; Lemon, S.C.; Schneider, K.L.; August, M.A.; Pagoto, S.L. Associations of mood and anxiety disorders with obesity: Comparisons by ethnicity. J. Psychosom. Res. 2011, 71, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Doczy, E.J.; Seroogy, K.; Harrison, C.R.; Herman, J.P. Hypothalamo-pituitary-adrenocortical axis, glucocorticoids, and neurologic disease. Immunol. Allergy Clin. N. Am. 2009, 29, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Black, P.H. Immune system-central nervous system interactions: Effect and immunomodulatory consequences of immune system mediators on the brain. Antimicrob. Agents Chemother. 1994, 38, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Wollman, E.; Vitkovic, L.; Yirmiya, R. Cytokines and depression: Fortuitous or causative association? Mol. Psychiatry 1999, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Cowen, P.J. Cortisol, serotonin and depression: All stressed out? Br. J. Psychiatry 2002, 180, 99–100. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Furay, A.R.; Bruestle, A.E.; Herman, J.P. The role of the forebrain glucocorticoid receptor in acute and chronic stress. Endocrinology 2008, 149, 5482–5490. [Google Scholar] [CrossRef] [PubMed]
- Karssen, A.M.; Meijer, O.C.; Berry, A.; Sanjuan Pinol, R.; de Kloet, E.R. Low doses of dexamethasone can produce a hypocorticosteroid state in the brain. Endocrinology 2005, 146, 5587–5595. [Google Scholar] [CrossRef] [PubMed]
- Lang, U.E.; Borgwardt, S. Molecular mechanisms of depression: Perspectives on new treatment strategies. Cell. Physiol. Biochem. 2013, 31, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Van Bogaert, T.; Vandevyver, S.; Dejager, L.; van Hauwermeiren, F.; Pinheiro, I.; Petta, I.; Engblom, D.; Kleyman, A.; Schutz, G.; Tuckermann, J.; et al. Tumor necrosis factor inhibits glucocorticoid receptor function in mice: A strong signal toward lethal shock. J. Biol. Chem. 2011, 286, 26555–26567. [Google Scholar] [CrossRef] [PubMed]
- Rider, C.F.; Shah, S.; Miller-Larsson, A.; Giembycz, M.A.; Newton, R. Cytokine-induced loss of glucocorticoid function: Effect of kinase inhibitors, long-acting β2-adrenoceptor agonist and glucocorticoid receptor ligands. PLoS ONE 2015, 10, e0116773. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.; Gaffey, K.; Kane, B.; Malia-Milanes, B.; Shah, R.; Bentley, A.; Ray, D.; Singh, D. Reduced glucocorticoid receptor expression and function in airway neutrophils. Int. Immunopharmacol. 2012, 12, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.; Hu, F.; Miller, A.H. Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav. Immun. 2007, 21, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000, 23, 477–501. [Google Scholar] [CrossRef]
- Van Rossum, E.F.; Binder, E.B.; Majer, M.; Koper, J.W.; Ising, M.; Modell, S.; Salyakina, D.; Lamberts, S.W.; Holsboer, F. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol. Psychiatry 2006, 59, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Schuld, A.; Schmid, D.A.; Haack, M.; Holsboer, F.; Friess, E.; Pollmacher, T. Hypothalamo-pituitary-adrenal function in patients with depressive disorders is correlated with baseline cytokine levels, but not with cytokine responses to hydrocortisone. J. Psychiatr. Res. 2003, 37, 463–470. [Google Scholar] [CrossRef]
- Himmerich, H.; Binder, E.B.; Kunzel, H.E.; Schuld, A.; Lucae, S.; Uhr, M.; Pollmacher, T.; Holsboer, F.; Ising, M. Successful antidepressant therapy restores the disturbed interplay between TNF-α system and hpa axis. Biol. Psychiatry 2006, 60, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.J. Relationship of neurotransmitters to the symptoms of major depressive disorder. J. Clin. Psychiatry 2008, 69 (Suppl. E1), 4–7. [Google Scholar] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1β and tumor necrosis factor-α activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [PubMed]
- Yamada, K.; Iida, R.; Miyamoto, Y.; Saito, K.; Sekikawa, K.; Seishima, M.; Nabeshima, T. Neurobehavioral alterations in mice with a targeted deletion of the tumor necrosis factor-α gene: Implications for emotional behavior. J. Neuroimmunol. 2000, 111, 131–138. [Google Scholar] [CrossRef]
- Zhu, C.B.; Carneiro, A.M.; Dostmann, W.R.; Hewlett, W.A.; Blakely, R.D. P38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J. Biol. Chem. 2005, 280, 15649–15658. [Google Scholar] [CrossRef] [PubMed]
- Van Heesch, F.; Prins, J.; Korte-Bouws, G.A.; Westphal, K.G.; Lemstra, S.; Olivier, B.; Kraneveld, A.D.; Korte, S.M. Systemic tumor necrosis factor-α decreases brain stimulation reward and increases metabolites of serotonin and dopamine in the nucleus accumbens of mice. Behav. Brain Res. 2013, 253, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Miura, H.; Ozaki, N.; Sawada, M.; Isobe, K.; Ohta, T.; Nagatsu, T. A link between stress and depression: Shifts in the balance between the kynurenine and serotonin pathways of tryptophan metabolism and the etiology and pathophysiology of depression. Stress 2008, 11, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Furtado, C.P.; Maller, J.J.; Fitzgerald, P.B. A magnetic resonance imaging study of the entorhinal cortex in treatment-resistant depression. Psychiatry Res. 2008, 163, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Rimol, L.M.; Hartberg, C.B.; Nesvag, R.; Fennema-Notestine, C.; Hagler, D.J., Jr.; Pung, C.J.; Jennings, R.G.; Haukvik, U.K.; Lange, E.; Nakstad, P.H.; et al. Cortical thickness and subcortical volumes in schizophrenia and bipolar disorder. Biol. Psychiatry 2010, 68, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, T.; Ullrich, C.; Sperner-Unterweger, B.; Humpel, C. Inflammatory stimuli reduce survival of serotonergic neurons and induce neuronal expression of indoleamine 2,3-dioxygenase in rat dorsal raphe nucleus organotypic brain slices. Neuroscience 2011, 184, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Kim, Y.K. Cytokine-serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med. Hypotheses 2003, 61, 519–525. [Google Scholar] [CrossRef]
- Reus, G.Z.; Jansen, K.; Titus, S.; Carvalho, A.F.; Gabbay, V.; Quevedo, J. Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: Evidences from animal and human studies. J. Psychiatr. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. The new “5-HT” hypothesis of depression: Cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 702–721. [Google Scholar]
- Mohamed, B.M.; Aboul-Fotouh, S.; Ibrahim, E.A.; Shehata, H.; Mansour, A.A.; Yassin, N.A.; El-Eraky, W.; Abdel-Tawab, A.M. Effects of pentoxifylline, 7-nitroindazole, and imipramine on tumor necrosis factor-α and indoleamine 2,3-dioxygenase enzyme activity in the hippocampus and frontal cortex of chronic mild-stress-exposed rats. Neuropsychiatr. Dis. Treat. 2013, 9, 697–708. [Google Scholar] [PubMed]
- Liu, Y.N.; Peng, Y.L.; Liu, L.; Wu, T.Y.; Zhang, Y.; Lian, Y.J.; Yang, Y.Y.; Kelley, K.W.; Jiang, C.L.; Wang, Y.X. TNF-α mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur. Cytokine Netw. 2015, 26, 15–25. [Google Scholar] [PubMed]
- Myint, A.M.; Kim, Y.K. Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 48, 304–313. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; Lawson, M.A.; Andre, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Pellicciari, R. Manipulation of brain kynurenines: Glial targets, neuronal effects, and clinical opportunities. J. Pharmacol. Exp. Ther. 2002, 303, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spalletta, G.; Bossu, P.; Ciaramella, A.; Bria, P.; Caltagirone, C.; Robinson, R.G. The etiology of poststroke depression: A review of the literature and a new hypothesis involving inflammatory cytokines. Mol. Psychiatry 2006, 11, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpe, S.; Maes, M. IDO and interferon-α-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatry 2005, 10, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Wichers, M.; Maes, M. The psychoneuroimmuno-pathophysiology of cytokine-induced depression in humans. Int. J. Neuropsychopharmacol. 2002, 5, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J. Immunological aspects of depressive disorders. Der Nervenarzt 2007, 78, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Bufalino, C.; Hepgul, N.; Aguglia, E.; Pariante, C.M. The role of immune genes in the association between depression and inflammation: A review of recent clinical studies. Brain Behav. Immun. 2013, 31, 31–47. [Google Scholar] [CrossRef] [PubMed]
- De Moor, M.H.; van den Berg, S.M.; Verweij, K.J.; Krueger, R.F.; Luciano, M.; Arias Vasquez, A.; Matteson, L.K.; Derringer, J.; Esko, T.; Amin, N.; et al. Meta-analysis of genome-wide association studies for neuroticism, and the polygenic association with major depressive disorder. JAMA Psychiatry 2015, 72, 642–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripke, S.; Wray, N.R.; Lewis, C.M.; Hamilton, S.P.; Weissman, M.M.; Breen, G.; Byrne, E.M.; Blackwood, D.H.; Boomsma, D.I.; Cichon, S.; et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 2013, 18, 497–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide snps. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baena, A.; Leung, J.Y.; Sullivan, A.D.; Landires, I.; Vasquez-Luna, N.; Quinones-Berrocal, J.; Fraser, P.A.; Uko, G.P.; Delgado, J.C.; Clavijo, O.P.; et al. TNF-α promoter single nucleotide polymorphisms are markers of human ancestry. Genes Immun. 2002, 3, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Merino, A.M.; Zhang, K.; Kaslow, R.A.; Aissani, B. Structure of tumor necrosis factor-α haploblocks in European populations. Immunogenetics 2013, 65, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, K.M.; Carville, K.S.; Abraham, L.J. The -308 tumor necrosis factor-α promoter polymorphism effects transcription. Mol. Immunol. 1997, 34, 391–399. [Google Scholar] [CrossRef]
- Louis, E.; Franchimont, D.; Piron, A.; Gevaert, Y.; Schaaf-Lafontaine, N.; Roland, S.; Mahieu, P.; Malaise, M.; de Groote, D.; Louis, R.; et al. Tumour necrosis factor (TNF) gene polymorphism influences TNF-α production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clin. Exp. Immunol. 1998, 113, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, J.; Cuchacovich, M.; Perez, C.; Ferreira, L.; Aguirre, A.; Schiattino, I.; Soto, L.; Cruzat, A.; Salazar-Onfray, F.; Aguillon, J.C. The -308 polymorphism in the tumour necrosis factor (TNF) gene promoter region and ex vivo lipopolysaccharide-induced TNF expression and cytotoxic activity in chilean patients with rheumatoid arthritis. Rheumatology (Oxford) 2003, 42, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Arosio, B.; Mundo, E.; Cattaneo, E.; Pozzoli, S.; Dell'osso, B.; Vergani, C.; Trabattoni, D.; Altamura, A.C. Cytokine polymorphisms in the pathophysiology of mood disorders. CNS Spectr. 2009, 14, 419–425. [Google Scholar] [PubMed]
- Kim, Y.K.; Hong, J.P.; Hwang, J.A.; Lee, H.J.; Yoon, H.K.; Lee, B.H.; Jung, H.Y.; Hahn, S.W.; Na, K.S. TNF-α -308g>a polymorphism is associated with suicide attempts in major depressive disorder. J. Affect. Disord. 2013, 150, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Jun, T.Y.; Pae, C.U.; Hoon, H.; Chae, J.H.; Bahk, W.M.; Kim, K.S.; Serretti, A. Possible association between -g308a tumour necrosis factor-α gene polymorphism and major depressive disorder in the Korean population. Psychiatr. Genet. 2003, 13, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Misener, V.L.; Gomez, L.; Wigg, K.G.; Luca, P.; King, N.; Kiss, E.; Daroczi, G.; Kapornai, K.; Tamas, Z.; Mayer, L.; et al. Cytokine genes TNF, IL1a, IL1b, IL6, IL1RN and IL10, and childhood-onset mood disorders. Neuropsychobiology 2008, 58, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Cerri, A.P.; Arosio, B.; Viazzoli, C.; Confalonieri, R.; Vergani, C.; Annoni, G. The -308 (G/A) single nucleotide polymorphism in the TNF-α gene and the risk of major depression in the elderly. Int. J. Geriatr. Psychiatry 2010, 25, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Lotrich, F.E.; Ferrell, R.E.; Rabinovitz, M.; Pollock, B.G. Labile anger during interferon α treatment is associated with a polymorphism in tumor necrosis factor α. Clin. Neuropharmacol. 2010, 33, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Bosker, F.J.; Hartman, C.A.; Nolte, I.M.; Prins, B.P.; Terpstra, P.; Posthuma, D.; van Veen, T.; Willemsen, G.; DeRijk, R.H.; de Geus, E.J.; et al. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol. Psychiatry 2011, 16, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Stewart, R.; Kim, S.W.; Shin, I.S.; Kim, J.T.; Park, M.S.; Park, S.W.; Kim, Y.H.; Cho, K.H.; Yoon, J.S. Associations of cytokine gene polymorphisms with post-stroke depression. World J. Biol. Psychiatry 2012, 13, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Haastrup, E.; Bukh, J.D.; Bock, C.; Vinberg, M.; Thorner, L.W.; Hansen, T.; Werge, T.; Kessing, L.V.; Ullum, H. Promoter variants in IL18 are associated with onset of depression in patients previously exposed to stressful-life events. J. Affect. Disord. 2012, 136, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, S.; Abbey, S.E.; Chan, C.; Bargman, J.M.; Stewart, D.E. A genetic predisposition to produce low levels of IL-10 is related to depressive symptoms: A pilot study of patients with end stage renal disease. Psychosomatics 2012, 53, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Valentini, G.; Ayala, F.; Cattaneo, A.; Valesini, G. New strategies to address the pharmacodynamics and pharmacokinetics of tumor necrosis factor (TNF) inhibitors: A systematic analysis. Autoimmun. Rev. 2015, 14, 812–829. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.M.; Trinh, H.; Le, J.; Siegel, S.; Shealy, D.; McDonough, M.; Scallon, B.; Moore, M.A.; Vilcek, J.; Daddona, P.; et al. Construction and initial characterization of a mouse-human chimeric anti-tnf antibody. Mol. Immunol. 1993, 30, 1443–1453. [Google Scholar] [CrossRef]
- Peppel, K.; Crawford, D.; Beutler, B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J. Exp. Med. 1991, 174, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Karson, A.; Demirtas, T.; Bayramgurler, D.; Balci, F.; Utkan, T. Chronic administration of infliximab (TNF-α inhibitor) decreases depression and anxiety-like behaviour in rat model of chronic mild stress. Basic Clin. Pharmacol. Toxicol. 2013, 112, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Bayramgurler, D.; Karson, A.; Ozer, C.; Utkan, T. Effects of long-term etanercept treatment on anxiety- and depression-like neurobehaviors in rats. Physiol. Behav. 2013, 119, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Krugel, U.; Fischer, J.; Radicke, S.; Sack, U.; Himmerich, H. Antidepressant effects of TNF-α blockade in an animal model of depression. J. Psychiatr. Res. 2013, 47, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Sahin, T.D.; Karson, A.; Balci, F.; Yazir, Y.; Bayramgurler, D.; Utkan, T. TNF-α inhibition prevents cognitive decline and maintains hippocampal bdnf levels in the unpredictable chronic mild stress rat model of depression. Behav. Brain Res. 2015, 292, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Bassukas, I.D.; Hyphantis, T.; Gamvroulia, C.; Gaitanis, G.; Mavreas, V. Infliximab for patients with plaque psoriasis and severe psychiatric comorbidity. J. Eur. Acad. Dermatol. Venereol. JEADV 2008, 22, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Augustin, M.; Signorovitch, J.; Yu, A.P.; Wu, E.Q.; Gupta, S.R.; Bao, Y.; Mulani, P. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: A randomized clinical trial. J. Am. Acad. Dermatol. 2010, 62, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, G.R.; Bala, M.; Han, C.; DeWoody, K.; Schaible, T. Infliximab improves quality of life in patients with Crohn’s disease. Inflamm. Bowel Dis. 2002, 8, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Persoons, P.; Vermeire, S.; Demyttenaere, K.; Fischler, B.; Vandenberghe, J.; van Oudenhove, L.; Pierik, M.; Hlavaty, T.; van Assche, G.; Noman, M.; et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment. Pharmacol. Ther. 2005, 22, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minderhoud, I.M.; Samsom, M.; Oldenburg, B. Crohn’s disease, fatigue, and infliximab: Is there a role for cytokines in the pathogenesis of fatigue? World J. Gastroenterol. WJG 2007, 13, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Arisoy, O.; Bes, C.; Cifci, C.; Sercan, M.; Soy, M. The effect of TNF-α blockers on psychometric measures in ankylosing spondylitis patients: A preliminary observation. Rheumatol. Int. 2013, 33, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Perera, T.D.; Dwork, A.J.; Keegan, K.A.; Thirumangalakudi, L.; Lipira, C.M.; Joyce, N.; Lange, C.; Higley, J.D.; Rosoklija, G.; Hen, R.; et al. Necessity of hippocampal neurogenesis for the therapeutic action of antidepressants in adult nonhuman primates. PLoS ONE 2011, 6, e17600. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Raison, C.L.; Woolwine, B.J.; Haroon, E.; Binder, E.B.; Miller, A.H.; Felger, J.C. Transcriptional signatures related to glucose and lipid metabolism predict treatment response to the tumor necrosis factor antagonist infliximab in patients with treatment-resistant depression. Brain Behav. Immun. 2013, 31, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Stellwagen, D.; Malenka, R.C.; Stryker, M.P. Tumor necrosis factor-α mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 2008, 58, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Eller, T.; Vasar, V.; Shlik, J.; Maron, E. Effects of bupropion augmentation on pro-inflammatory cytokines in escitalopram-resistant patients with major depressive disorder. J. Psychopharmacol. 2009, 23, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Murata, T.; Takahashi, T.; Kosaka, H.; Omata, N.; Wada, Y. Plasma levels of adiponectin and tumor necrosis factor-α in patients with remitted major depression receiving long-term maintenance antidepressant therapy. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Kraus, T.; Haack, M.; Schuld, A.; Hinze-Selch, D.; Koethe, D.; Pollmacher, T. Body weight, the tumor necrosis factor system, and leptin production during treatment with mirtazapine or venlafaxine. Pharmacopsychiatry 2002, 35, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Piletz, J.E.; Halaris, A.; Iqbal, O.; Hoppensteadt, D.; Fareed, J.; Zhu, H.; Sinacore, J.; Devane, C.L. Pro-inflammatory biomakers in depression: Treatment with venlafaxine. World J. Biol. Psychiatry 2009, 10, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qi, D.; Chen, J.; Zhang, C.; Yi, Z.; Yuan, C.; Wang, Z.; Hong, W.; Yu, S.; Cui, D.; et al. Venlafaxine inhibits the upregulation of plasma tumor necrosis factor-α (TNF-α) in the Chinese patients with major depressive disorder: A prospective longitudinal study. Psychoneuroendocrinology 2013, 38, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Herane Vives, A.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Duseja, R.; Heir, R.; Lewitus, G.M.; Altimimi, H.F.; Stellwagen, D. Astrocytic TNF-α regulates the behavioral response to antidepressants. Brain Behav. Immun. 2015, 44, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Malynn, S.; Campos-Torres, A.; Moynagh, P.; Haase, J. The pro-inflammatory cytokine TNF-α regulates the activity and expression of the serotonin transporter (sert) in astrocytes. Neurochem. Res. 2013, 38, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Aloe, L.; Properzi, F.; Probert, L.; Akassoglou, K.; Kassiotis, G.; Micera, A.; Fiore, M. Learning abilities, NGF and BDNF brain levels in two lines of TNF-α transgenic mice, one characterized by neurological disorders, the other phenotypically normal. Brain Res. 1999, 840, 125–137. [Google Scholar] [CrossRef]
- Cai, X.; Kallarackal, A.J.; Kvarta, M.D.; Goluskin, S.; Gaylor, K.; Bailey, A.M.; Lee, H.K.; Huganir, R.L.; Thompson, S.M. Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat. Neurosci. 2013, 16, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, J.F.; Raison, C.L.; Rye, D.B.; Montague, A.R.; Woolwine, B.J.; Felger, J.C.; Haroon, E.; Miller, A.H. Inhibition of tumor necrosis factor improves sleep continuity in patients with treatment resistant depression and high inflammation. Brain Behav. Immun. 2015, 47, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Richardson-Jones, J.W.; Craige, C.P.; Guiard, B.P.; Stephen, A.; Metzger, K.L.; Kung, H.F.; Gardier, A.M.; Dranovsky, A.; David, D.J.; Beck, S.G.; et al. 5-HT 1A autoreceptor levels determine vulnerability to stress and response to antidepressants. Neuron 2010, 65, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Tang, C.S.; Ho, R.C. Serum tumour necrosis factor-α is associated with poor health-related quality of life and depressive symptoms in patients with systemic lupus erythematosus. Lupus 2013, 22, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Postal, M.; Pelicari, K.O.; Sinicato, N.A.; Marini, R.; Costallat, L.T.; Appenzeller, S. Th1/Th2 cytokine profile in childhood-onset systemic lupus erythematosus. Cytokine 2013, 61, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Kahl, K.G.; Kruse, N.; Faller, H.; Weiss, H.; Rieckmann, P. Expression of tumor necrosis factor-α and interferon-γ mRNA in blood cells correlates with depression scores during an acute attack in patients with multiple sclerosis. Psychoneuroendocrinology 2002, 27, 671–681. [Google Scholar] [CrossRef]
- Ding, C.Z.; Yao, Y.; Fang, Y.; Sun, L.Y.; Wang, Y. Effects of zhengqing fengtongning tablet and methotrexate on the serum OPG/RANKL and IL-17 of collagen-induced arthritis rats. Zhongguo Zhong Xi Yi Jie He Za Zhi 2013, 33, 256–260. [Google Scholar] [PubMed]

{kind=link}
| Study (Year) | Type of Study | SNP | Ethnicity | Sample (Case/Control) | Genotyping | Diagnostic Criteria | Main Conclusion |
|---|---|---|---|---|---|---|---|
| Jun et al. [116] | Case-control | -308G/A | Korea | 108/125 | PCR-RFLP | DSM-IV | TNF-α -308 A allele affected MDD susceptibility |
| Misener et al. [117] | Family-based association of COMD | -238A/G -308G/A -857C/T -1031T/C | Caucasian Roma African | 460 children from 384 families | TaqMan® 5′ nuclease Assay | DSM-IV ISCA-D | No evidence supported TNF-α as genetic risk factors contributing to COMD |
| Cerri et al. [118] | Case-control | -308G/A | Italy | 50/240 | PCR-SSP | DSM-IV GDS ≥ 15 MMSE ≥ 24 | TNF-α -308 G/G genotype related with a greater risk of developing MDD |
| Clerici et al. [114] | Case-control | -308G/A | Italy | 32 MDD 32BD I 20BD II/363 | PCR-SSP | DSM-IV Physical examination | No association of TNF-α polymorphisms with MDD |
| Lotrich et al. [119] | Prospective study | -308G/A | NR | 105 IFN-α induced depression cases in HCV patients | TaqMan® 5′ nuclease Assay | DSM-IV BDI-II AIAQ TNF-α levels | TNF-α -308 A allele associated with labile anger and fatigue but not with MDD |
| Bosker et al. [120] | GWAS | 55 gene 92 SNP | Multi-ethnic | 1738/1802 | NR | GAIN | TNF-α was identified as the only gene associated with MDD (rs76917) |
| Kim et al. [121] | Case-control | -238A/G -308G/A | Korea | 29 major post-stroke depression cases/199 | PCR-SSP | DSM-IV MINI | Higher frequencies of TNF-α-308 A in major post stroke depression |
| Haastrup et al. [122] | Case-control | -238A/G -308G/A -857C/T -1031T/C | Denmark | 288/335 | Qiagen® FlexiGene kit | SCAN IRLE | None of the examined TNF-α alleles were differently distributed among MDD |
| Holtzman et al. [123] | Cross-sectional study | -308G/A | Canada | 93 MDD cases with end stage renal disease | Sequenom iPLEX assay | BDI-II | No associations with depression were found |
| Kim et al. [115] | Case-control | -308G/A | Korea | 204 MDD cases with attempted suicide/97 | PCR-SSP | DSM-IV | TNF-α-308G/A polymorphism was an independent risk factor for suicide attempts in MDD |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.; Zhang, H.; Baloch, Z. Pathogenetic and Therapeutic Applications of Tumor Necrosis Factor-α (TNF-α) in Major Depressive Disorder: A Systematic Review. Int. J. Mol. Sci. 2016, 17, 733. https://doi.org/10.3390/ijms17050733
Ma K, Zhang H, Baloch Z. Pathogenetic and Therapeutic Applications of Tumor Necrosis Factor-α (TNF-α) in Major Depressive Disorder: A Systematic Review. International Journal of Molecular Sciences. 2016; 17(5):733. https://doi.org/10.3390/ijms17050733
Chicago/Turabian StyleMa, Ke, Hongxiu Zhang, and Zulqarnain Baloch. 2016. "Pathogenetic and Therapeutic Applications of Tumor Necrosis Factor-α (TNF-α) in Major Depressive Disorder: A Systematic Review" International Journal of Molecular Sciences 17, no. 5: 733. https://doi.org/10.3390/ijms17050733