
Design of Acceptors with Suitable Frontier Molecular Orbitals to Match Donors via Substitutions on Perylene Diimide for Organic Solar Cells

Abstract
:
1. Introduction
2. Results and Discussion
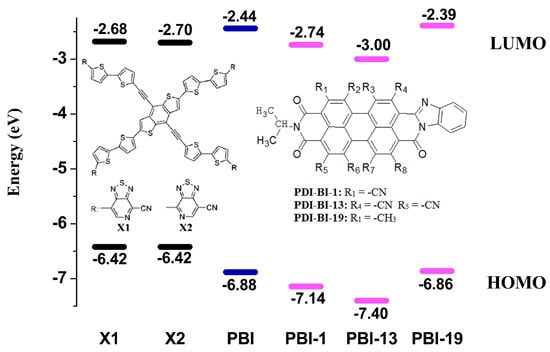
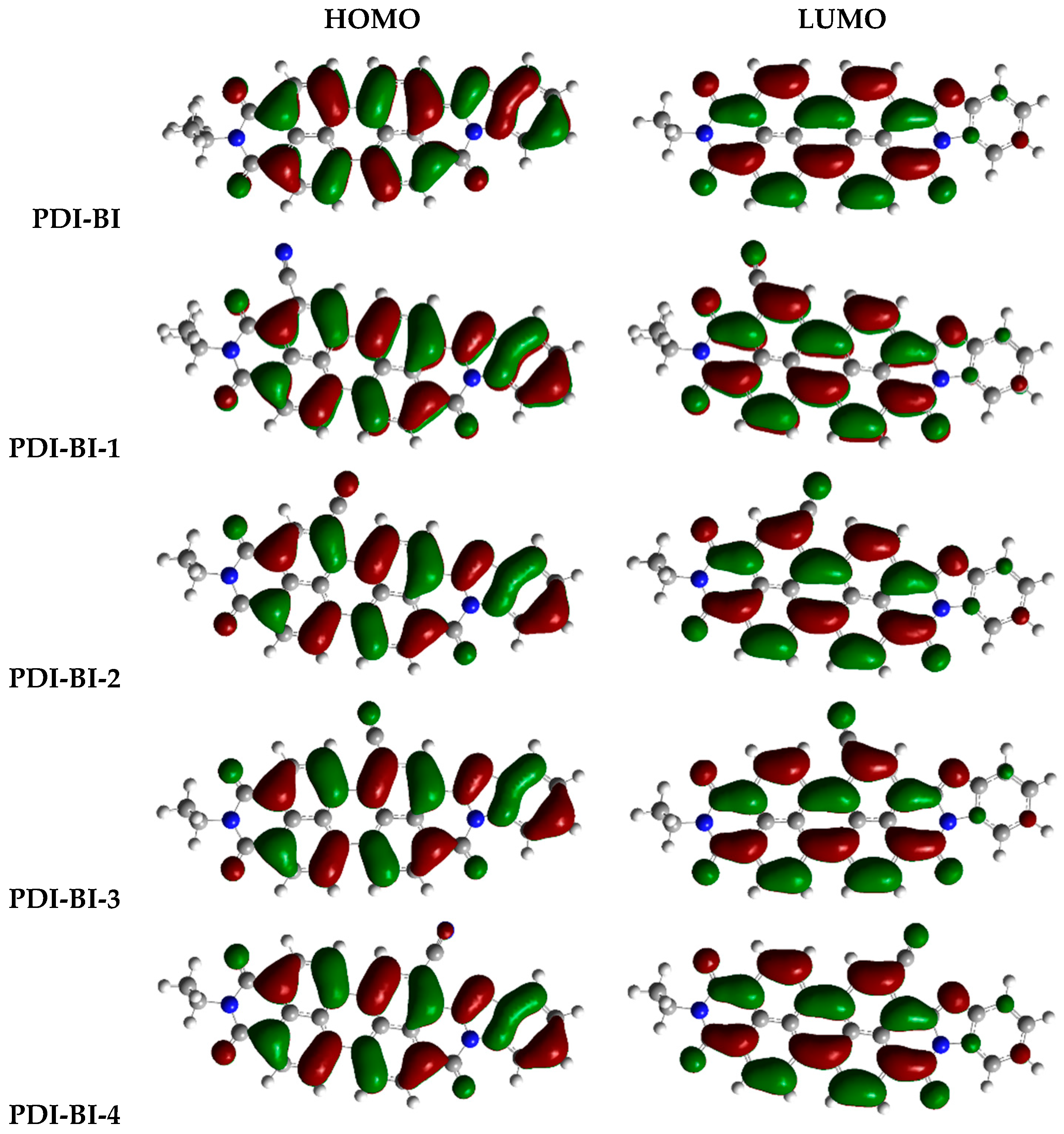
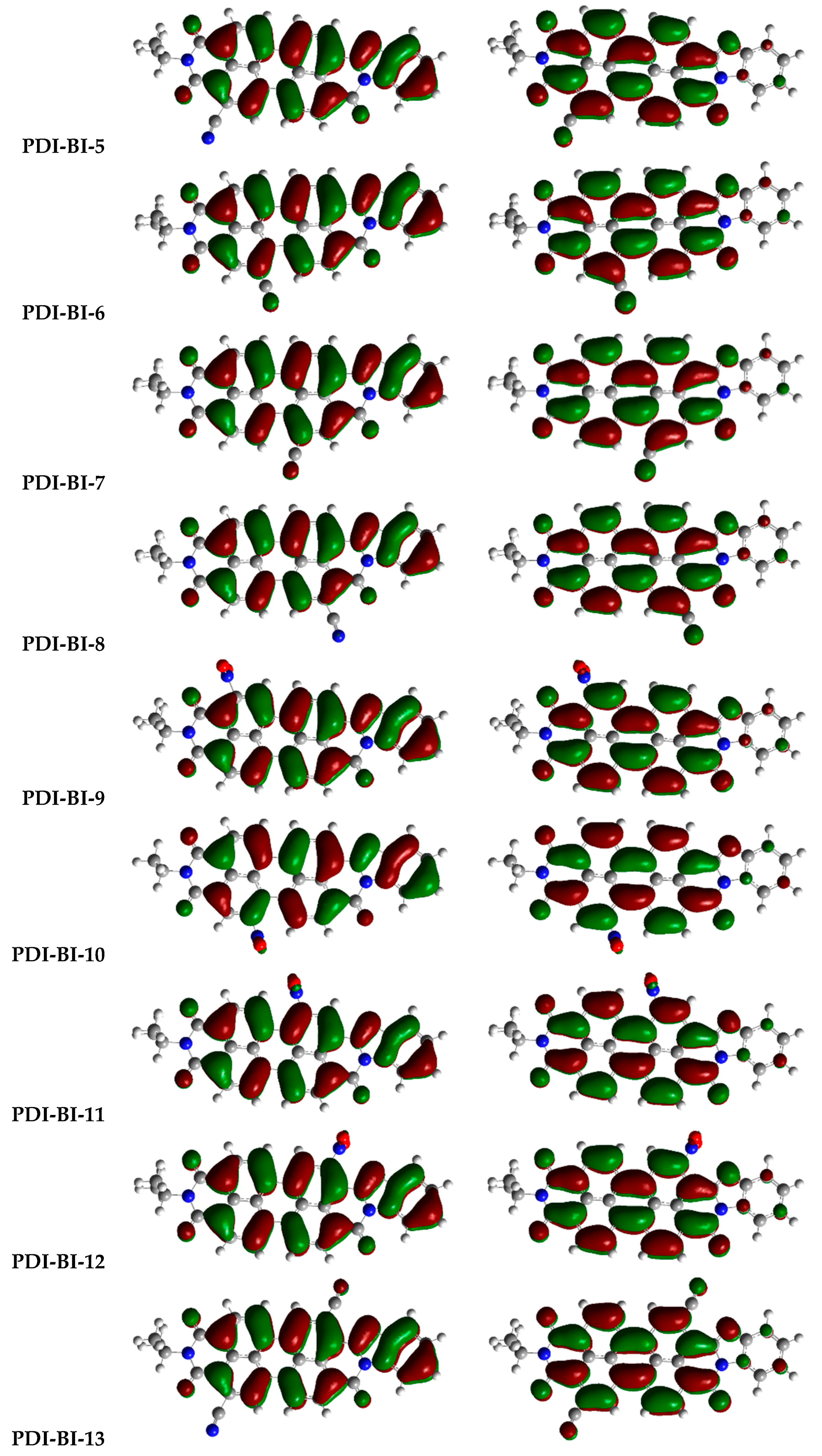
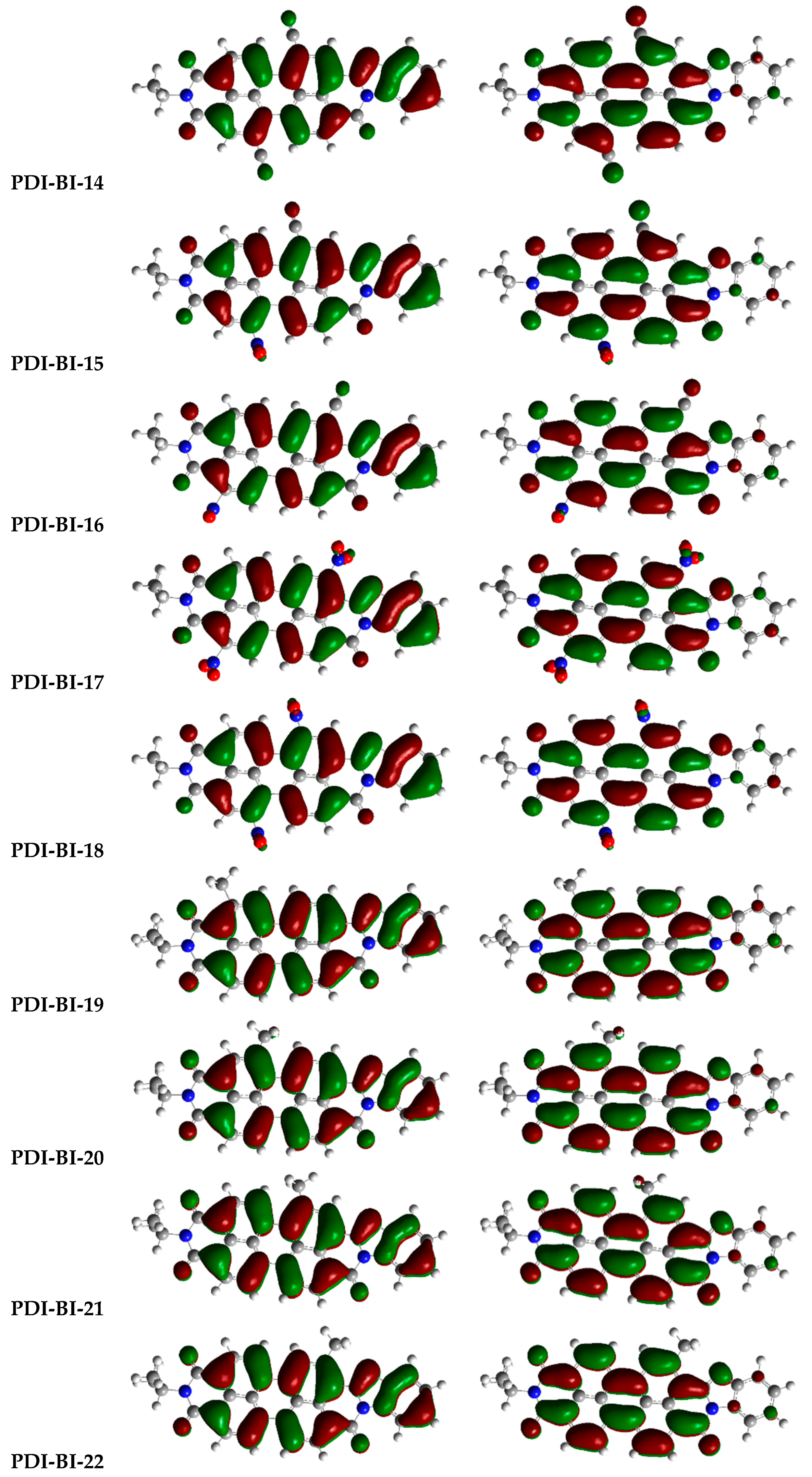
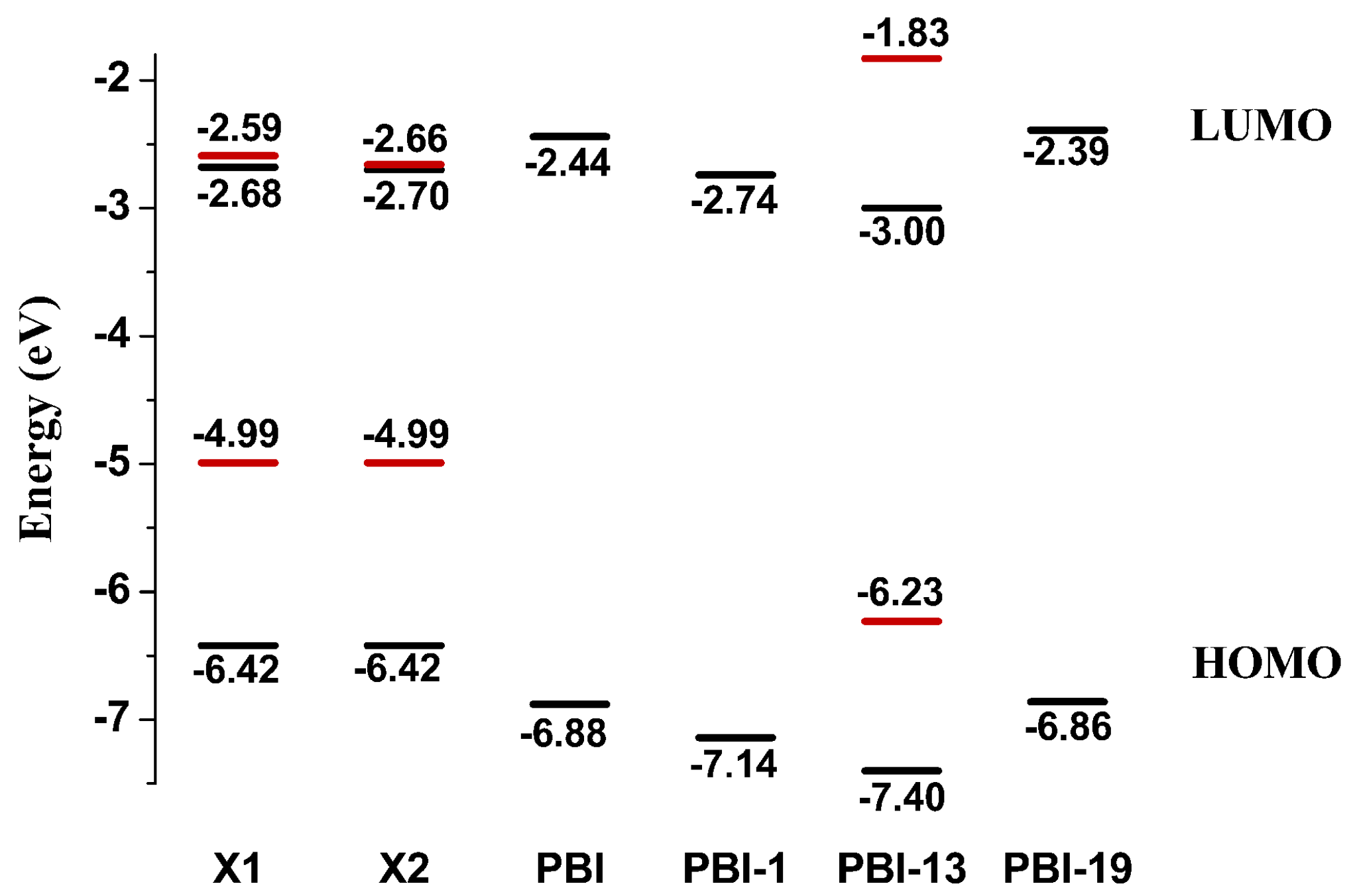
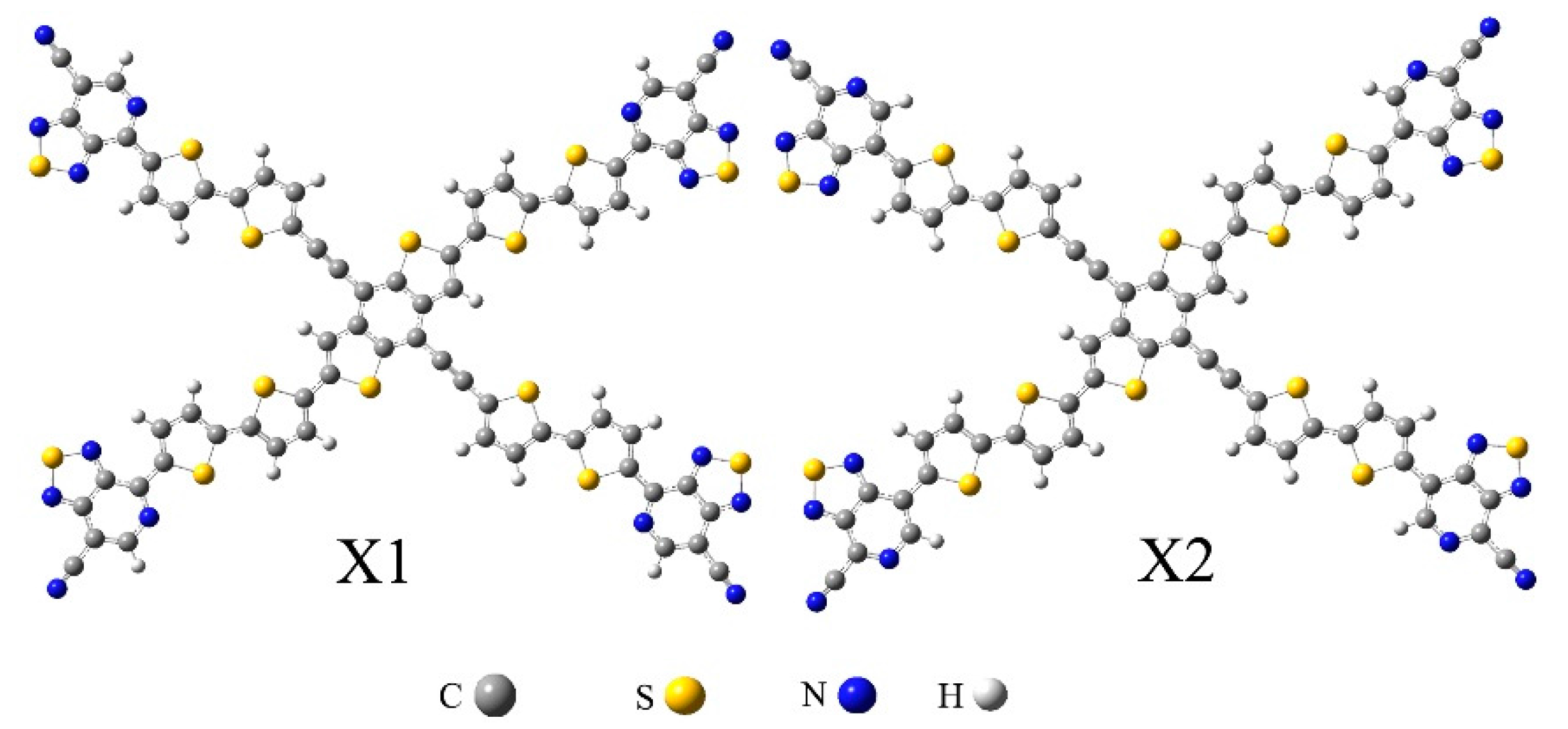
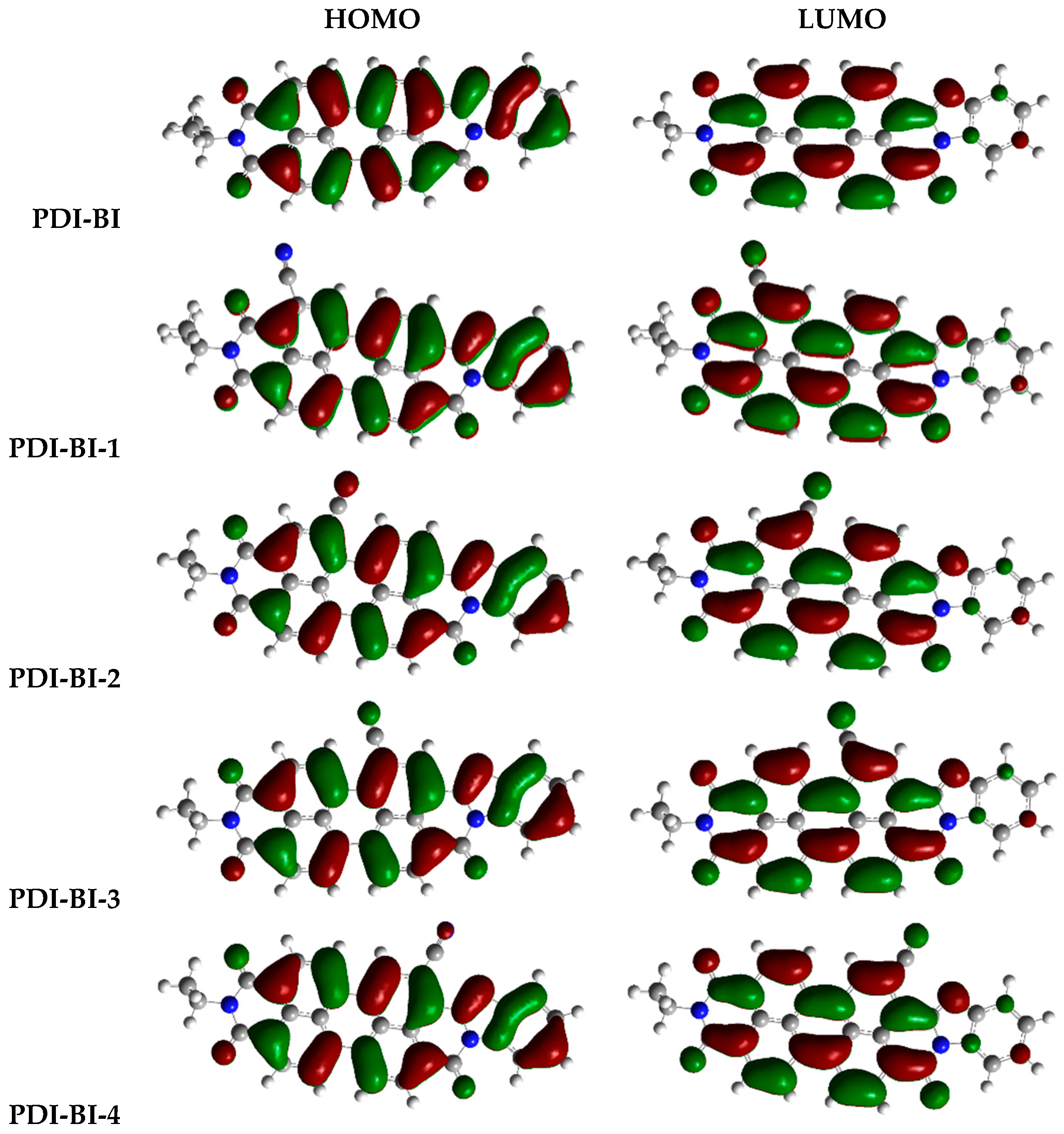
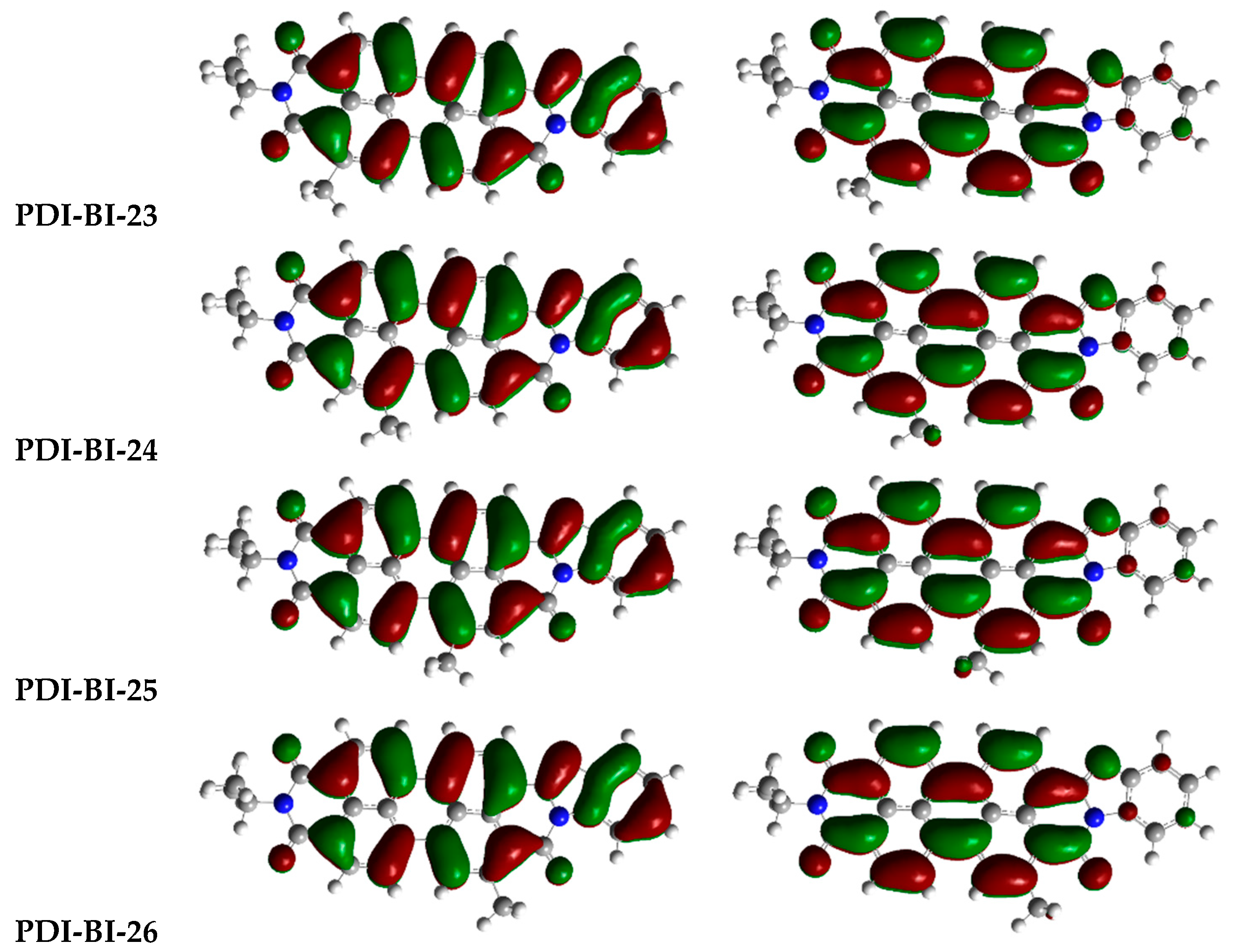
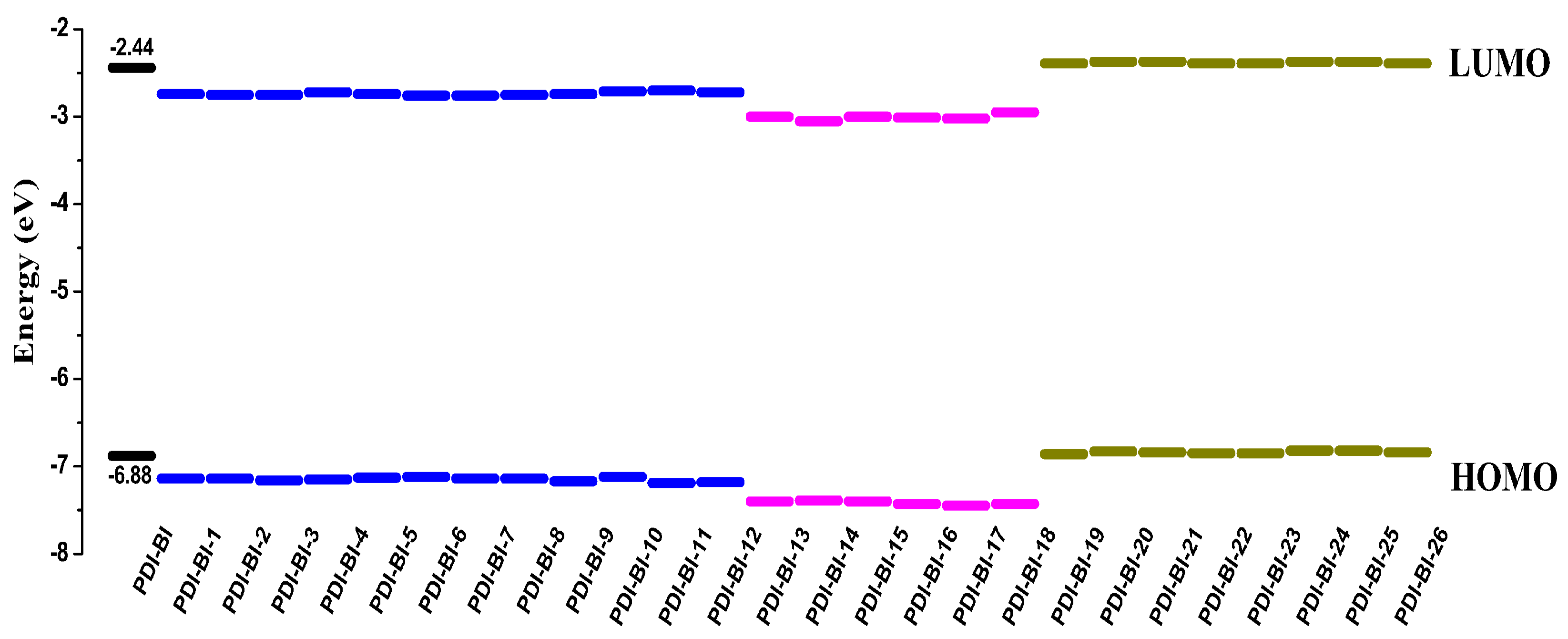
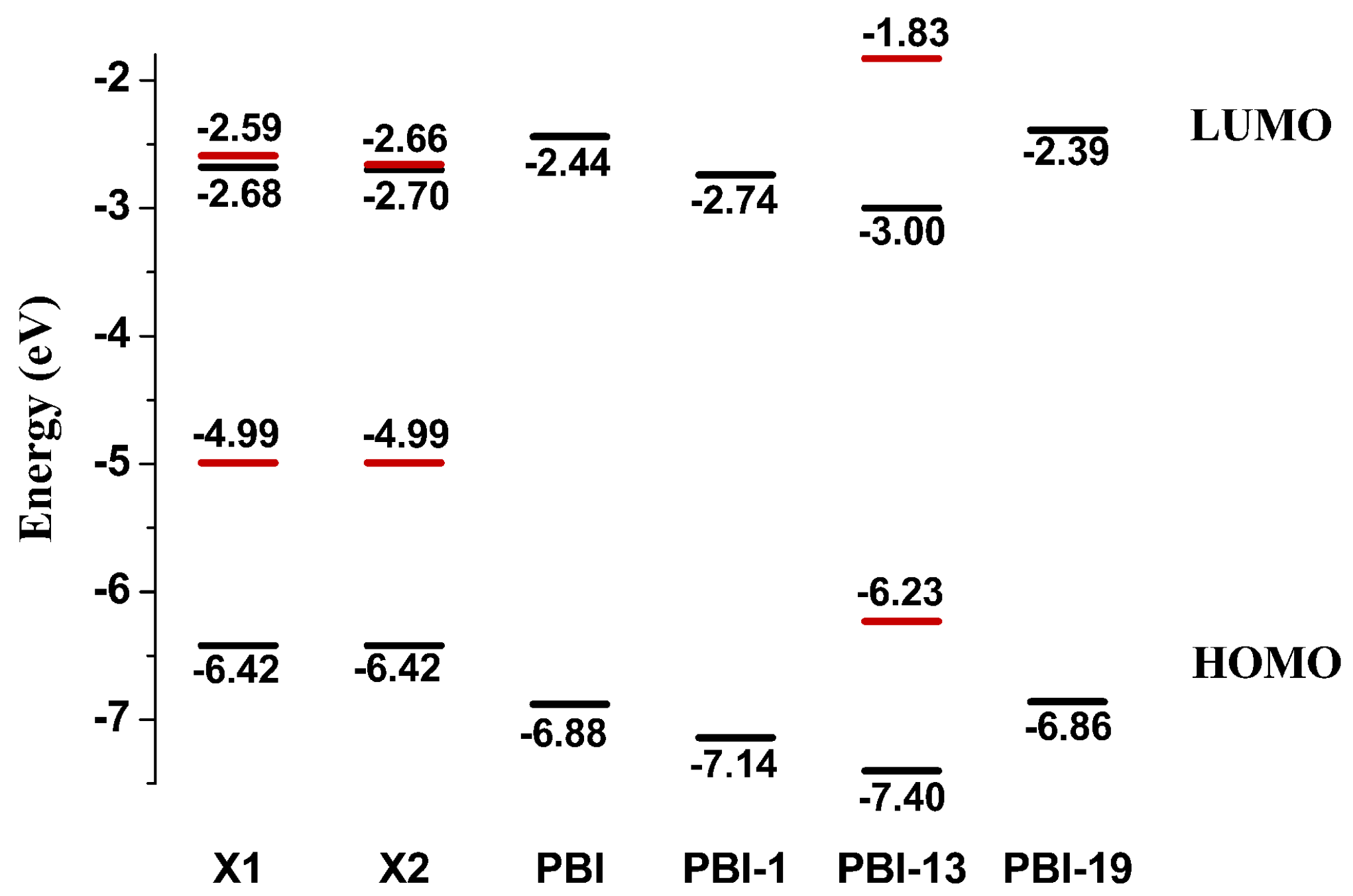
2.1. Frontier Molecular Orbitals
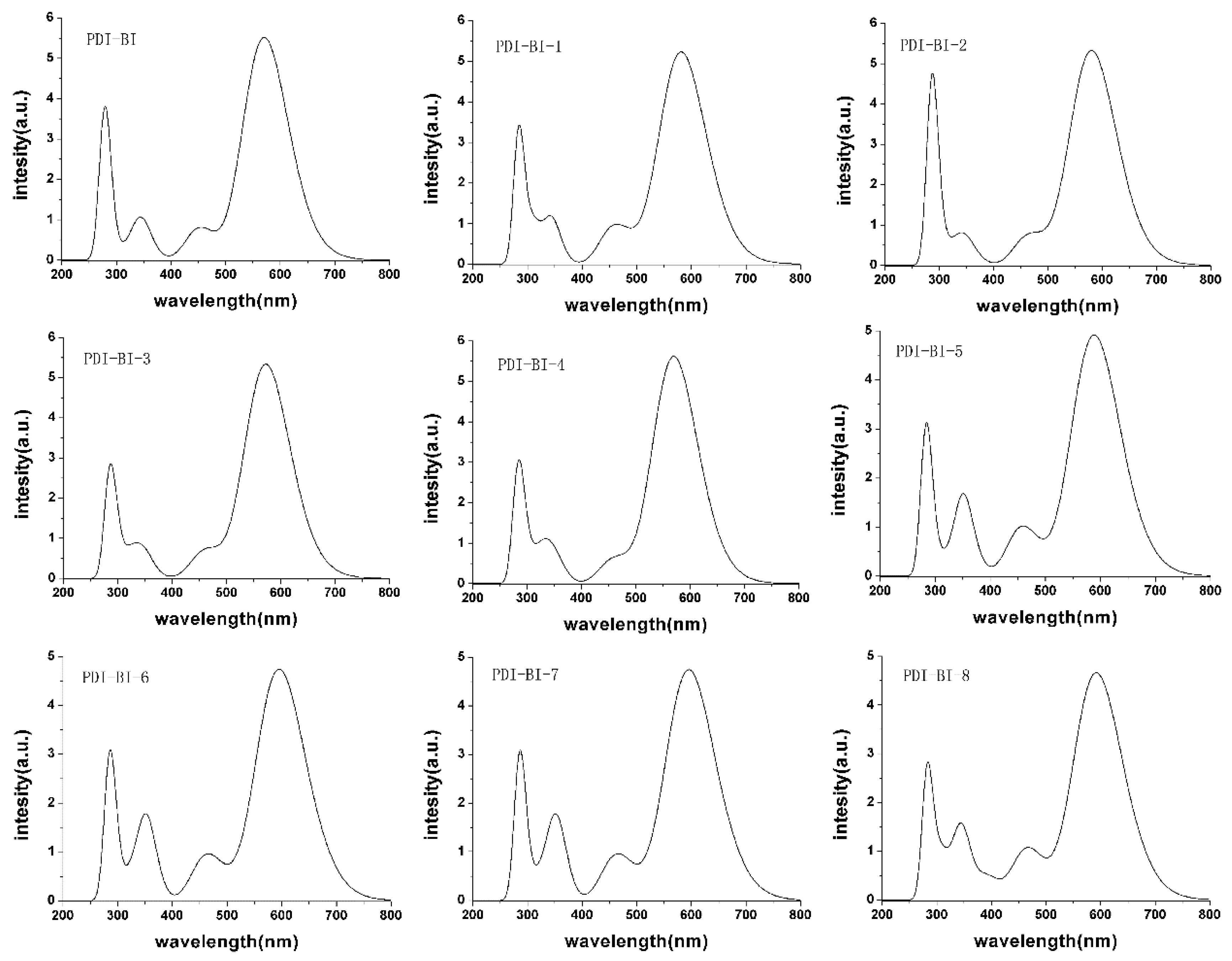
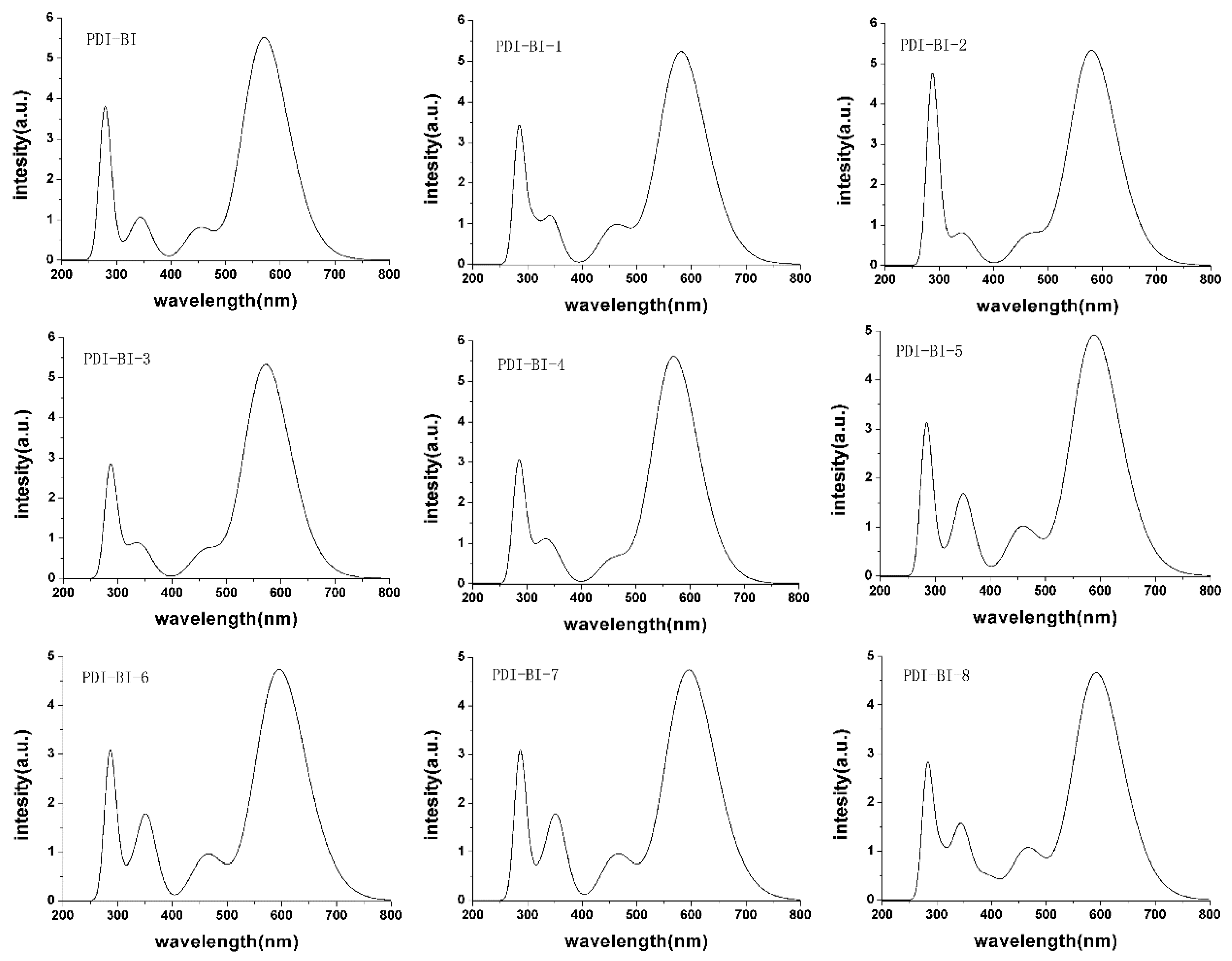
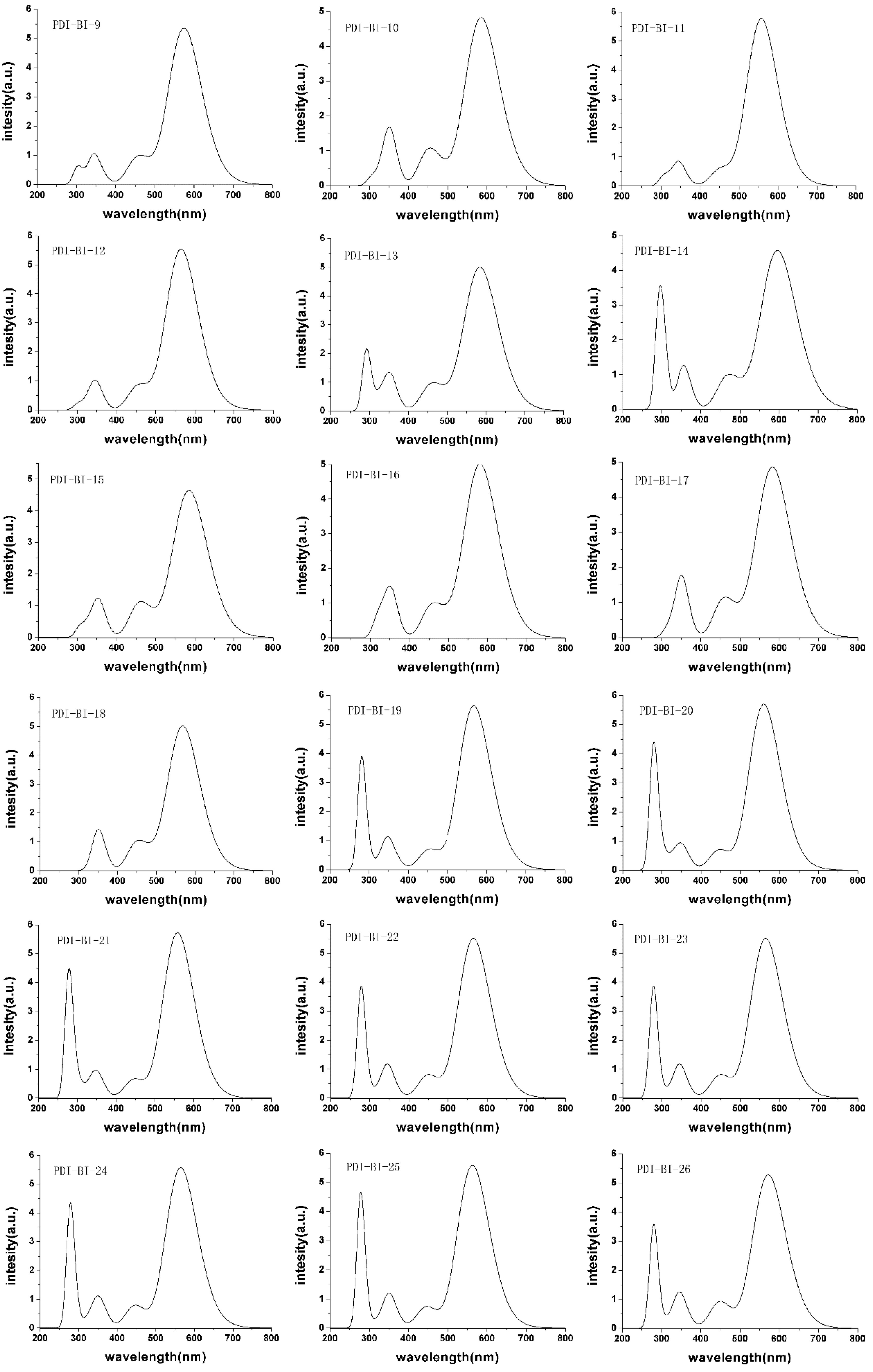
2.2. Absorption Spectra
2.3. Reorganization Energy
3. Materials and Methods
Computational Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PDI | Perylene diimide |
| OSCs | Organic solar cells |
| PCEs | Power conversion efficiencies |
| FMOs | Frontier molecular orbital energies |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| DFT | Density function theory |
References
- Sharenko, A.; Gehrig, D.; Laquai, F.; Nguyen, T.Q. The effect of solvent additive on the charge generation and photovoltaic performance of a solution-processed small molecule: Perylene diimide bulk heterojunction solar cell. Chem. Mater. 2014, 26, 4109–4118. [Google Scholar] [CrossRef]
- Darling, S.B.; You, F. The case for organic photovoltaics. RSC Adv. 2013, 3, 17633–17648. [Google Scholar] [CrossRef]
- Pelzera, K.M.; Darling, S.B. Charge generation in organic photovoltaics: A review of theory and computation. Mol. Syst. Des. Eng. 2016. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells. Nat. Commun. 2014, 5, 5293–5293. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-D.; Cui, C.; Li, Y.-Q.; Zhou, L.; Ou, Q.-D.; Li, C.; Li, Y.; Tang, J.-X. Polymer solar cells: Single-junction polymer solar cells exceeding 10% power conversion efficiency. Adv. Mater. 2015, 27, 1035–1041. [Google Scholar]
- Brunetti, F.G.; Gong, X.; Tong, M.; Heeger, A.J.; Wudl, F. Strain and hückel aromaticity: Driving forces for a promising new generation of electron acceptors in organic electronics. Angew. Chem. Int. Ed. 2010, 49, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Kim, J.-H.; Suh, H.; Kwak, J.; Kim, D.; Grimsdale, A.C.; Yoon, S.C.; Hwang, D.-H. High open circuit voltage organic photovoltaic cells fabricated using 9,9′-bifluorenylidene as a non-fullerene type electron acceptor. Chem. Commun. 2013, 49, 10950–10952. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.L.; Jia, T.; Kang, B.N.; Li, F.H.; Fahlman, M.; Wang, Y. Nitrile-substituted QA derivatives: New acceptor materials for solution-processable organic bulk heterojunction solar cells. Adv. Energy Mater. 2011, 1, 431–439. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Cheng, P.; Li, Y.F.; Zhan, X.W. A 3D star-shaped non-fullerene acceptor for solution-processed organic solar cells with a high open-circuit voltage of 1.18 V. Chem. Commun. 2012, 48, 4773–4775. [Google Scholar] [CrossRef] [PubMed]
- Sonar, P.; Ng, G.-M.; Lin, T.T.; Dodabalapur, A.; Chen, Z.-K. Solution processable low bandgap diketopyrrolopyrrole (DPP) based derivatives: Novel acceptors for organic solar cells. J. Mater. Chem. 2010, 20, 3626–3636. [Google Scholar]
- Woo, C.H.; Holcombe, T.W.; Unruh, D.A.; Sellinger, A.; Frechet, J.M.J. Phenyl vs. alkyl polythiophene: A solar cell comparison using a vinazene derivative as acceptor. Chem. Mater. 2010, 22, 1673–1679. [Google Scholar] [CrossRef]
- Bloking, J.T.; Han, X.; Higgs, A.T.; Kastrop, J.P.; Pandey, L.; Norton, J.E.; Risko, C.; Chen, C.E.; Bredas, J.-L.; Mc Gehee, M.D.; et al. Solution-processed organic solar cells with power conversion efficiencies of 2.5% using benzothiadiazole/imide-based acceptors. Chem. Mater. 2011, 23, 5484–5490. [Google Scholar] [CrossRef]
- Zhou, Y.; Ding, L.; Shi, K.; Dai, Y.-Z.; Ai, N.; Wang, J.; Pei, J. A non-fullerene small molecule as efficient electron acceptor in organic bulk heterojunction solar cells. Adv. Mater. 2012, 24, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dai, Y.-Z.; Zheng, Y.-Q.; Wang, X.-Y.; Wang, J.-Y.; Pei, J. Non-fullerene acceptors containing fluoranthene-fused imides for solution-processed inverted organic solar cells. Chem. Commun. 2013, 49, 5802–5804. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Ahmed, E.; Jenekhe, S.A. Non-fullerene acceptor-based bulk heterojunction polymer solar cells: Engineering the nanomorphology via processing additives. Adv. Energy Mater. 2011, 1, 946–953. [Google Scholar] [CrossRef]
- Ahmed, E.; Ren, G.; Kim, F.S.; Hollenbeck, E.C.; Jenekhe, S.A. Design of new electron acceptor materials for organic photovoltaics: Synthesis, electron transport, photophysics, and photovoltaic properties of oligothiophene-functionalized naphthalene diimides. Chem. Mater. 2011, 23, 4563–4571. [Google Scholar] [CrossRef]
- Shu, Y.; Lim, Y.-F.; Li, Z.; Purushothaman, B.; Hallani, R.; Kim, J.E.; Parkin, S.R.; Malliaras, G.G.; Anthony, J.E. A survey of electron-deficient pentacenes as acceptors in polymer bulk heterojunction solar cells. Chem. Sci. 2011, 2, 363–368. [Google Scholar] [CrossRef]
- Schmidt-Mende, L.; Fechtenkötter, A.; Müllen, K.; Moons, E.; Friend, R.H.; MacKenzie, J.D. Self-organized discotic liquid crystals for high-efficiency organic photovoltaics. Science 2001, 293, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Oku, T.; Takeda, A.; Nagata, A.; Kidowaki, H.; Kumada, K.; Fujimoto, K.; Suzuki, A.; Akiyama, T.; Yamasaki, Y.; Ōsawa, E. Microstructures and photovoltaic properties of C60 based solar cells with copper oxides, CuInS2, phthalocyanines, porphyrin, PVK, nanodiamond, germanium and exciton diffusion blocking layers. Mater. Technol. 2013, 28, 21–39. [Google Scholar] [CrossRef]
- Sharenko, A.; Proctor, C.M.; van der Poll, T.S.; Henson, Z.B.; Nguyen, T.-Q.; Bazan, G.C. A high-performing solution-processed small molecule: Perylene diimide bulk heterojunction solar cell. Adv. Mater. 2013, 25, 4403–4406. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.H.; Jiang, B.; Zhang, X.; Tang, A.L.; Chen, L.L.; Zhan, C.L.; Yao, J.N. Perylene-diimide based non-fullerene solar cells with 4.34% efficiency through engineering surface donor/acceptor compositions. Chem. Mater. 2014, 26, 2907–2914. [Google Scholar] [CrossRef]
- Würthner, F. Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures. Chem. Commun. 2004, 1564–1579. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Sun, K.; Jiang, W.; Zhang, S.Q.; Zhao, W.C.; Yao, H.F.; Wang, Z.H.; Hou, J.H. Enhanced efficiency in fullerene-free polymer solar cell by incorporating fine-designed donor and acceptor materials. ACS Appl. Mater. Interfaces 2015, 7, 9274–9280. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.C.R.; Kim, E.-G.; da Silva Filho, D.A.; Bredas, J.-L. Tuning the charge-transport parameters of perylene diimide single crystals via end and/or core functionalization: A density functional theory investigation. J. Am. Chem. Soc. 2010, 132, 3375–3387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, Y.; Liu, Y. 25th anniversary article: Recent advances in n-type and ambipolar organic field-effect transistors. Adv. Mater. 2013, 25, 5372–5391. [Google Scholar] [CrossRef] [PubMed]
- Weitz, R.T.; Amsharov, K.; Zschieschang, U.; Burghard, M.; Jansen, M.; Kelsch, M.; Rhamati, B.; van Aken, P.A.; Kern, K.; Klauk, H. The importance of grain boundaries for the time-dependent mobility degradation in organic thin-film transistors. Chem. Mater. 2009, 21, 4949–4954. [Google Scholar] [CrossRef]
- Schmidt, R.D.; Oh, J.H.; Sun, Y.-S.; Deppisch, M.; Krause, A.-M.; Radacki, K.; Braunschweig, H.; Könemann, M.; Erk, P.; Bao, Z.N.; et al. High-performance air-stable n-channel organic thin film transistors based on halogenated perylene bisimide semiconductors. J. Am. Chem. Soc. 2009, 131, 6215–6220. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.S.; Jeong, H.-H.; Kim, M.-K.; Jin, S.-H.; Kim, M.-R.; Lee, J.-K.; Lee, J.W.; Gal, Y.-S. Effects of functional groups at perylene diimide derivatives on organic photovoltaic device application. J. Mater. Chem. 2006, 16, 384–390. [Google Scholar] [CrossRef]
- Bibi, S.; Li, P.; Zhang, J.P. X-Shaped donor molecules based on benzo[2,1-b:3,4-b′]dithiophene as organic solar cell materials with PDIs as acceptors. J. Mater. Chem. A 2013, 1, 13828–13841. [Google Scholar] [CrossRef]
- Yong, X.; Zhang, J.P. Theoretical investigations for organic solar cells. Mater. Technol. 2013, 28, 40–64. [Google Scholar] [CrossRef]
- Tang, S.S.; Liang, D.D.; Chen, G.; Jin, R.F. Design of acceptors based on perylene diimide toward organic solar cell materials. Mater. Technol. 2015, 30, 230–240. [Google Scholar] [CrossRef]
- Yong, X.; Zhang, J.P. A rational design strategy for donors in organic solar cells: The conjugated planar molecules possessing anisotropic multibranches and intramolecular charge transfer. J. Mater. Chem. 2011, 21, 11159–11166. [Google Scholar] [CrossRef]
- Tang, S.S.; Zhang, J.P. Design of donors with broad absorption regions and suitable frontier molecular orbitals to match typical acceptors via substitution on oligo(thienylenevinylene) toward solar cells. J. Comput. Chem. 2012, 33, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University: Oxford, UK, 1989. [Google Scholar]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic ecitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Rao, A.; Chow, P.C.Y.; Gélinas, S.; Schlenker, C.W.; Li, C.Z.; Yip, H.L.; Jen, A.K.Y.; Ginger, D.S.; Friend, R.H. The role of spin in the kinetic control of recombination in organic photovoltaics. Nature 2013, 500, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Schlenker, C.W.; Chen, K.S.; Yip, H.L.; Li, C.Z.; Bradshaw, L.R.; Ochsenbein, S.T.; Ding, F.; Li, X.S.; Gamelin, D.R.; Jen, A.K.Y.; et al. Polymer triplet energy levels need not limit photocurrent collection in organic solar cells. J. Am. Chem. Soc. 2012, 134, 19661–19668. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, M.; Sperlich, A.; Kraus, H.; Baumann, A.; Deibel, C.; Wirix, M.J.M.; Loos, J.; Cardona, C.M.; Dyakonov, V. Triplet exciton generation in bulk-heterojunction solar cells based on endohedral fullerenes. J. Am. Chem. Soc. 2011, 133, 9088–9094. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Vos, J.G. GaussSum 1.0; Dublin City University: Dublin, Ireland, 2003. [Google Scholar]
- Lin, B.C.; Cheng, C.P.; You, Z.Q.; Hsu, C.P. Charge transport properties of tris(8-hydroxyquinolinato)aluminum(III): Why it is an electron transporter. J. Am. Chem. Soc. 2005, 127, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Gruhn, N.E.; da Silva Filho, D.A.; Bill, T.G.; Malagoli, M.; Coropceanu, V.; Kahn, A.; Brédas, J.L. The vibrational reorganization energy in pentacene: Molecular influences on charge transport. J. Am. Chem. Soc. 2002, 124, 7918–7919. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Suite of Programs; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wang, L.J.; Nan, G.J.; Yang, X.D.; Peng, Q.; Li, Q.K.; Shuai, Z.G. Computational methods for design of organic materials with high charge mobility. Chem. Soc. Rev. 2010, 39, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Köse, M.E.; Mitchell, W.J.; Kopidakis, N.; Chang, C.H.; Shaheen, S.E.; Kim, K.; Rumbles, G. Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 2007, 129, 14257–14270. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | R-Groups | Molecules | R-Groups |
|---|---|---|---|
| PDI-BI-1 | R1 = –CN | PDI-BI-14 | R3 = –CN R6 = –CN |
| PDI-BI-2 | R2 = –CN | PDI-BI-15 | R3 = –CN R6 = –NO2 |
| PDI-BI-3 | R3 = –CN | PDI-BI-16 | R4 = –CN R5 = –NO2 |
| PDI-BI-4 | R4 = –CN | PDI-BI-17 | R4 = –NO2 R5 = –NO2 |
| PDI-BI-5 | R5 = –CN | PDI-BI-18 | R3 = –NO2 R6 = –NO2 |
| PDI-BI-6 | R6 = –CN | PDI-BI-19 | R1 = –CH3 |
| PDI-BI-7 | R7 = –CN | PDI-BI-20 | R2 = –CH3 |
| PDI-BI-8 | R8 = –CN | PDI-BI-21 | R3 = –CH3 |
| PDI-BI-9 | R1 = –NO2 | PDI-BI-22 | R4 = –CH3 |
| PDI-BI-10 | R2 = –NO2 | PDI-BI-23 | R5 = –CH3 |
| PDI-BI-11 | R3 = –NO2 | PDI-BI-24 | R6 = –CH3 |
| PDI-BI-12 | R4 = –NO2 | PDI-BI-25 | R7 = –CH3 |
| PDI-BI-13 | R4 = –CN R5 = –CN | PDI-BI-26 | R8 = –CH3 |
| EHOMO | ELOMO | Eg | λabs-max | λabs-min | R | |
|---|---|---|---|---|---|---|
| PDI-BI | −6.88 | −2.44 | 4.44 | 570.40 | 269.50 | 300.90 |
| PDI-BI-1 | −7.14 | −2.74 | 4.40 | 581.24 | 282.68 | 298.56 |
| PDI-BI-2 | −7.14 | −2.75 | 4.39 | 580.46 | 281.54 | 298.92 |
| PDI-BI-3 | −7.16 | −2.75 | 4.41 | 572.27 | 281.43 | 290.84 |
| PDI-BI-4 | −7.15 | −2.72 | 4.43 | 569.68 | 278.77 | 290.91 |
| PDI-BI-5 | −7.13 | −2.74 | 4.39 | 588.55 | 278.64 | 309.91 |
| PDI-BI-6 | −7.12 | −2.76 | 4.36 | 595.24 | 280.40 | 314.84 |
| PDI-BI-7 | −7.14 | −2.76 | 4.38 | 590.79 | 281.67 | 309.12 |
| PDI-BI-8 | −7.14 | −2.75 | 4.39 | 592.00 | 280.06 | 311.94 |
| PDI-BI-9 | −7.17 | −2.74 | 4.43 | 574.08 | 297.82 | 276.26 |
| PDI-BI-10 | −7.12 | −2.71 | 4.41 | 584.98 | 300.92 | 284.06 |
| PDI-BI-11 | −7.19 | −2.70 | 4.49 | 557.27 | 298.59 | 258.68 |
| PDI-BI-12 | −7.18 | −2.72 | 4.46 | 564.87 | 298.98 | 265.89 |
| PDI-BI-13 | −7.40 | −3.00 | 4.40 | 584.00 | 289.49 | 294.51 |
| PDI-BI-14 | −7.39 | −3.05 | 4.34 | 596.19 | 291.72 | 304.47 |
| PDI-BI-15 | −7.40 | −3.00 | 4.40 | 585.19 | 305.80 | 279.39 |
| PDI-BI-16 | −7.43 | −3.01 | 4.42 | 581.03 | 307.99 | 273.04 |
| PDI-BI-17 | −7.45 | −3.02 | 4.43 | 582.91 | 311.98 | 270.93 |
| PDI-BI-18 | −7.43 | −2.95 | 4.48 | 567.95 | 343.18 | 224.77 |
| PDI-BI-19 | −6.86 | −2.39 | 4.47 | 564.68 | 270.26 | 294.42 |
| PDI-BI-20 | −6.83 | −2.37 | 4.46 | 559.84 | 271.85 | 287.99 |
| PDI-BI-21 | −6.84 | −2.37 | 4.47 | 557.89 | 271.51 | 286.38 |
| PDI-BI-22 | −6.85 | −2.39 | 4.46 | 564.76 | 269.00 | 295.76 |
| PDI-BI-23 | −6.85 | −2.39 | 4.46 | 567.34 | 270.18 | 297.16 |
| PDI-BI-24 | −6.82 | −2.37 | 4.45 | 564.37 | 272.33 | 292.04 |
| PDI-BI-25 | −6.82 | −2.37 | 4.45 | 562.73 | 272.19 | 290.54 |
| PDI-BI-26 | −6.84 | −2.39 | 4.45 | 571.83 | 270.17 | 301.66 |
| λe | λh | |
|---|---|---|
| PDI-BI | 0.298 | 0.210 |
| PDI-BI-1 | 0.278 | 0.222 |
| PDI-BI-2 | 0.277 | 0.215 |
| PDI-BI-3 | 0.278 | 0.221 |
| PDI-BI-4 | 0.272 | 0.222 |
| PDI-BI-5 | 0.282 | 0.226 |
| PDI-BI-6 | 0.285 | 0.224 |
| PDI-BI-7 | 0.286 | 0.230 |
| PDI-BI-8 | 0.278 | 0.232 |
| PDI-BI-9 | 0.296 | 0.225 |
| PDI-BI-10 | 0.360 | 0.236 |
| PDI-BI-11 | 0.290 | 0.222 |
| PDI-BI-12 | 0.320 | 0.234 |
| PDI-BI-13 | 0.265 | 0.240 |
| PDI-BI-14 | 0.266 | 0.240 |
| PDI-BI-15 | 0.343 | 0.249 |
| PDI-BI-16 | 0.279 | 0.245 |
| PDI-BI-17 | 0.312 | 0.264 |
| PDI-BI-18 | 0.476 | 0.250 |
| PDI-BI-19 | 0.297 | 0.201 |
| PDI-BI-20 | 0.296 | 0.201 |
| PDI-BI-21 | 0.296 | 0.195 |
| PDI-BI-22 | 0.298 | 0.200 |
| PDI-BI-23 | 0.299 | 0.205 |
| PDI-BI-24 | 0.298 | 0.206 |
| PDI-BI-25 | 0.298 | 0.200 |
| PDI-BI-26 | 0.300 | 0.213 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Li, Z.; Li, S.; Luan, G.; Liang, D.; Tang, S.; Jin, R. Design of Acceptors with Suitable Frontier Molecular Orbitals to Match Donors via Substitutions on Perylene Diimide for Organic Solar Cells. Int. J. Mol. Sci. 2016, 17, 721. https://doi.org/10.3390/ijms17050721
Lv X, Li Z, Li S, Luan G, Liang D, Tang S, Jin R. Design of Acceptors with Suitable Frontier Molecular Orbitals to Match Donors via Substitutions on Perylene Diimide for Organic Solar Cells. International Journal of Molecular Sciences. 2016; 17(5):721. https://doi.org/10.3390/ijms17050721
Chicago/Turabian StyleLv, Xiaoli, Zhuoxin Li, Songyang Li, Guoyou Luan, Dadong Liang, Shanshan Tang, and Ruifa Jin. 2016. "Design of Acceptors with Suitable Frontier Molecular Orbitals to Match Donors via Substitutions on Perylene Diimide for Organic Solar Cells" International Journal of Molecular Sciences 17, no. 5: 721. https://doi.org/10.3390/ijms17050721