
Genome-Wide Transcriptome Profiling of Mycobacterium smegmatis MC2 155 Cultivated in Minimal Media Supplemented with Cholesterol, Androstenedione or Glycerol

Abstract
:1. Introduction
2. Results
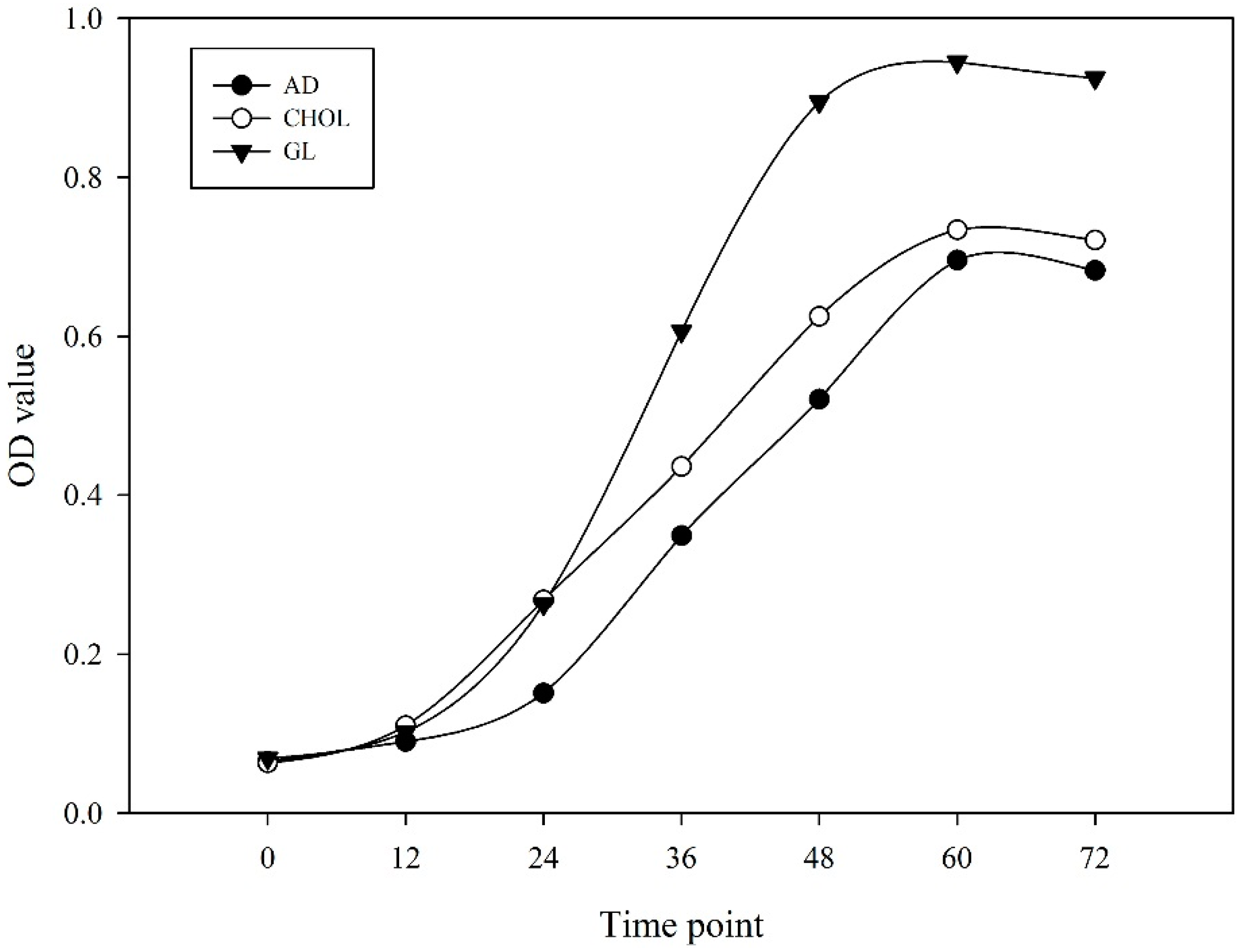
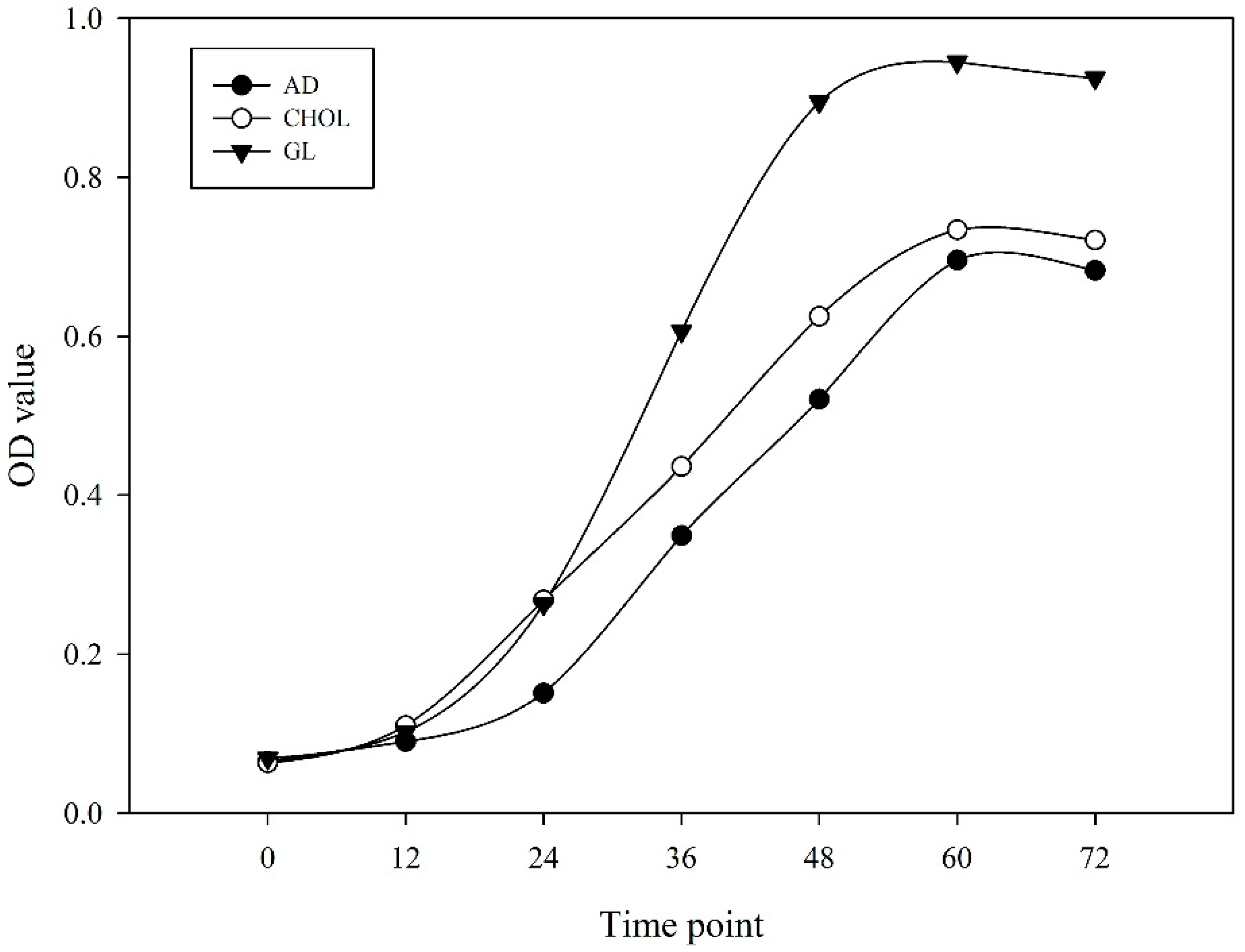
2.1. RNA Sequencing and Global Expression Patterns
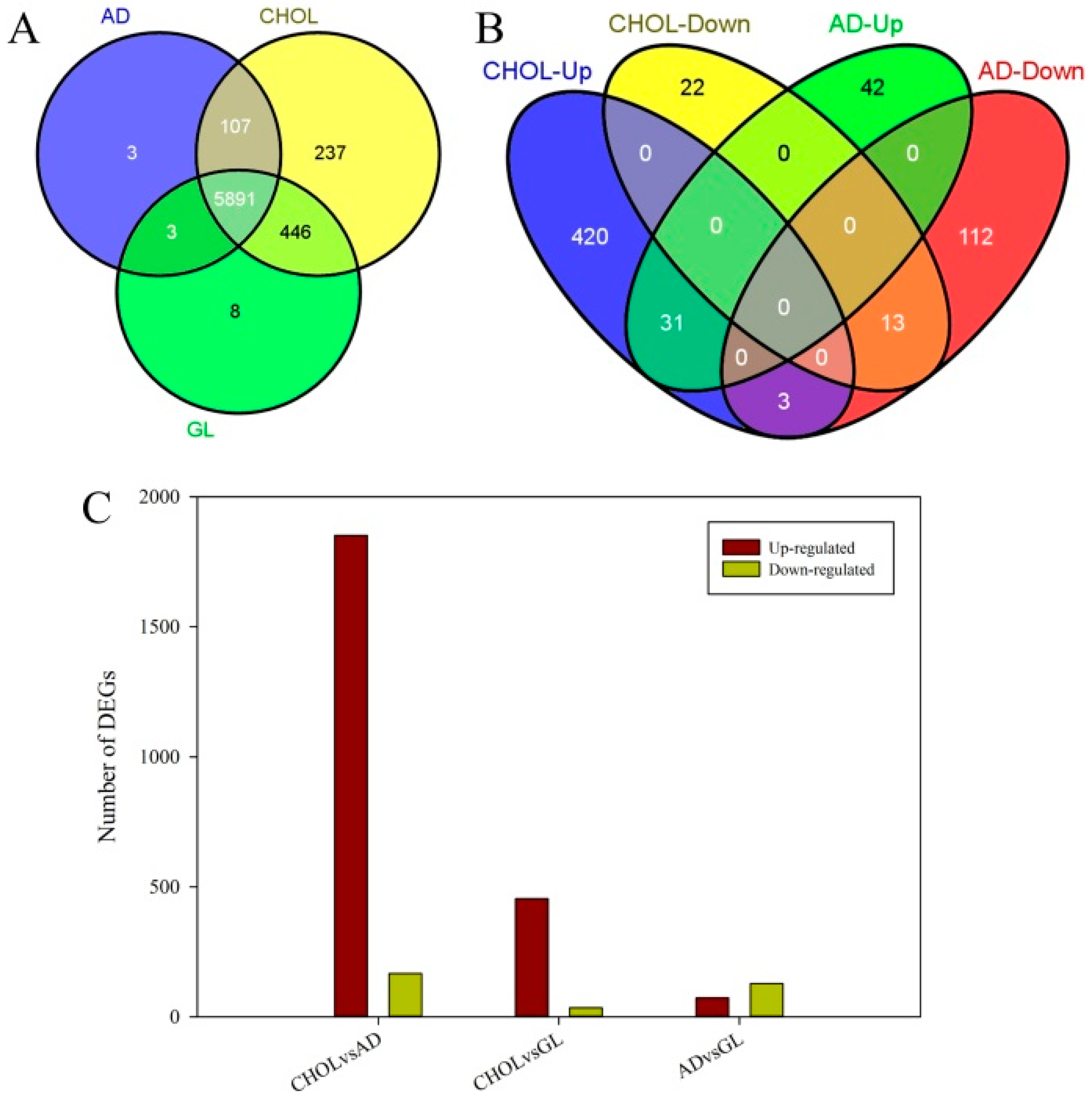
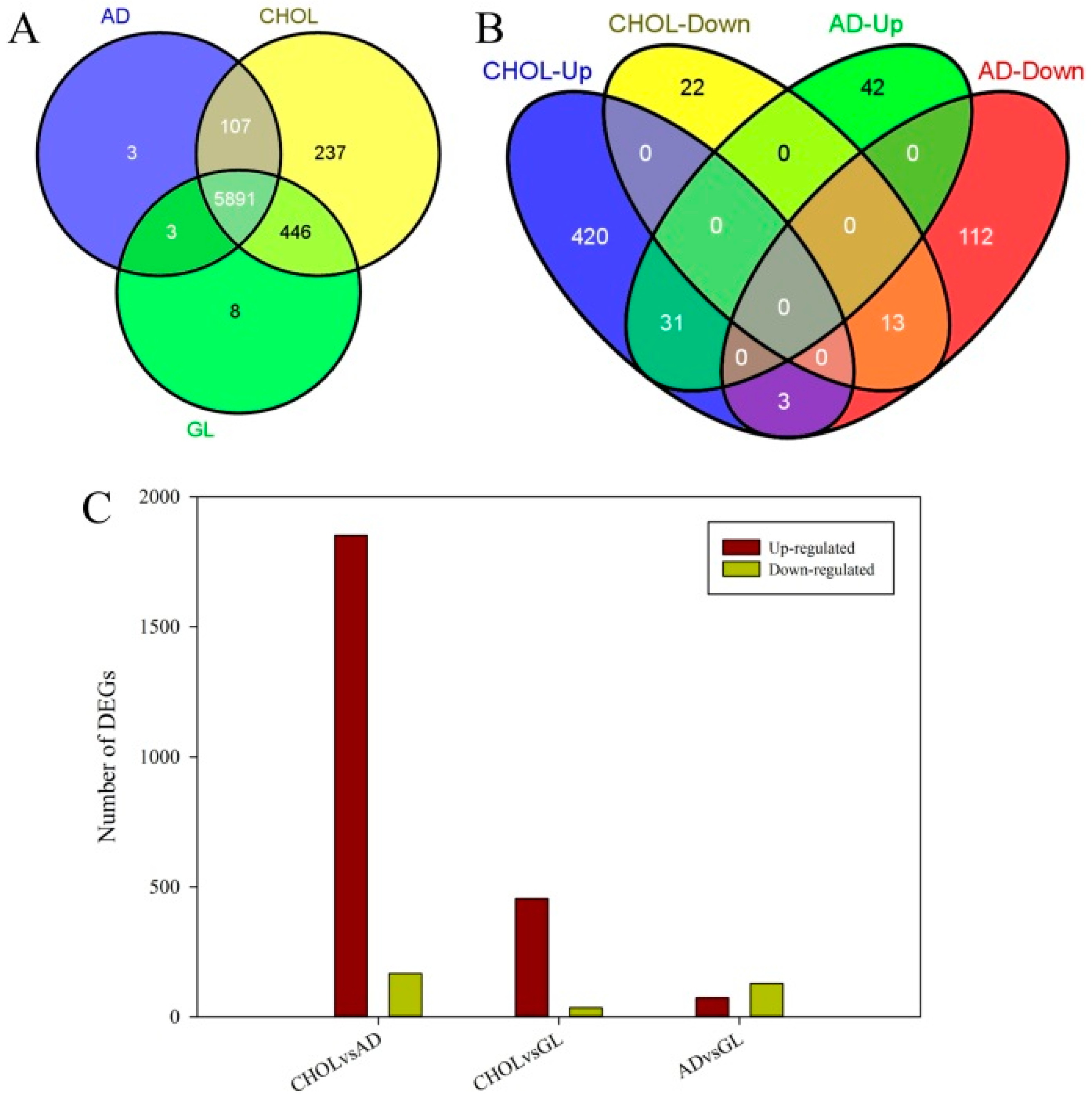
2.2. Differentially Expressed Genes and Functional Enrichment Analyses
2.3. Nitrogen and Carbon Metabolism Related Genes
2.4. Glycerol and Androstenedione Metabolism
2.5. Cholesterol Metabolism Related Genes
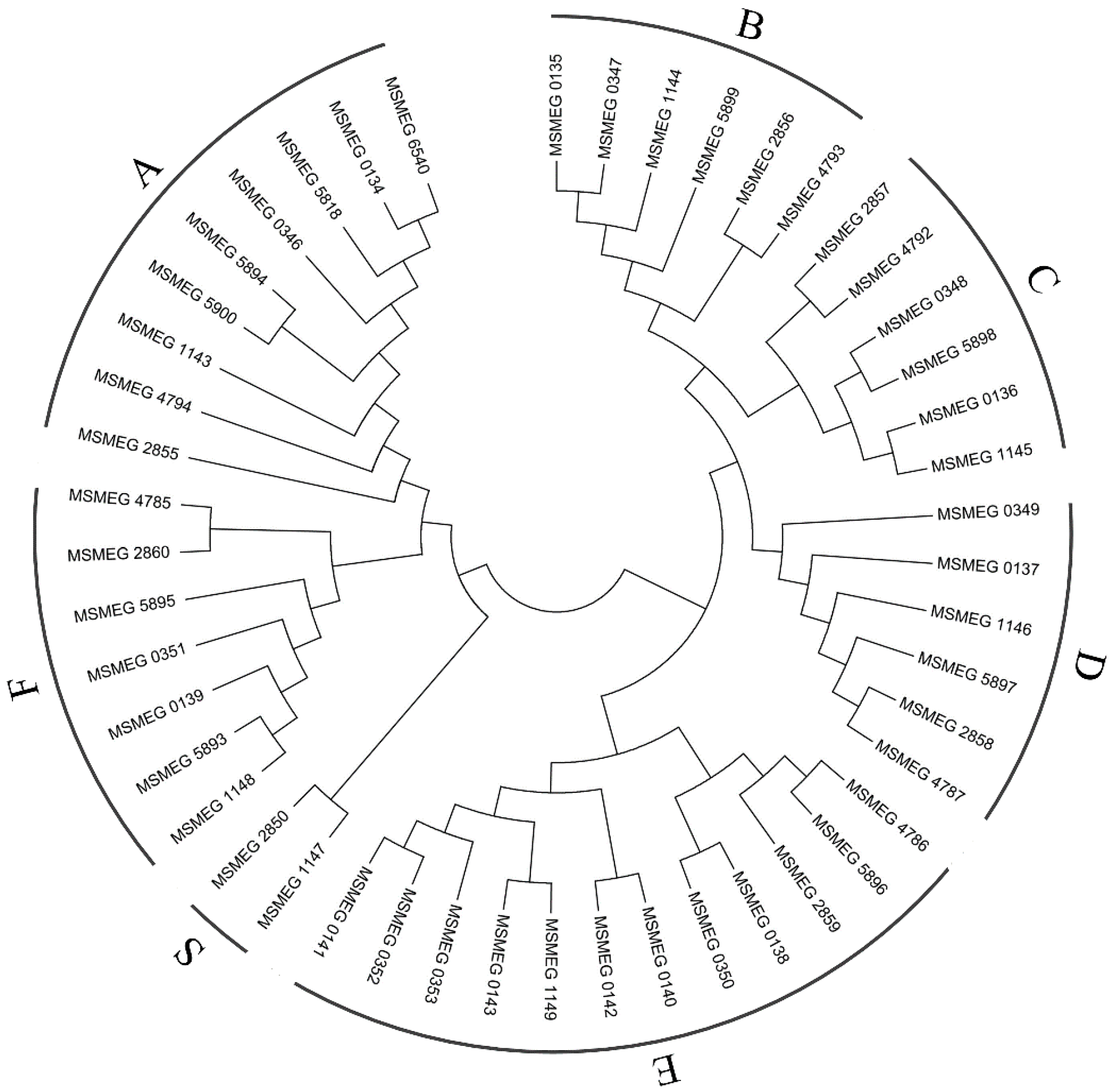
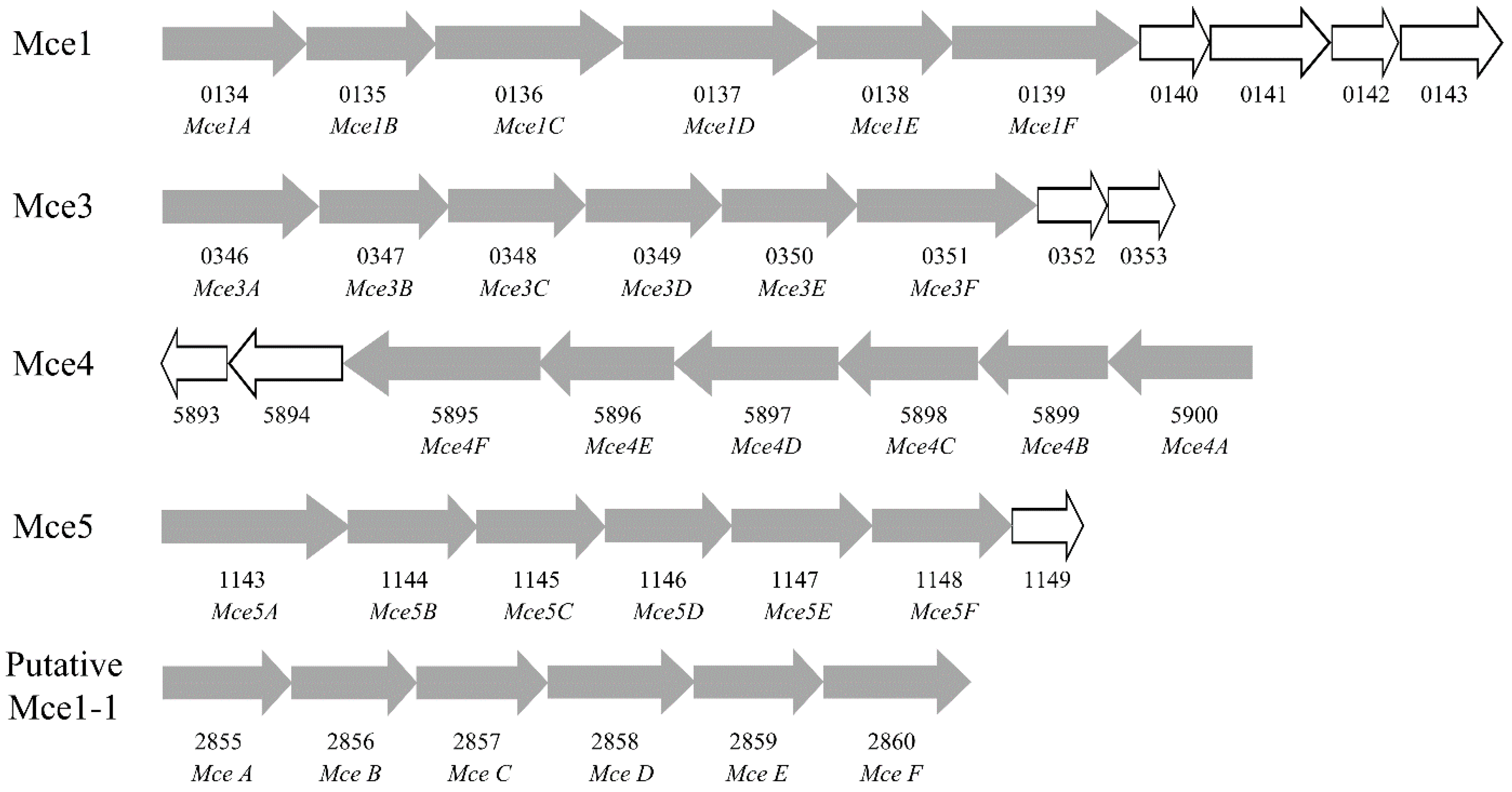
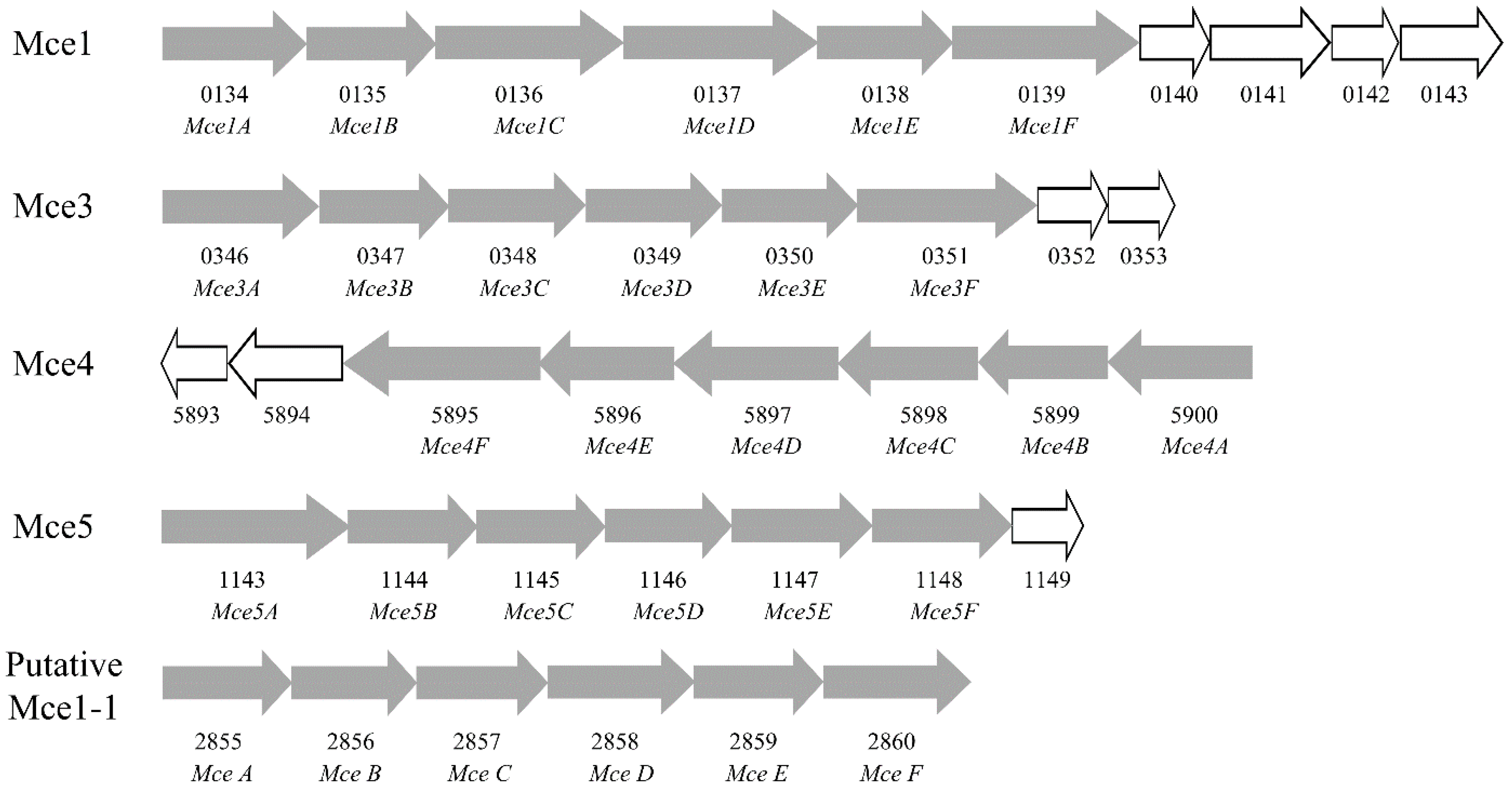
2.6. Mammalian Cell Entry Related Genes
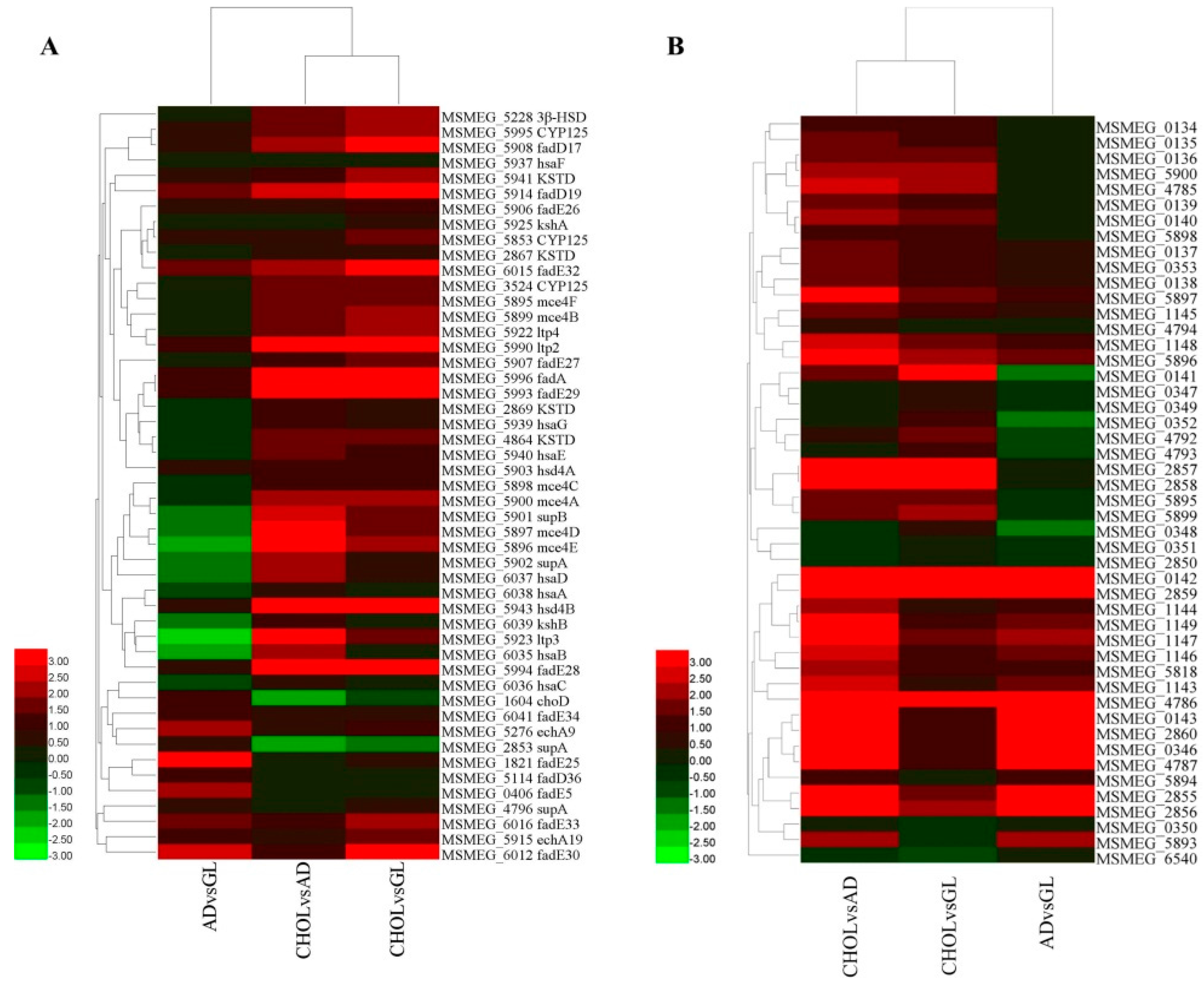
2.7. Gene Clusters Induced by Cholesterol
3. Discussion
3.1. Genome-Wide Transcriptome Changes Response to Different Supplements
3.2. Cholesterol Catabolism and Mammalian Cell Entry Related Genes
4. Materials and Methods
4.1. Bacterial Strains
4.2. Bacterial Cultivation and Sampling
4.3. RNA Extraction and Library Construction
4.4. RNA Sequencing and Data Analyzing
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Slaytor, M.; Bloch, K. Metabolic transformation of cholestenediols. J. Biol. Chem. 1965, 240, 4598–4602. [Google Scholar] [PubMed]
- Van der Geize, R.; Dijkhuizen, L. Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr. Opin. Microbiol. 2004, 7, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.L.; Uhia, I.; Galan, B. Catabolism and biotechnological applications of cholesterol degrading bacteria. Microb. Biotechnol. 2012, 5, 679–699. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.C. Use of HeLa cells infected with tubercle bacilli for the study of anti-tuberculous drugs. J. Bacteriol. 1957, 73, 494–498. [Google Scholar] [PubMed]
- Kress, G. Combining dental training with medical training. J. Dent. Educ. 1995, 59, 1061. [Google Scholar] [PubMed]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef] [PubMed]
- Turfitt, G. Microbiological agencies in the degradation of steroids: I. The cholesterol-decomposing organisms of soils. J. Bacteriol. 1944, 47, 487. [Google Scholar] [PubMed]
- Turfitt, G. The microbiological degradation of steroids: 4. Fission of the steroid molecule. Biochem. J. 1948, 42, 376. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, J.; Bloom, B.R.; Rubin, E.J. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. USA 2005, 102, 8327–8332. [Google Scholar] [CrossRef] [PubMed]
- Van der Geize, R.; Yam, K.; Heuser, T.; Wilbrink, M.H.; Hara, H.; Anderton, M.C.; Sim, E.; Dijkhuizen, L.; Davies, J.E.; Mohn, W.W.; et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Arruda, S.; Bomfim, G.; Knights, R.; Huima-Byron, T.; Riley, L.W. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science 1993, 261, 1454–1457. [Google Scholar] [CrossRef] [PubMed]
- Mohn, W.W.; van der Geize, R.; Stewart, G.R.; Okamoto, S.; Liu, J.; Dijkhuizen, L.; Eltis, L.D. The actinobacterial MCE4 locus encodes a steroid transporter. J. Biol. Chem. 2008, 283, 35368–35374. [Google Scholar] [CrossRef] [PubMed]
- van der Geize, R.; de Jong, W.; Hessels, G.I.; Grommen, A.W.; Jacobs, A.A.; Dijkhuizen, L. A novel method to generate unmarked gene deletions in the intracellular pathogen Rhodococcus equi using 5-fluorocytosine conditional lethality. Nucleic Acids Res. 2008, 36, e151. [Google Scholar] [CrossRef] [PubMed]
- Kreit, J.; Sampson, N.S. Cholesterol oxidase: Physiological functions. FEBS J. 2009, 276, 6844–6856. [Google Scholar] [CrossRef] [PubMed]
- De las Heras, L.F.; Mascaraque, V.; Fernandez, E.G.; Navarro-Llorens, J.M.; Perera, J.; Drzyzga, O. ChoG is the main inducible extracellular cholesterol oxidase of Rhodococcus sp. strain CECT3014. Microbiol. Res. 2011, 166, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Capyk, J.K.; Kalscheuer, R.; Stewart, G.R.; Liu, J.; Kwon, H.; Zhao, R.; Okamoto, S.; Jacobs, W.R., Jr.; Eltis, L.D.; Mohn, W.W. Mycobacterial cytochrome P450 125 (CYP125) catalyzes the terminal hydroxylation of c27 steroids. J. Biol. Chem. 2009, 284, 35534–35542. [Google Scholar] [CrossRef] [PubMed]
- Rosloniec, K.Z.; Wilbrink, M.H.; Capyk, J.K.; Mohn, W.W.; Ostendorf, M.; van der Geize, R.; Dijkhuizen, L.; Eltis, L.D. Cytochrome P450 125 (CYP125) catalyses c26-hydroxylation to initiate sterol side-chain degradation in Rhodococcus jostii rha1. Mol. Microbiol. 2009, 74, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, M.D.; McLean, K.J.; Levy, C.; Mast, N.; Pikuleva, I.A.; Lafite, P.; Rigby, S.E.J.; Leys, D.; Munro, A.W. Structural and biochemical characterization of Mycobacterium tuberculosis CYP142. J. Biol. Chem. 2010, 285, 38270–38282. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Ouellet, H.; Ortiz de Montellano, P.R. Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J. Biol. Chem. 2010, 285, 36352–36360. [Google Scholar] [CrossRef] [PubMed]
- Sih, C.J.; Wang, K.-C.; Tai, H.-H. Mechanisms of steroid oxidation by microorganisms. XIII. C22 acid intermediates in the degradation of the cholesterol side chain. Biochemistry 1968, 7, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Sih, C.J.; Tai, H.-H.; Tsong, Y.Y.; Lee, S.S.; Coombe, R.G. Mechanisms of steroid oxidation by microorgansism. XIV. Pathway of cholesterol side-chain degradation. Biochemistry 1968, 7, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Brzostek, A.; Sliwinski, T.; Rumijowska-Galewicz, A.; Korycka-Machala, M.; Dziadek, J. Identification and targeted disruption of the gene encoding the main 3-ketosteroid dehydrogenase in Mycobacterium smegmatis. Microbiology 2005, 151, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Van der Geize, R.; Hessels, G.I.; van Gerwen, R.; Vrijbloed, J.W.; van der Meijden, P.; Dijkhuizen, L. Targeted disruption of the kstD gene encoding a 3-ketosteroid Δ1-dehydrogenase isoenzyme of Rhodococcus erythropolis strain SQ1. Appl. Environ. Microbiol. 2000, 66, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Van der Geize, R.; Hessels, G.I.; Dijkhuizen, L. Molecular and functional characterization of the kstD2 gene of Rhodococcus erythropolis SQ1 encoding a second 3-ketosteroid Δ1-dehydrogenase isoenzyme. Microbiology 2002, 148, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- King, G.M. Uptake of carbon monoxide and hydrogen at environmentally relevant concentrations by mycobacteria. Appl. Environ. Microb. 2003, 69, 7266–7272. [Google Scholar] [CrossRef]
- Snapper, S.B.; Melton, R.E.; Mustafa, S.; Kieser, T.; Jacobs, W.R., Jr. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 1990, 4, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Uhia, I.; Galan, B.; Kendall, S.L.; Stoker, N.G.; Garcia, J.L. Cholesterol metabolism in Mycobacterium smegmatis. Environ. Microbiol. Rep. 2012, 4, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Hurd, P.J.; Nelson, C.J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief. Funct. Genom. 2009, 8, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- ’t Hoen, P.A.C.; Ariyurek, Y.; Thygesen, H.H.; Vreugdenhil, E.; Vossen, R.H.; de Menezes, R.X.; Boer, J.M.; van Ommen, G.-J.B.; den Dunnen, J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36, e141. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online: http://bioinfogp.cnb.csic.es/tools/venny/ (accessed on 8 January 2015).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar]
- Richardson, D.J.; Watmough, N.J. Inorganic nitrogen metabolism in bacteria. Curr. Opin. Chem. Biol. 1999, 3, 207–219. [Google Scholar] [CrossRef]
- Reitzer, L. Nitrogen assimilation and global regulation in Escherichia coli. Annu. Rev. Microbiol. 2003, 57, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Whitesides, G.M. Glycerol kinase: Synthesis of dihydroxyacetone phosphate, sn-glycerol-3-phosphate, and chiral analogs. J. Am. Chem. Soc. 1985, 107, 7019–7027. [Google Scholar] [CrossRef]
- Abbad-Andaloussi, S.; Guedon, E.; Spiesser, E.; Petitdemange, H. Glycerol dehydratase activity: The limiting step for 1,3-propanediol production by Clostridium butyricum DSM 5431. Lett. Appl. Microbiol. 1996, 22, 311–314. [Google Scholar] [CrossRef]
- Boenigk, R.; Bowien, S.; Gottschalk, G. Fermentation of glycerol to 1,3-propanediol in continuous cultures of Citrobacter freundii. Appl. Microbiol. Biotechnol. 1993, 38, 453–457. [Google Scholar] [CrossRef]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xie, J.P. Mammalian cell entry gene family of Mycobacterium tuberculosis. Mol. Cell. Biochem. 2011, 352, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, P.; Brutnell, T.P. Exploring plant transcriptomes using ultra high-throughput sequencing. Brief. Funct. Genom. 2010, 9, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.N.; Schlueter, J.; Spooner, D.M. Applications of next-generation sequencing in plant biology. Am. J. Bot. 2012, 99, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, V.A.; Barton, G.R.; Robertson, B.D.; Williams, K.J. Genome wide analysis of the complete GlnR nitrogen-response regulon in Mycobacterium smegmatis. BMC Genom. 2013, 14, 301. [Google Scholar] [CrossRef] [PubMed]
- Tak, J. On bacteria decomposing cholesterol. Anton. Leeuw. 1942, 8, 32–40. [Google Scholar] [CrossRef]
- Turfitt, G. Microbiological agencies in the degradation of steroids: I. Steroid utilization by the microflora of soils. J. Bacteriol. 1947, 54, 557. [Google Scholar] [PubMed]
- De la Paz Santangelo, M.; Klepp, L.; Nunez-Garcia, J.; Blanco, F.C.; Soria, M.; Garcia-Pelayo, M.C.; Bianco, M.V.; Cataldi, A.A.; Golby, P.; Jackson, M.; et al. Mce3R, a TetR-type transcriptional repressor, controls the expression of a regulon involved in lipid metabolism in Mycobacterium tuberculosis. Microbiology 2009, 155, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ge, F.; Zhang, Q.; Ren, Y.; Yuan, J.; He, J.; Chen, G.; Zhang, G.; Zhuang, Y.; Xu, L. Identification of gene expression profiles in the actinomycete gordonia neofelifaecis grown with different steroids. Genome 2014, 57, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Casali, N.; White, A.M.; Riley, L.W. Regulation of the Mycobacterium tuberculosis MCE1 operon. J. Bacteriol. 2006, 188, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bose, M.; Brahmachari, V. Analysis of expression profile of mammalian cell entry (MCE) operons of Mycobacterium tuberculosis. Infect. Immun. 2003, 71, 6083–6087. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Nesbitt, N.M.; Dubnau, E.; Smith, I.; Sampson, N.S. Cholesterol metabolism increases the metabolic pool of propionate in Mycobacterium tuberculosis. Biochemistry 2009, 48, 3819–3821. [Google Scholar] [CrossRef] [PubMed]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages insights into the phagosomal environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Uhia, I.; Galan, B.; Morales, V.; Garcia, J.L. Initial step in the catabolism of cholesterol by Mycobacterium smegmatis MC2 155. Environ. Microbiol. 2011, 13, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.X.; Dubnau, E.; Smith, I.; Sampson, N.S. Rv1106c from Mycobacterium tuberculosis is a 3 β-hydroxysteroid dehydrogenase. Biochemistry 2007, 46, 9058–9067. [Google Scholar] [CrossRef] [PubMed]
- Capyk, J.K.; D’Angelo, I.; Strynadka, N.C.; Eltis, L.D. Characterization of 3-ketosteroid 9α-hydroxylase, a rieske oxygenase in the cholesterol degradation pathway of Mycobacterium tuberculosis. J. Biol. Chem. 2009, 284, 9937–9946. [Google Scholar] [CrossRef] [PubMed]
- Yam, K.C.; D’Angelo, I.; Kalscheuer, R.; Zhu, H.; Wang, J.X.; Snieckus, V.; Ly, L.H.; Converse, P.J.; Jacobs, W.R., Jr.; Strynadka, N.; et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009, 5, e1000344. [Google Scholar] [CrossRef] [PubMed]
- Van der Geize, R.; Hessels, G.I.; van Gerwen, R.; van der Meijden, P.; Dijkhuizen, L. Unmarked gene deletion mutagenesis of KstD, encoding 3-ketosteroid Δ1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counter-selectable marker. FEMS Microbiol. Lett. 2001, 205, 197–202. [Google Scholar] [CrossRef]
- Van der Geize, R.; Hessels, G.; van Gerwen, R.; van der Meijden, P.; Dijkhuizen, L. Molecular and functional characterization of kshA and kshB, encoding two components of 3-ketosteroid 9α-hydroxylase, a class IA monooxygenase, in Rhodococcus erythropolis strain SQ1. Mol. Microbiol. 2002, 45, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.; Wang, K.; Sih, C.J.; Whitlock, H. Mechanisms of steroid oxidation by microorganisms IX. On the mechanism of ring a cleavage in the degradation of 9,10-seco steroids by microorganisms. J. Biol. Chem. 1966, 241, 551–559. [Google Scholar] [PubMed]
- Kendall, S.L.; Withers, M.; Soffair, C.N.; Moreland, N.J.; Gurcha, S.; Sidders, B.; Frita, R.; Ten Bokum, A.; Besra, G.S.; Lott, J.S.; et al. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol. Microbiol. 2007, 65, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, E.; Medrano, F.J.; Galan, B.; Garcia, J.L. Deciphering the transcriptional regulation of cholesterol catabolic pathway in mycobacteria: Identification of the inducer of KstR repressor. J. Biol. Chem. 2014, 289, 17576–17588. [Google Scholar] [CrossRef] [PubMed]
- Mycobacterium smegmatis MC2 155 Genome. Available online: http://www.ncbi.nlm.nih.gov/nuccore/NC_008596.1 (accessed on 8 January 2015).
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Read Classification | AD | CHOL | GL |
|---|---|---|---|
| Total Reads | 4,727,962 | 5,524,220 | 5,432,638 |
| Mapped | 4,630,566 | 5,403,240 | 5,325,615 |
| Unmapped | 97,396 | 120,980 | 107,023 |
| rRNA Mapped | 38,769 | 28,174 | 19,557 |
| Gene Name | Gene_ID | Expression Level | ||
|---|---|---|---|---|
| Myco_1 | Myco_2 | Myco_3 | ||
| ammonium transporter | MSMEG_4635 | 22.86 | 28.85 | 28.80 |
| nitrate reductase | MSMEG_2837 | 806.92 | 166.85 | 351.58 |
| nitrate reductase | MSMEG_5139 | 107.44 | 326.17 | 183.73 |
| nitrate reductase | MSMEG_5140 | 210.30 | 614.29 | 442.95 |
| carbamoyl phosphate synthase | MSMEG_3046 | 6.86 | 37.22 | 12.91 |
| carbamoyl phosphate synthase | MSMEG_3047 | 34.29 | 73.60 | 35.75 |
| carbamoyl phosphate synthase | MSMEG_4726 | 0.00 | 0.42 | 0.00 |
| glutamine synthetase | MSMEG_3561 | 393.17 | 392.24 | 640.58 |
| glutamine synthetase | MSMEG_4290 | 416.03 | 477.55 | 685.27 |
| glutamine synthetase | MSMEG_4294 | 3744.30 | 990.23 | 1818.46 |
| glutamate dehydrogenase | MSMEG_4699 | 1769.28 | 1946.59 | 1900.89 |
| glutamate synthase | MSMEG_6263 | 29.72 | 36.80 | 23.84 |
| glutamate synthase | MSMEG_6458 | 4.57 | 23.00 | 5.96 |
| glutamate synthase | MSMEG_3226 | 240.02 | 117.92 | 295.96 |
| glutamate synthase | MSMEG_3225 | 985.22 | 484.24 | 1505.62 |
| glutamate synthase | MSMEG_5594 | 22.86 | 82.38 | 26.82 |
| glutamate synthase | MSMEG_6459 | 25.14 | 70.67 | 30.79 |
| hexokinase | MSMEG_5577 | 313.17 | 225.81 | 397.26 |
| 6-phosphofructokinase | MSMEG_2366 | 1430.97 | 288.54 | 746.85 |
| pyruvate kinase | MSMEG_3227 | 1312.10 | 1222.31 | 2115.41 |
| citrate synthase | MSMEG_5676 | 598.90 | 155.14 | 395.27 |
| isocitrate dehydrogenase | MSMEG_1654 | 1675.56 | 1218.55 | 1752.91 |
| alpha-ketoglutarate decarboxylase | MSMEG_5049 | 1398.97 | 2698.88 | 3475.03 |
| dihydrolipoamide dehydrogenase | MSMEG_0903 | 612.62 | 425.70 | 414.14 |
| glycerol kinase | MSMEG_6759 | 25.14 | 8.36 | 456.85 |
| glycerol kinase | MSMEG_6229 | 294.88 | 271.81 | 195.65 |
| glycerol kinase | MSMEG_6756 | 0.00 | 12.55 | 12.91 |
| glycerol-3-phosphate dehydrogenase | MSMEG_1736 | 651.48 | 705.87 | 637.60 |
| glycerol-4-phosphate dehydrogenase | MSMEG_6761 | 64.01 | 38.05 | 302.91 |
| glycerol-5-phosphate dehydrogenase | MSMEG_1140 | 777.20 | 1007.79 | 1057.71 |
| glycerol-6-phosphate dehydrogenase | MSMEG_2393 | 182.87 | 110.82 | 244.32 |
| glycerol dehydratase | MSMEG_0497 | 0.00 | 30.11 | 8.94 |
| glycerol dehydratase | MSMEG_1547 | 246.88 | 221.63 | 262.19 |
| glycerol dehydratase | MSMEG_6321 | 48.00 | 120.85 | 57.60 |
| 3-ketosteroid-δ-1-dehydrogenase | MSMEG_2867 | 25.14 | 35.54 | 17.88 |
| 3-ketosteroid-δ-2-dehydrogenase | MSMEG_2869 | 9.14 | 18.40 | 10.92 |
| 3-ketosteroid-δ-3-dehydrogenase | MSMEG_4864 | 4.57 | 17.15 | 5.96 |
| 3-ketosteroid-δ-4-dehydrogenase | MSMEG_5941 | 41.15 | 115.42 | 20.86 |
| CHOLvsAD | CHOLvsGL | ||
|---|---|---|---|
| Start | End | Start | End |
| MSMEG_0132 | MSMEG_0144 | MSMEG_0132 | MSMEG_0144 |
| MSMEG_0329 | MSMEG_0346 | MSMEG_2705 | MSMEG_2714 |
| MSMEG_0500 | MSMEG_0518 | ||
| MSMEG_0638 | MSMEG_0649 | ||
| MSMEG_1141 | MSMEG_1150 | MSMEG_1141 | MSMEG_1150 |
| MSMEG_1364 | MSMEG_1375 | ||
| MSMEG_1435 | MSMEG_1448 | ||
| MSMEG_2705 | MSMEG_2713 | MSMEG_6115 | MSMEG_6125 |
| MSMEG_2854 | MSMEG_2865 | MSMEG_2854 | MSMEG_2861 |
| MSMEG_3997 | MSMEG_4005 | ||
| MSMEG_4414 | MSMEG_4427 | MSMEG_4419 | MSMEG_4427 |
| MSMEG_4835 | MSMEG_4843 | MSMEG_4461 | MSMEG_4468 |
| MSMEG_5953 | MSMEG_5966 | MSMEG_4864 | MSMEG_4873 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Ge, F.; Tan, Y.; Zhang, G.; Li, W. Genome-Wide Transcriptome Profiling of Mycobacterium smegmatis MC2 155 Cultivated in Minimal Media Supplemented with Cholesterol, Androstenedione or Glycerol. Int. J. Mol. Sci. 2016, 17, 689. https://doi.org/10.3390/ijms17050689
Li Q, Ge F, Tan Y, Zhang G, Li W. Genome-Wide Transcriptome Profiling of Mycobacterium smegmatis MC2 155 Cultivated in Minimal Media Supplemented with Cholesterol, Androstenedione or Glycerol. International Journal of Molecular Sciences. 2016; 17(5):689. https://doi.org/10.3390/ijms17050689
Chicago/Turabian StyleLi, Qun, Fanglan Ge, Yunya Tan, Guangxiang Zhang, and Wei Li. 2016. "Genome-Wide Transcriptome Profiling of Mycobacterium smegmatis MC2 155 Cultivated in Minimal Media Supplemented with Cholesterol, Androstenedione or Glycerol" International Journal of Molecular Sciences 17, no. 5: 689. https://doi.org/10.3390/ijms17050689