
Pathogens Use and Abuse MicroRNAs to Deceive the Immune System

Abstract
:
{kind=link}
{kind=link}
{kind=link}
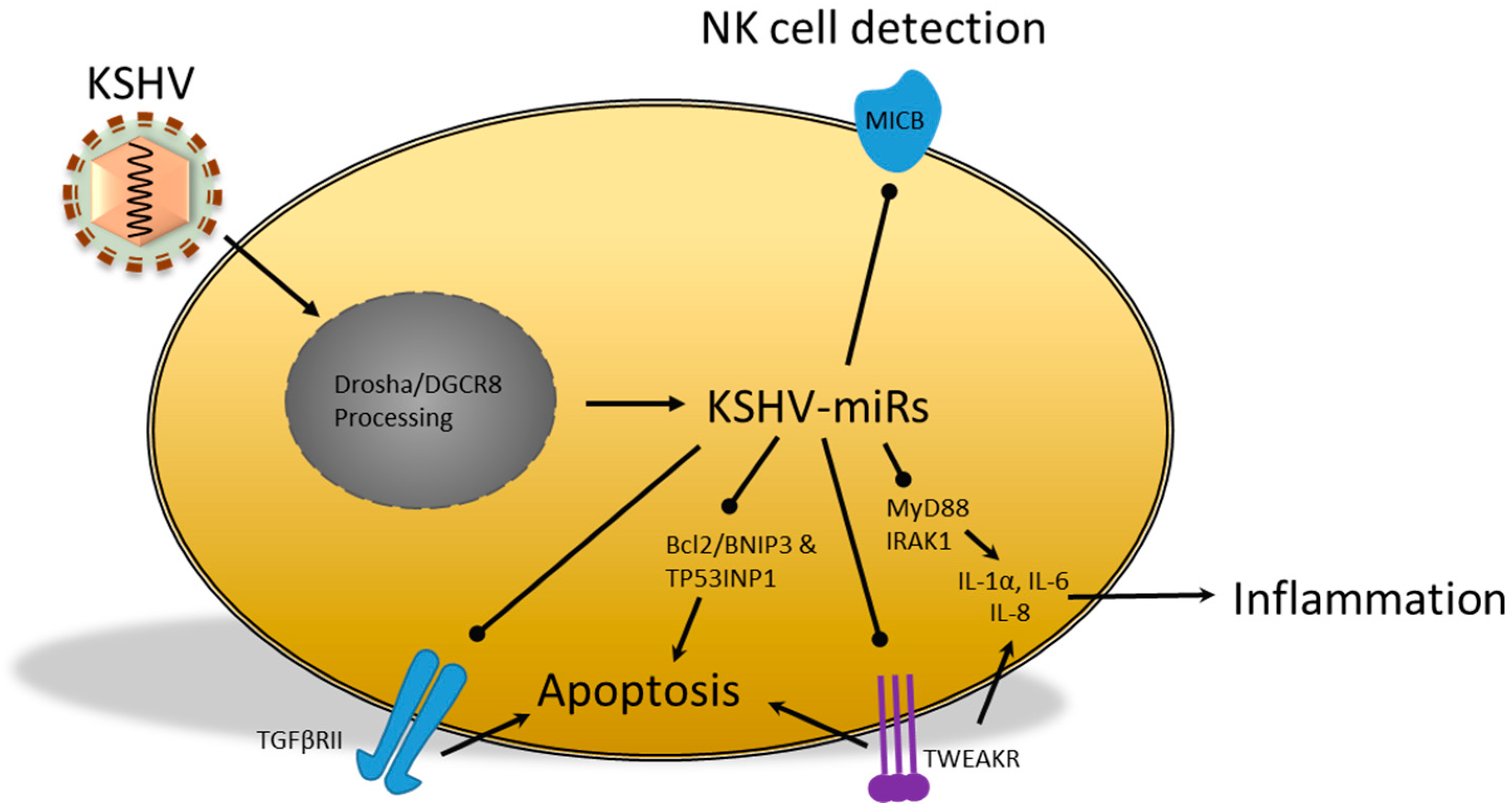{kind=link}
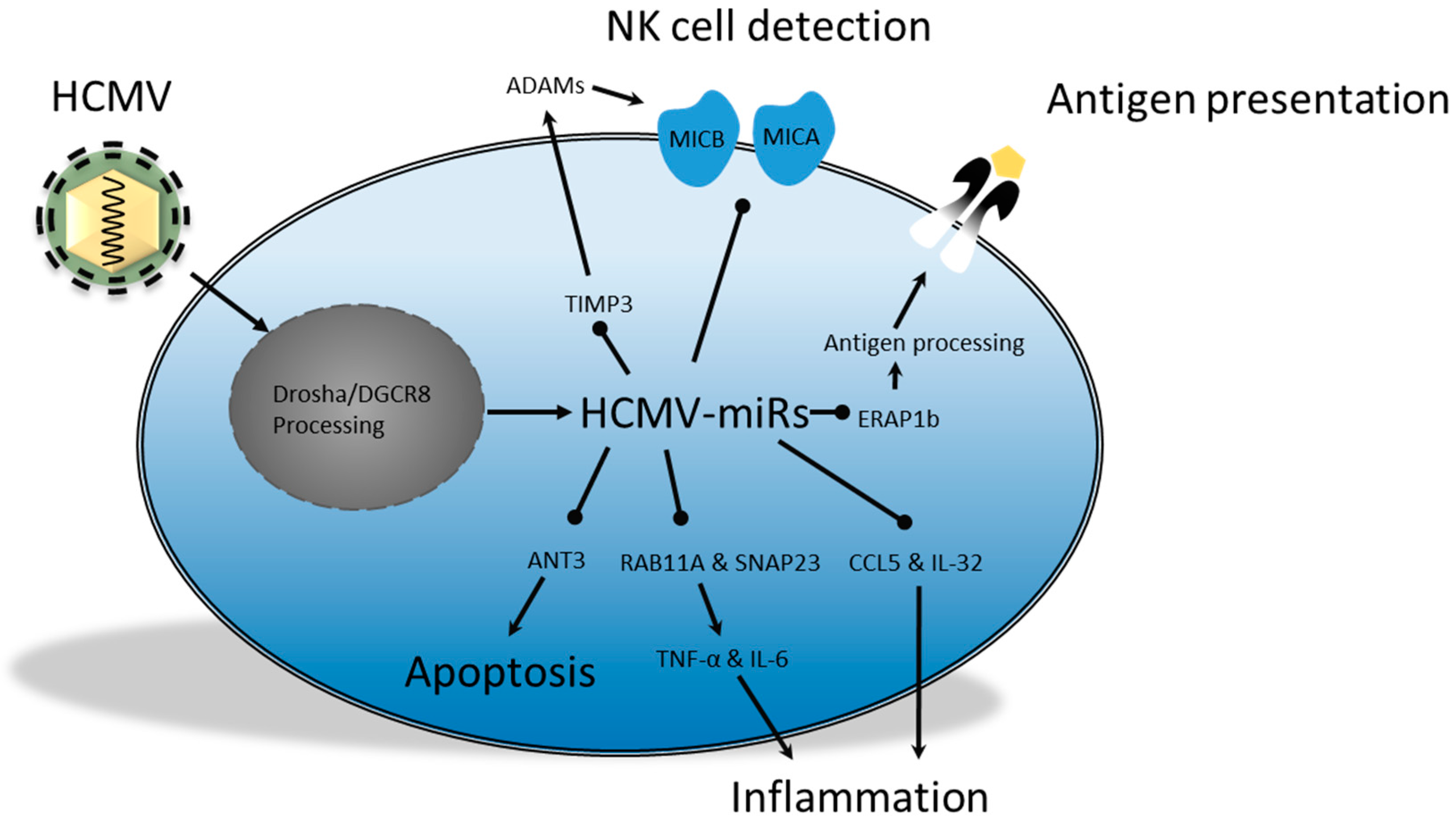{kind=link}
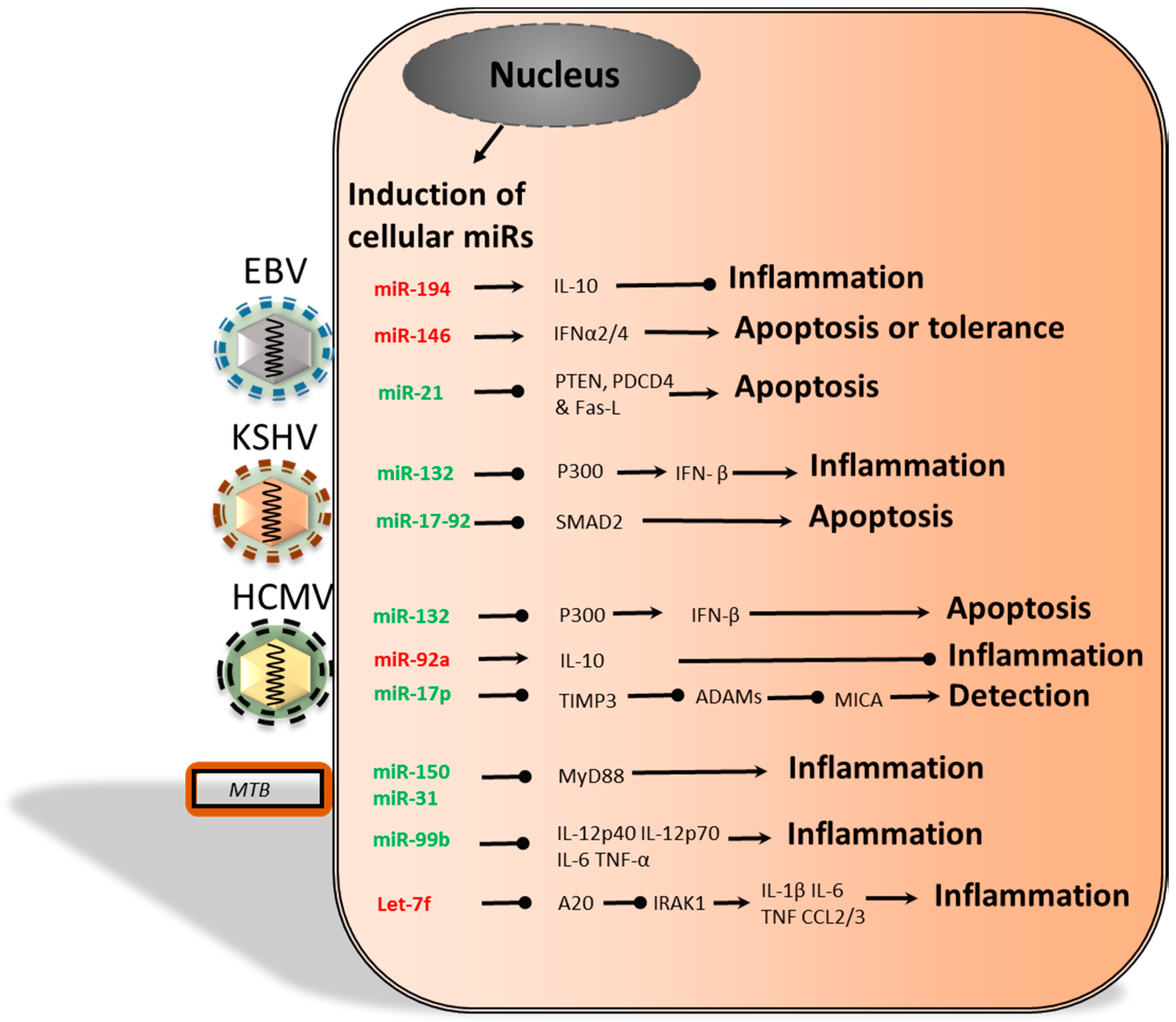{kind=link}
1. Introduction
1.1. MicroRNAs (miRs)
1.2. Immune Responses
1.3. Scope of the Review
2. Viral miRs
2.1. Are miRs Relevant during Infection with HIV-1?
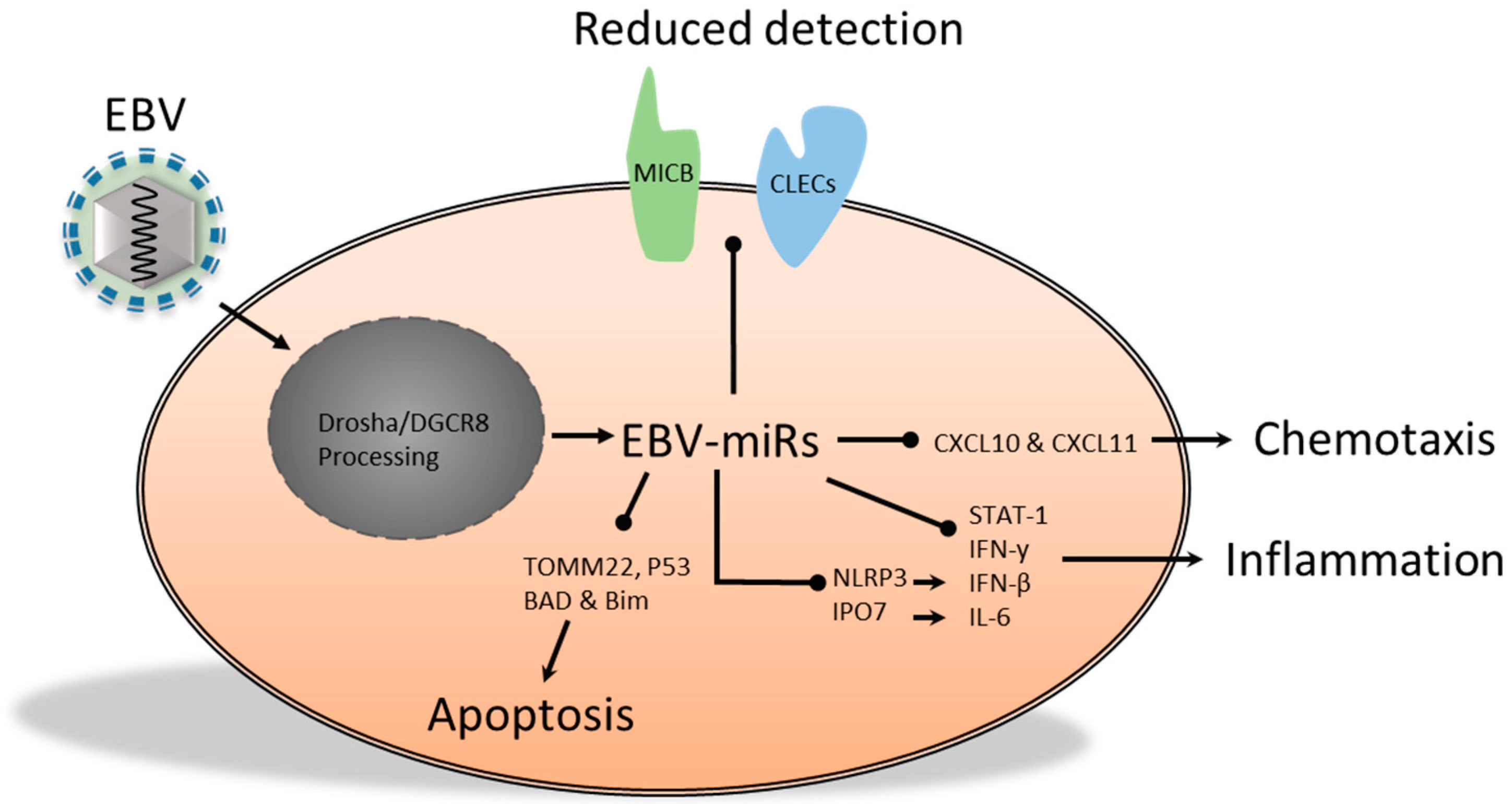
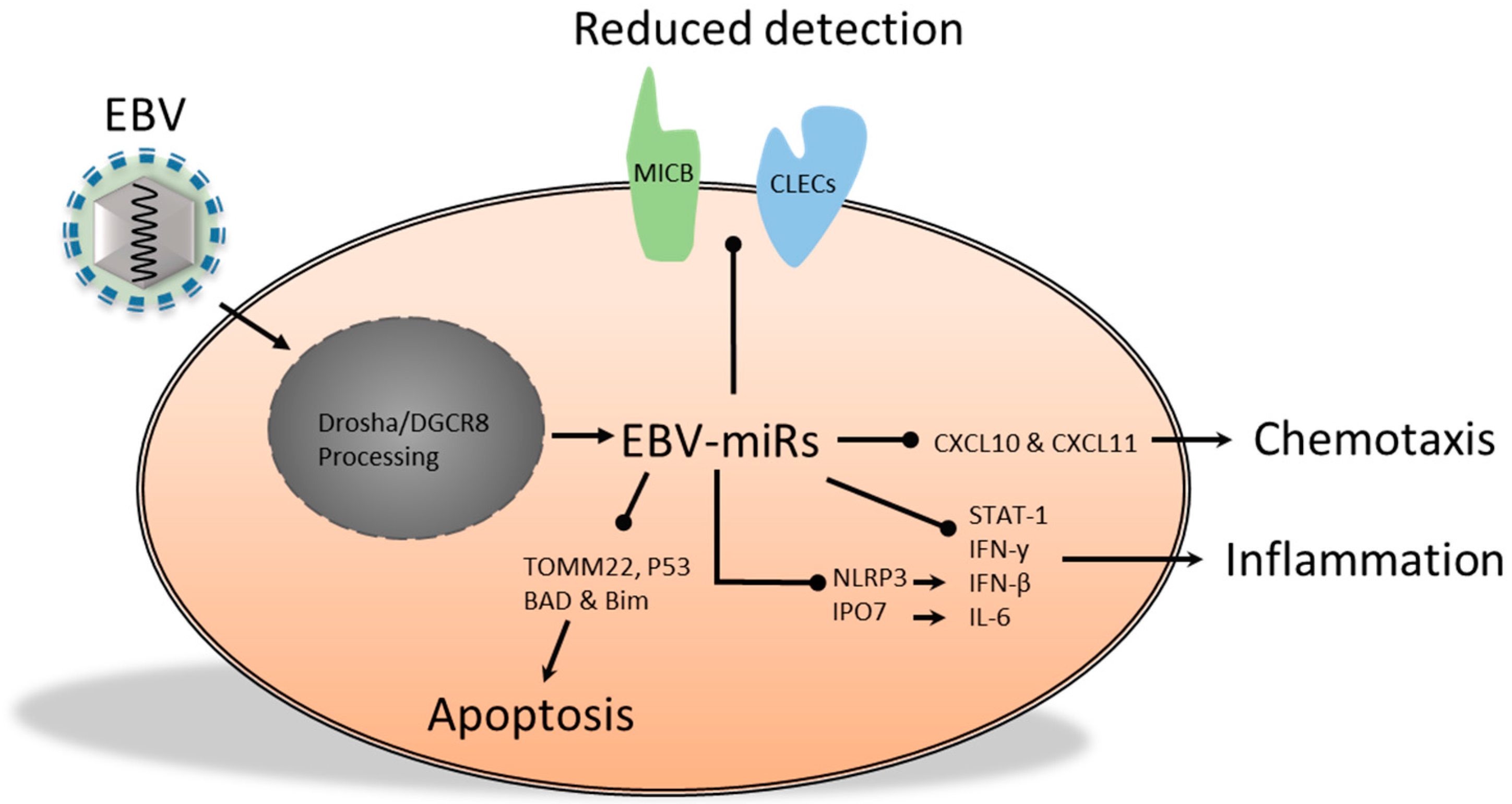
2.2. The Functional Role of Epstein-Barr Virus-Encoded miRs
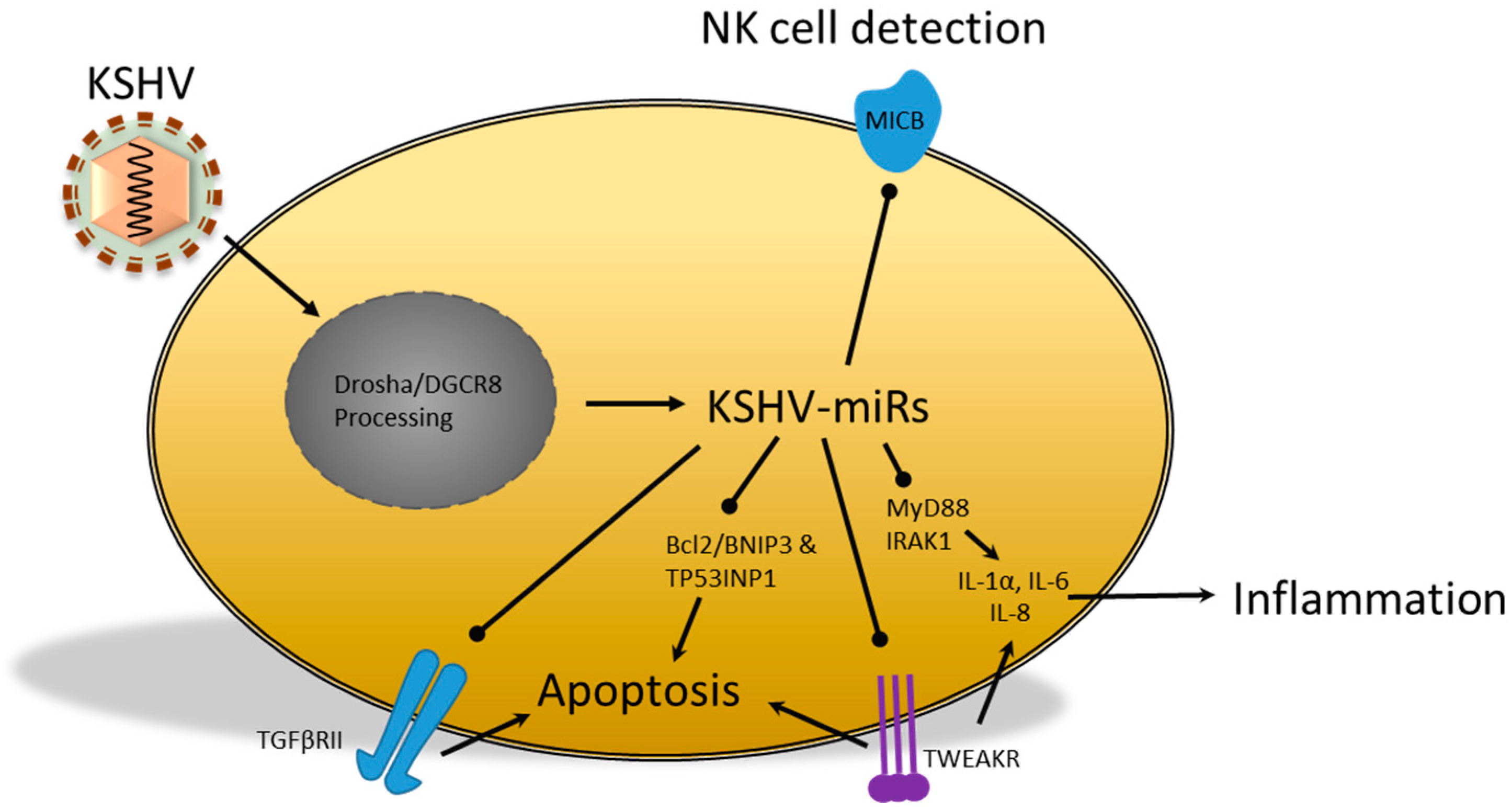
2.3. miRs Produced by the Kaposi’s Sarcoma-Associated Herpesvirus
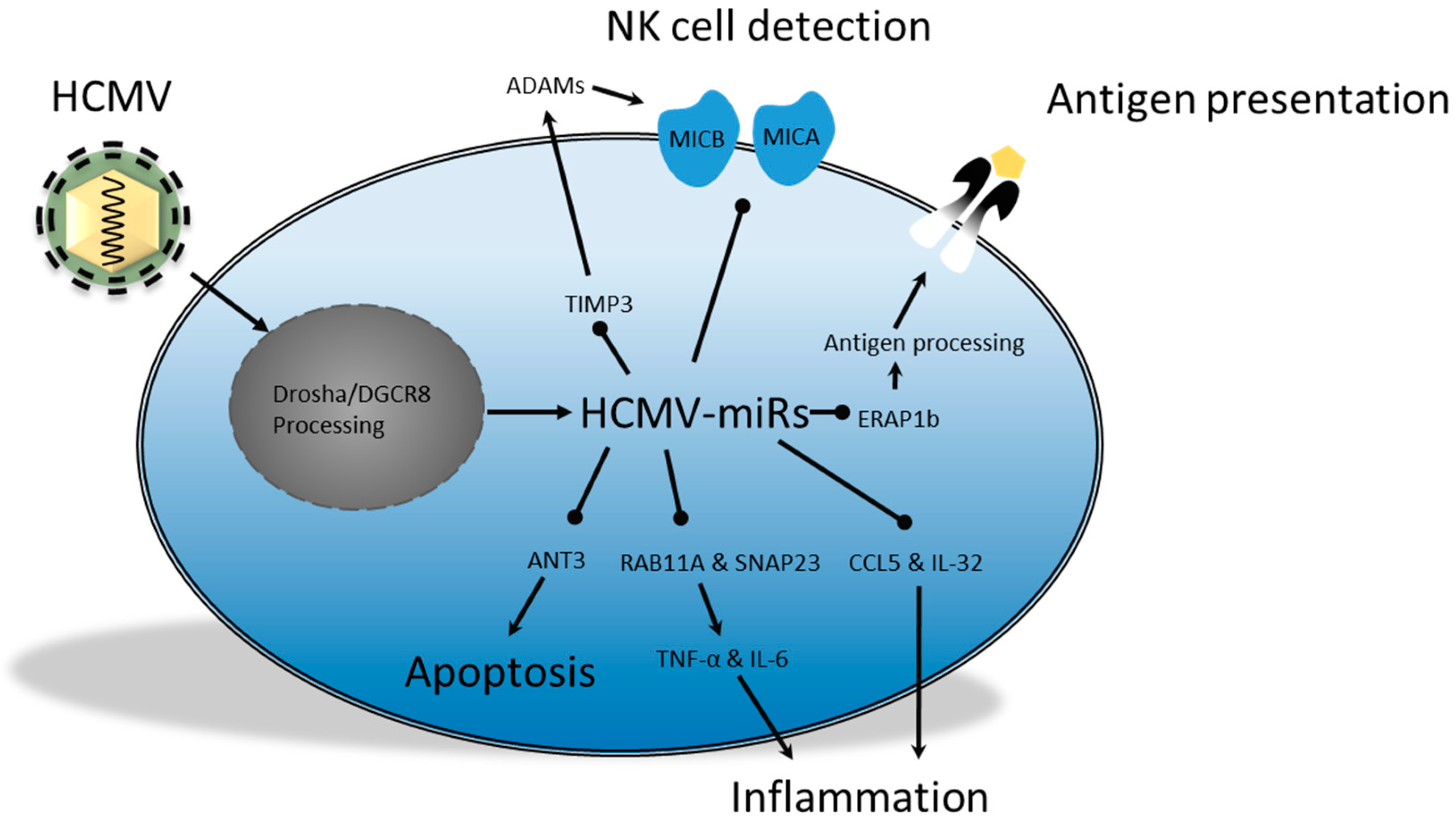
2.4. Human Cytomegalovirus; miRs in Persistence and Immunomodulation
3. Host Cell miRs: A Means for Viral Immune Modulation?
3.1. Cellular miR Expression; a Way for HIV-1 to Sustain Latency?
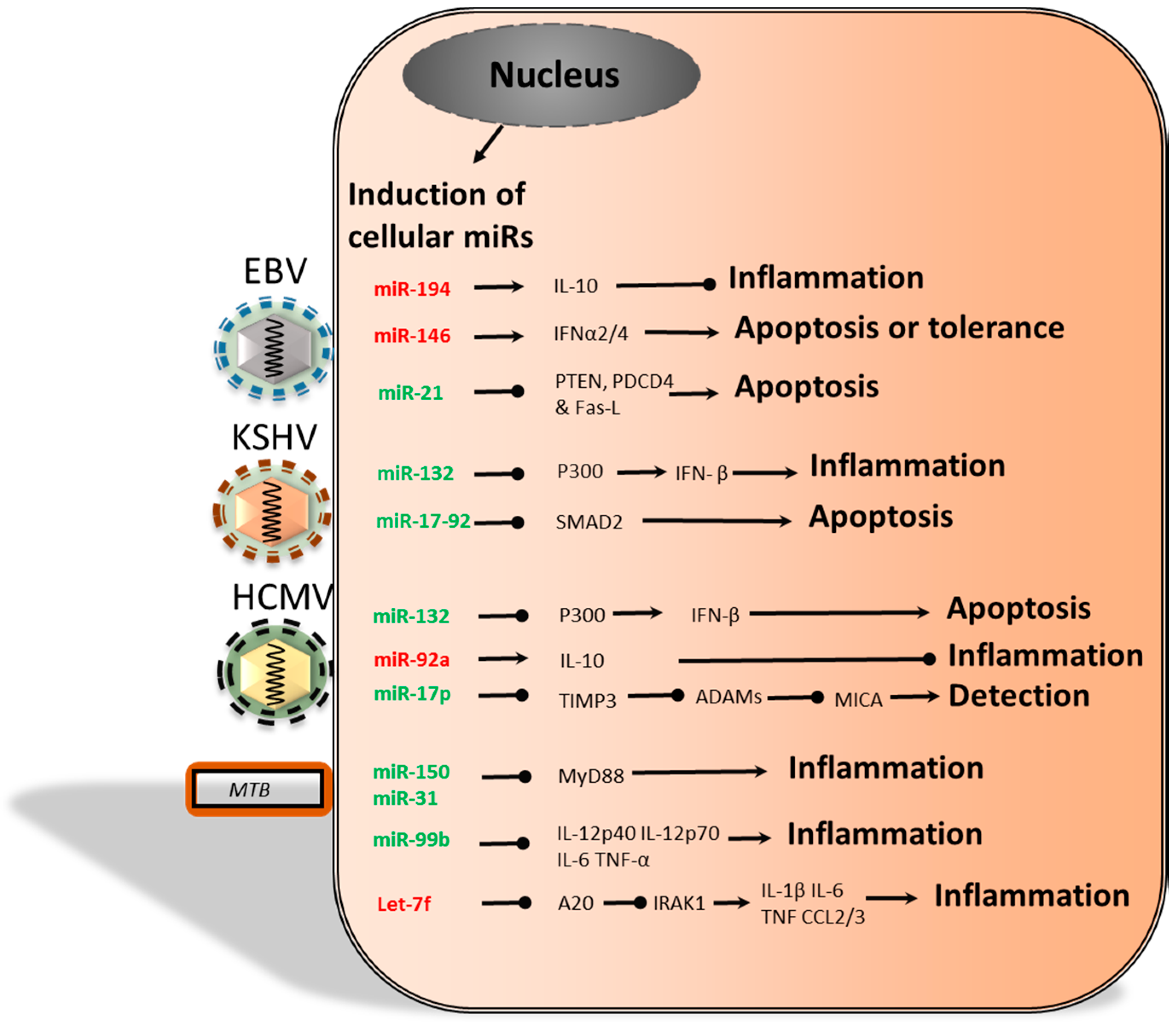
3.2. Herpersviridae and Dysregulation of Cellular miRs
4. Bacterial and Cellular miRs
Cellular miRs Induced by Bacteria
5. Concluding Remarks
- The combination of PAR-CLIP analysis and reporter assays provides clear indications that miR is functionally relevant.
- Evidence to support the claim that HIV-1 encodes functionally relevant vmiRs is limited.
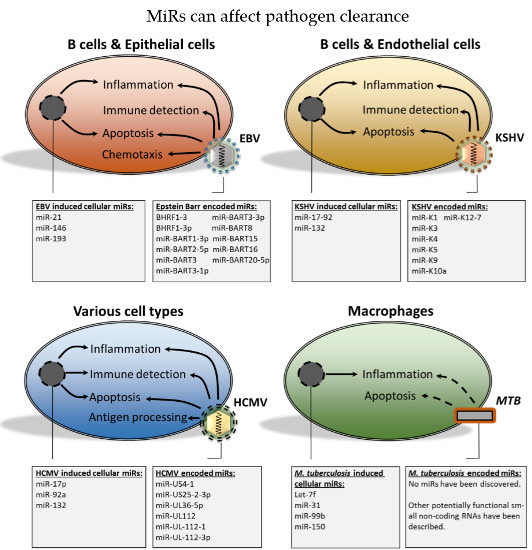
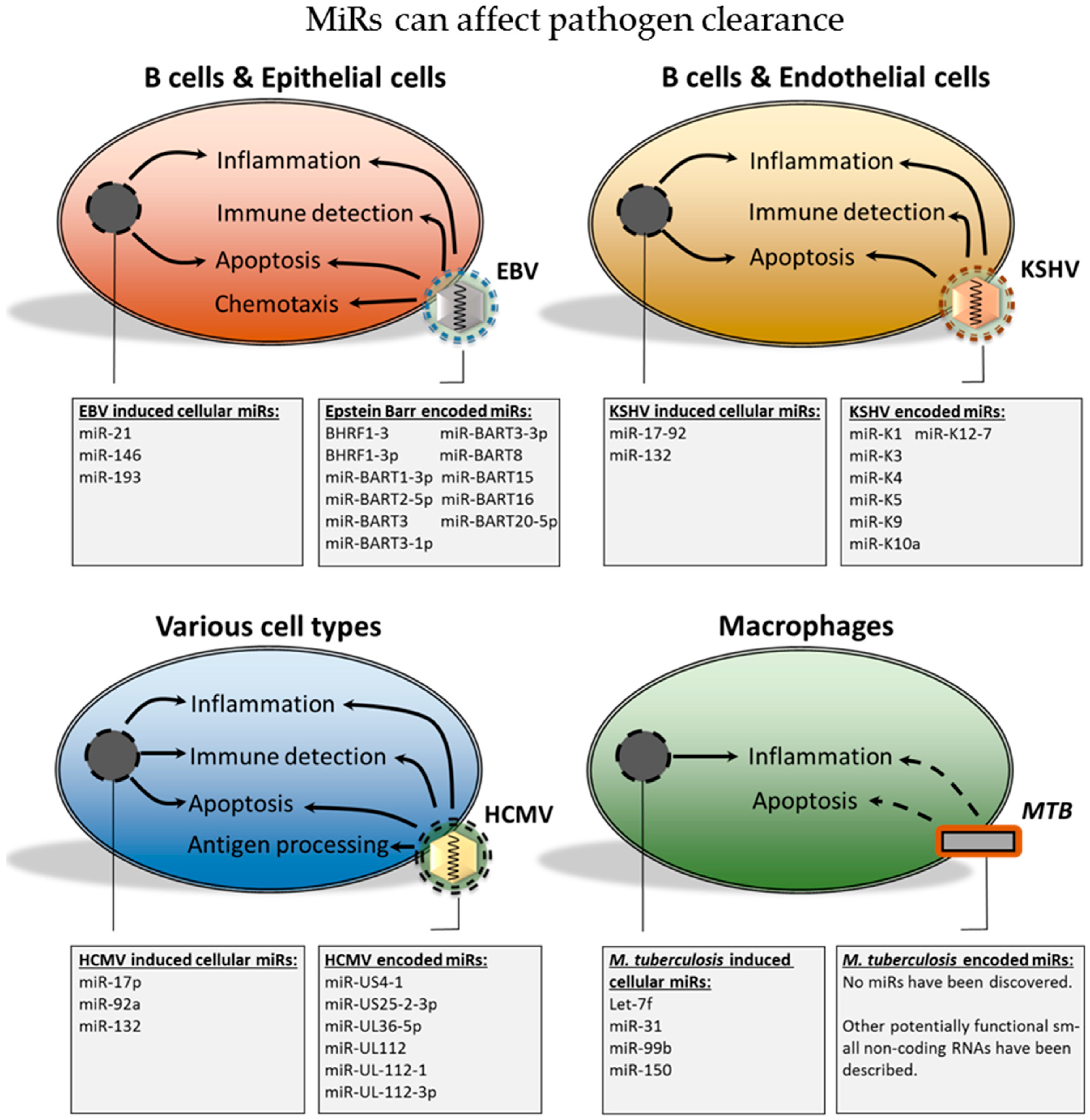
- EBV, KSHV and HCMV-encoded miRs contribute, to varying degrees, to immune evasion and survival of the pathogen.
- Alterations in host cell miR expression during EBV, KSHV and HCMV infections may contribute to a cellular miR expression profile that promotes immune evasion and survival, thereby contributing to viral persistence.
- MTB infection affects expression of cellular miRs that decrease immune function, which may in some cases be pathogen specific and hence relevant for therapeutic targeting.
Conflicts of Interest
References
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.D.; Alder, M.N. The evolution of adaptive immune systems. Cell 2006, 124, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Cold Spring Harb. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Abraham, E. MicroRNAs in immune response and macrophage polarization. Arter. Thromb. Vasc. Biol. 2013, 33, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′ UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Berninger, P.; Rothballer, A.M.A., Jr.; Munschauer, M.; Ulrich, A.; Wardle, G.S.; Dewell, S.; et al. Transcritpome wide identification of RNA binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Corcoran, D.L.; Mukherjee, N.; Skalsky, R.L.; Hafner, M.; Nusbaum, J.D.; Shamulailatpam, P.; Love, C.L.; Dave, S.S.; Tuschl, T.; et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 2011, 10, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Ursula Klingmüller, K.M.-D. Cellular Signal Processing: An Introduction to the Molecular Mechanisms of Signal Transduction; Garland Science, Taylor & Francis Group: New York, NY, USA, 2009. [Google Scholar]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Sorensen, D.L.; Booth, S.A. MicroRNA-146a: A dominant, negative regulator of the innate immune response. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Karrich, J.J.; Jachimowski, L.C.M.; Libouban, M.; Iyer, A.; Brandwijk, K.; Taanman-kueter, E.W.; Nagasawa, M.; de Jong, E.C.; Uittenbogaart, C.H.; Blom, B. MicroRNA-146a regulates survival and maturation of human plasmacytoid dendritic cells. Blood 2013, 122, 3001–3010. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Hashimi, S.T.; Fulcher, J.A.; Chang, M.H.; Gov, L.; Wang, S.; Lee, B. MicroRNA profiling identifies miR34a and miR21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood 2009, 114, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yu, M.; Lee, W.-W.; Tsang, M.; Krishnan, E.; Weyand, C.M.; Goronzy, J.J. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat. Med. 2012, 18, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-J.; Chau, J.; Ebert, P.J.R.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 2007, 129, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Schambach, F.; Dejong, C.S.; Hammond, S.M.; Steven, L. Micro-RNA-155 inhibits IFN-γ signaling in CD4+ T cells. Eur. J. Immunol. 2011, 40, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signaling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.S.; McCoy, C.E.; Lloyd, A.T.; O’Farrelly, C.; Stevenson, N.J. miR-19a: An effective regulator of SOCS3 and enhancer of JAK-STAT signaling. PLoS ONE 2013, 8, e69090. [Google Scholar]
- Goergen, D.; Niepmann, M. Stimulation of Hepatitis C Virus RNA translation by microRNA-122 occurs under different conditions in vivo and in vitro. Virus Res. 2012, 167, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, B.M.; Trono, D. Hide, shield and strike back: How HIV-infected cells avoid immune eradication. Nat. Rev. Immunol. 2003, 3, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Bennasser, Y.; Le, S.; Yeung, L.M.; Jeang, J.-K. HIV-1 encoded candidate micro-RNAs and their cellular targets. Retrovirology 2004, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grässer, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. MicroRNAs as mediators of viral evasion of the immune system. Nat. Immunol. 2013, 14, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Navas-Martín, S.; Martín-García, J. MicroRNAs and HIV-1 infection: Antiviral activities and beyond. J. Mol. Biol. 2014, 426, 1178–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, M.; Geng, G.; Liu, B.; Huang, Z.; Luo, H.; Zhou, J.; Guo, X.; Cai, W.; Zhang, H. A novel HIV-1-encoded microRNA enhances its viral replication by targeting the TATA box region. Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cullen, B.R. Analysis of the interaction of primate retroviruses with the human RNA interference machinery. J. Virol. 2007, 81, 12218–12226. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Plante, I.; Landry, P.; Barat, C.; Janelle, M.E.; Flamand, L.; Tremblay, M.J.; Provost, P. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Ito, M.; Tsutsumi, Y.; Ichikawa, Y.; Okuyama, H.; Brisibe, E.A.; Saksena, N.K.; Fujii, Y.R. HIV-1 nef suppression by virally encoded microRNA. Retrovirology 2004, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Vigneault-Edwards, J.; Létourneau, K.; Gobeil, L.-A.; Plante, I.; Burnett, J.C.; Rossi, J.J.; Provost, P. Regulation of host gene expression by HIV-1 TAR microRNAs. Retrovirology 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Berkhout, B.; Das, A. Characterization of microRNAs derived from the HIV-1 TAR RNA hairpin. Retrovirology 2013, 10. [Google Scholar] [CrossRef]
- Schopman, N.C.T.; Willemsen, M.; Liu, Y.P.; Bradley, T.; van Kampen, A.; Baas, F.; Berkhout, B.; Haasnoot, J. Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012, 40, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Berkhout, B.; Das, A.T. Deep sequencing of small TAR-derived RNAs in HIV-1 producing cells. Retrovirology 2011, 8. [Google Scholar] [CrossRef]
- Bennasser, Y.; Le, S.Y.; Benkirane, M.; Jeang, K.T. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 2005, 22, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Whisnant, A.W.; Bogerd, H.P.; Flores, O.; Ho, P.; Powers, J.G.; Sharova, N.; Stevenson, M.; Chen, C.-H.; Cullen, B.R. In-depth analysis of the interaction of HIV-1 with cellular microRNA biogenesis and effector mechanisms. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Vongrad, V.; Imig, J.; Mohammadi, P.; Kishore, S.; Jaskiewicz, L.; Hall, J.; Günthard, H.F.; Beerenwinkel, N.; Metzner, K.J. HIV-1 RNAs are not part of the Argonaute 2 associated RNA interference pathway in macrophages. PLoS ONE 2015, 10, e0132127. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef] [PubMed]
- Kempkes, B.R.E. Epstein-Barr virus latency: Current and future perspectives. Curr. Opin. Virol. 2015, 14, 138–144. [Google Scholar] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Dölken, L.; Malterer, G.; Erhard, F.; Kothe, S.; Friedel, C.C.; Suffert, G.; Marcinowski, L.; Motsch, N.; Barth, S.; Beitzinger, M.; et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human γ-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 2010, 7, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Lung, R.W.-M.; Tong, J.H.-M.; Sung, Y.-M.; Leung, P.-S.; Ng, D.C.-H.; Chau, S.-L.; Chan, A.W.-H.; Ng, E.K.-O.; Lo, K.-W.; To, K.-F. Modulation of LMP2A expression by a newly identified Epstein-Barr virus-encoded microRNA miR-BART22. Neoplasia 2009, 11. [Google Scholar] [CrossRef]
- Lo, A.K.-F.; Dawson, C.W.; Jin, D.-Y.; Lo, K.-W. The pathological roles of BART miRNAs in nasopharyngeal carcinoma. J. Pathol. 2012, 227, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; To, K.F.; Lo, K.W.; Lung, R.W.; Hui, J.W.; Liao, G.; Hayward, S.D. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc. Natl. Acad. Sci. USA 2007, 104, 16164–16169. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.-J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yee, C.; Beavo, J.A. CD3- and CD28-dependent induction of PDE7 required for T cell activation. Science 1999, 283, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.-A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.J.; Masters, S.L. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1 production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [PubMed]
- Yang, I.V.; Wade, C.M.; Kang, H.M.; Alper, S.; Rutledge, H.; Lackford, B.; Eskin, E.; Daly, M.J.; Schwartz, D.A. Identification of novel genes that mediate innate immunity using inbred mice. Genetics 2009, 183, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; O’Hara, A.; Araujo, I.; Barreto, J.; Carvalho, E.; Sapucaia, J.B.; Ramos, J.C.; Luz, E.; Pedroso, C.; Manrique, M.; et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by EBV-miR-BHRF1-3. Cancer Res. 2008, 68, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-T.; Lin, C.-W. EBV-encoded miR-BART20-5p and miR-BART8 inhibit the IFN-γ-STAT1 pathway associated with disease progression in nasal NK-cell lymphoma. Am. J. Pathol. 2014, 184, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Murayama, T.; Li, Y.; Takahashi, T.; Yamada, R.; Matsubara, K.; Tuchida, Y.; Li, Z.; Sadanari, H. Anti-cytomegalovirus effects of tricin are dependent on CXCL11. Microbes Infect. 2012, 14, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.J.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Würdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011, 89, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Lu, B.; Gerard, C.; Iwasaki, A. CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature 2009, 462, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jäker, C.; Höck, J.; Meister, G.; Grässer, F.A. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-J.; Choi, H.; Kim, H.; Lee, S.K. MicroRNA miR-BART20-5p stabilizes Epstein-Barr virus latency by directly targeting BZLF1 and BRLF1. J. Virol. 2014, 88, 9027–9037. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Moosmann, A.; Grömminger, S.; Walz, N.; Grundhoff, A.; Hammerschmidt, W. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010, 6, e1001063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, X.; Li, L.; Liu, S.; Yang, L.; Ma, X.; Tang, M.; Bode, A.M.; Dong, Z.; Sun, L.; Cao, Y. EBV encoded miR-BHRF1-1 potentiates viral lytic replication by downregulating host p53 in nasopharyngeal carcinoma. Int. J. Biochem. Cell Biol. 2012, 44, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Skalsky, R.L.; Cullen, B.R. EBV BART microRNAs target multiple pro-apoptotic cellular genes to promote epithelial cell survival. PLoS Pathog. 2015, 11, e1004979. [Google Scholar] [CrossRef]
- Bellot, G.; Cartron, P.-F.; Er, E.; Oliver, L.; Juin, P.; Armstrong, L.C.; Bornstein, P.; Mihara, K.; Manon, S.; Vallette, F.M. TOM22, a core component of the mitochondria outer membrane protein translocation pore, is a mitochondrial receptor for the proapoptotic protein Bax. Cell Death Differ. 2007, 14, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, H.; Lee, S.K. Epstein-Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 2015, 356, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Marquitz, A.R.; Mathur, A.; Nam, C.S.; Raab-Traub, N. The Epstein–Barr Virus BART microRNAs target the pro-apoptotic protein Bim. Virology 2011, 412, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.Y.-W.; Siu, K.-L.; Kok, K.-H.; Lung, R.W.-M.; Tsang, C.M.; To, K.-F.; Kwong, D.L.-W.; Tsao, S.W.; Jin, D.-Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, T.; Yuen, K.-S.; Xu, R.; Tsao, S.W.; Chen, H.; Li, M.; Kok, K.-H.; Jin, D.-Y. Targeting of DICE1 tumor suppressor by Epstein-Barr virus-encoded miR-BART3* microRNA in nasopharyngeal carcinoma. Int. J. Cancer 2013, 133, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV and the pathogenesis of Kaposi sarcoma: Listening to human biology and medicine. J. Clin. Investig. 2010, 120, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Abend, J.R.; Ramalingam, D.; Kieffer-Kwon, P.; Uldrick, T.S.; Yarchoan, R.; Ziegelbauer, J.M. Kaposi’s sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J. Virol. 2012, 86, 11663–11674. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liang, D.; Sun, R.; Jia, B.; Xia, T.; Xiao, H.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. J. Virol. 2015, 89, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Abend, J.R.; Uldrick, T.; Ziegelbauer, J.M. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi’s sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J. Virol. 2010, 84, 12139–12151. [Google Scholar] [CrossRef] [PubMed]
- Papers, J.B.C.; Doi, M.; Polek, T.C.; Talpaz, M.; Darnay, B.G.; Spivak-kroizman, T. TWEAK mediates signal transduction and differentiation of RAW264.7 cells in the absence of Fn14/TweakR. J. Biol. Chem. 2003, 278, 32317–32323. [Google Scholar]
- Wiley, S.R.; Cassiano, L.; Lofton, T.; Davis-Smith, T.; Winkles, J.A.; Lindner, V.; Liu, H.; Daniel, T.O.; Smith, C.A.; Fanslow, W.C. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 2001, 15, 837–846. [Google Scholar] [CrossRef]
- Lei, X.; Zhu, Y.; Jones, T.; Bai, Z.; Huang, Y.; Gao, S.-J. A Kaposi’s sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor β pathway to promote cell survival. J. Virol. 2012, 86, 11698–11711. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [PubMed]
- Loenen, W.A.; Bruggeman, C.A.; Wiertz, E.J. Immune evasion by human cytomegalovirus: Lessons in immunology and cell biology. Semin. Immunol. 2001, 13, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.; Shin, J.; Kim, Y.; Evnouchidou, I.; Kim, D.; Kim, Y.-K.; Kim, Y.-E.; Ahn, J.-H.; Riddell, S.R.; et al. Human cytomegalovirus microRNA miR-US4-1 inhibits CD8+ T cell responses by targeting the aminopeptidase ERAP1. Nat. Immunol. 2011, 12, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Esteso, G.; Luzón, E.; Sarmiento, E.; Gómez-Caro, R.; Steinle, A.; Murphy, G.; Carbone, J.; Valés-Gómez, M.; Reyburn, H.T. Altered microRNA expression after infection with human cytomegalovirus leads to TIMP3 downregulation and increased shedding of metalloprotease substrates, including MICA. J. Immunol. 2014, 193, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host immune system gene targeting by a viral miRNA. Science 2007, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Landais, I.; Pelton, C.; Streblow, D.; DeFilippis, V.; McWeeney, S.; Nelson, J.A. Human cytomegalovirus miR-UL112-3p targets TLR2 and modulates the TLR2/IRAK1/NFκB signaling pathway. PLoS Pathog. 2015, 11, e1004881. [Google Scholar] [CrossRef] [PubMed]
- Szomolanyi-Tsuda, E.; Liang, X.; Welsh, R.M.; Kurt-Jones, E.A.; Finberg, R.W. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J. Virol. 2006, 80, 4286–4291. [Google Scholar] [CrossRef] [PubMed]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in Trans acting as the viral fusion protein rather than as a receptor-binding protein. MBio 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.; Kim, S.; Kim, D.; Ahn, J.-H.; Ahn, K. Human cytomegalovirus clinical strain-specific microRNA miR-UL148D targets the human chemokine RANTES during infection. PLoS Pathog. 2012, 8, e1002577. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Qi, Y.; Ma, Y.; He, R.; Ji, Y.; Sun, Z.; Ruan, Q. The expression of interleukin-32 is activated by human cytomegalovirus infection and down regulated by hcmv-miR-UL112-1. Virol. J. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Hook, L.M.; Grey, F.; Grabski, R.; Tirabassi, R.; Doyle, T.; Hancock, M.; Landais, I.; Jeng, S.; McWeeney, S.; Britt, W.; et al. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 2014, 15, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.G.; Pandeya, A.; Verma, N.; Shukla, N.; Kumar, R.V.; Saxena, S. Role of HCMV miR-UL70-3p and miR-UL148D in overcoming the cellular apoptosis. Mol. Cell. Biochem. 2014, 393, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Huang, Y.; Qi, Y.; Liu, Z.; Ma, Y.; Shao, Y.; Jiang, S.; Sun, Z.; Ruan, Q. Human cytomegalovirus miR-UL36-5p inhibits apoptosis via downregulation of adenine nucleotide translocator 3 in cultured cells. Arch. Virol. 2015, 160, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Mari, B.; Lin, Y.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef]
- Chable-Bessia, C.; Meziane, O.; Latreille, D.; Triboulet, R.; Zamborlini, A.; Wagschal, A.; Jacquet, J.-M.; Reynes, J.; Levy, Y.; Saib, A.; et al. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 2009, 6. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, J.K.; Khan, S.Z.; Soni, K.; Rawat, P.; Gupta, A.; Hariharan, M.; Scaria, V.; Lalwani, M.; Pillai, B.; Mitra, D.; et al. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology 2008, 5. [Google Scholar] [CrossRef] [PubMed]
- Nathans, R.; Chu, C.-Y.; Serquina, A.K.; Lu, C.-C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Adoro, S.; Cubillos-Ruiz, J.R.; Chen, X.; Deruaz, M.; Vrbanac, V.D.; Song, M.; Park, S.; Murooka, T.T.; Dudek, T.E.; Luster, A.D.; et al. IL-21 induces antiviral microRNA-29 in CD4 T cells to limit HIV-1 infection. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Chan, J.K.; Oh, E.; Heidersbach, A.J.; Hebbeler, A.M.; Chavez, L.; Verdin, E.; Rape, M.; Greene, W.C. MicroRNA-155 reinforces HIV latency. J. Biol. Chem. 2015, 290, 13736–13748. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.E.; Yin, Q.; Fewell, C.; Lacey, M.; McBride, J.; Wang, X.; Lin, Z.; Schaefer, B.C.; Flemington, E.K. Epstein-Barr virus latent membrane protein 1 induces cellular microRNA miR-146a, a modulator of lymphocyte signaling pathways. J. Virol. 2008, 82, 1946–1958. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.; Anastasiadou, E.; Garg, N.; Lenze, D.; Boccellato, F.; Vincenti, S.; Severa, M.; Coccia, E.M.; Bigi, R.; Cirone, M.; et al. Differential regulation of miR-21 and miR-146a by Epstein-Barr virus-encoded EBNA2. Leukemia 2012, 26, 2343–2352. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Sato, M.; Lamphier, M.S.; Nozawa, H.; Oda, E.; Noguchi, S.; Schreiber, R.D.; Tsujimoto, Y.; Taniguchi, T. Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells 1998, 3, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Harris-Arnold, A.; Arnold, C.P.; Schaffert, S.; Hatton, O.; Krams, S.M.; Esquivel, C.O.; Martinez, O.M. Epstein-Barr virus modulates host cell microRNA-194 to promote IL-10 production and B lymphoma cell survival. Am. J. Transplant. 2015, 15, 2814–2824. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-D.; Huang, T.-J.; Peng, L.-X.; Yang, C.-F.; Liu, R.-Y.; Huang, H.-B.; Chu, Q.-Q.; Yang, H.-J.; Huang, J.-L.; Zhu, Z.-Y.; et al. Epstein-Barr Virus_Encoded LMP1 upregulates microRNA-21 to promote the resistance of nasopharyngeal carcinoma cells to cisplatin-induced Apoptosis by suppressing PDCD4 and Fas-L. PLoS ONE 2013, 8, e78355. [Google Scholar] [CrossRef] [PubMed]
- Catrina Ene, A.M.; Borze, I.; Guled, M.; Costache, M.; Leen, G.; Sajin, M.; Ionica, E.; Chitu, A.; Knuutila, S. MicroRNA expression profiles in Kaposi’s sarcoma. Pathol. Oncol. Res. 2013, 1, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Jain, V.; Krueger, B.; Marshall, V.; Kim, C.H.; Shisler, J.L.; Whitby, D.; Renne, R. Kaposi’s sarcoma-associated herpesvirus (KSHV) induces the oncogenic miR-17-92 cluster and down-regulates TGF-β signaling. PLOS Pathog. 2015, 11, e1005255. [Google Scholar] [CrossRef] [PubMed]
- Lagos, D.; Pollara, G.; Henderson, S.; Gratrix, F.; Fabani, M.; Milne, R.S.B.; Gotch, F.; Boshoff, C. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 2010, 12, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Avdic, S.; Hodkinson, J.; Jackson, S.; Wills, M.; Slobedman, B.; Sinclair, J. Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J. Virol. 2014, 88, 13947–13955. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.M.; Poole, E.; Sissons, J.G.P.; Wills, M.R.; Sinclair, J.H. Human cytomegalovirus latency alters the cellular secretome, inducing cluster of differentiation (CD)4+ T-cell migration and suppression of effector function. Proc. Natl. Acad. Sci. USA 2012, 109, 14538–14543. [Google Scholar] [CrossRef] [PubMed]
- Furuse, Y.; Finethy, R.; Saka, H.A.; Xet-Mull, A.M.; Sisk, D.M.; Smith, K.L.J.; Lee, S.; Coers, J.; Valdivia, R.H.; Tobin, D.M.; et al. Search for microRNAs expressed by intracellular bacterial pathogens in infected mammalian cells. PLoS ONE 2014, 9, e106434. [Google Scholar] [CrossRef] [PubMed]
- Obregón-Henao, A.; Duque-Correa, M.A.; Rojas, M.; García, L.F.; Brennan, P.J.; Ortiz, B.L.; Belisle, J.T. Stable extracellular RNA fragments of Mycobacterium tuberculosis induce early apoptosis in human monocytes via a caspase-8 dependent mechanism. PLoS ONE 2012, 7, e29970. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, C.P.; Dorrington, M.G.; Novakowski, K.E.; Kaiser, J.; Radford, K.; Nair, P.; Anipindi, V.; Kaushic, C.; Surette, M.G.; Bowdish, D.M.E. MicroRNA-155 is required for clearance of Streptococcus pneumoniae from the nasopharynx. Infect. Immun. 2014, 82, 4824–4833. [Google Scholar] [CrossRef] [PubMed]
- Oertli, M.; Engler, D.B.; Kohler, E.; Koch, M.; Meyer, T.F.; Müller, A. MicroRNA-155 is essential for the T cell-mediated control of Helicobacter pylori infection and for the induction of chronic Gastritis and Colitis. J. Immunol. 2011, 187, 3578–3586. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Xu, S.; Liu, X.; Zhang, Q.; Xu, X.; Liu, M.; Hua, M.; Li, N.; Yao, H.; Cao, X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ. Nat. Immunol. 2011, 12, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; John, C.M.; Jarvis, G.A. Induction of endotoxin tolerance by pathogenic Neisseria is correlated with the inflammatory potential of lipooligosaccharides and regulated by microRNA-146a. J. Immunol. 2014, 192, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.J.; Ravneberg, D.H.; Clay, C.D.; Piper-Hunter, M.G.; Marsh, C.B.; Elton, T.S.; Gunn, J.S.; Amer, A.; Kanneganti, T.-D.; Schlesinger, L.S.; et al. miR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS ONE 2009, 4, e8508. [Google Scholar] [CrossRef] [PubMed]
- Pamer, E.G. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 2004, 4, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Melton-Witt, J.A.; Rafelski, S.M.; Portnoy, D.A.; Bakardjiev, A.I. Oral infection with signature-tagged Listeria monocytogenes reveals organ-specific growth and dissemination routes in guinea pigs. Infect. Immun. 2012, 80, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Köster, S.; van Pee, K.; Hudel, M.; Leustik, M.; Rhinow, D.; Kühlbrandt, W.; Chakraborty, T.; Yildiz, Ö. Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Izar, B.; Mannala, G.K.; Mraheil, M.A.; Chakraborty, T.; Hain, T. MicroRNA response to Listeria monocytogenes infection in epithelial cells. Int. J. Mol. Sci. 2012, 13, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Maudet, C.; Mano, M.; Eulalio, A. MicroRNAs in the interaction between host and bacterial pathogens. FEBS Lett. 2014, 588, 4140–4147. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, Y.; Li, M.; Deng, G.; Wu, X.; Zeng, J.; Hao, X.; Wang, X.; Liu, J.; Cho, W.C.S.; et al. MicroRNA-124 negatively regulates TLR signaling in alveolar macrophages in response to mycobacterial infection. Mol. Immunol. 2014, 62, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, G.; Deng, X.; Yu, Q.; Hu, Y.; Sun, H.; Wang, Z.; Chen, H.; Jia, C.; Wang, D. Analysis of miRNA expression profiling in human macrophages responding to Mycobacterium infection: Induction of the immune regulator miR-146a. J. Infect. 2014, 68, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yue, Y.; Xu, W.; Xiong, S. MicroRNA-146a represses mycobacteria-induced inflammatory response and facilitates bacterial replication via targeting IRAK-1 and TRAF-6. PLoS ONE 2013, 8, e81438. [Google Scholar]
- Ni, B.; Rajaram, M.V.S.; Lafuse, W.P.; Landes, M.B.; Schlesinger, L.S. Mycobacterium tuberculosis decreases human macrophage IFN-γ responsiveness through miR-132 and miR-26a. J. Immunol. 2014, 193, 4537–4547. [Google Scholar] [CrossRef] [PubMed]
- Ghorpade, D.S.; Holla, S.; Kaveri, S.V.; Bayry, J.; Patil, S.A.; Balaji, K.N. Sonic hedgehog-dependent induction of microRNA 31 and microRNA 150 regulates Mycobacterium bovis BCG-driven toll-like receptor 2 signaling. Mol. Cell. Biol. 2013, 33, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Kaul, V.; Mehra, A.; Chatterjee, S.; Tousif, S.; Dwivedi, V.P.; Suar, M.; van Kaer, L.; Bishai, W.R.; Das, G. Mycobacterium tuberculosis controls microRNA-99b (miR-99b) expression in infected murine dendritic cells to modulate host immunity. J. Biol. Chem. 2013, 288, 5056–5061. [Google Scholar] [CrossRef] [PubMed]
- Pathway, N.-B.; Kumar, M.; Sahu, S.K.; Kumar, R.; Subuddhi, A.; Maji, R.K.; Jana, K. MicroRNA let-7 modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-κB pathway. Cell Host Microbe 2015, 17, 1–12. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flór, T.B.; Blom, B. Pathogens Use and Abuse MicroRNAs to Deceive the Immune System. Int. J. Mol. Sci. 2016, 17, 538. https://doi.org/10.3390/ijms17040538
Flór TB, Blom B. Pathogens Use and Abuse MicroRNAs to Deceive the Immune System. International Journal of Molecular Sciences. 2016; 17(4):538. https://doi.org/10.3390/ijms17040538
Chicago/Turabian StyleFlór, Thomas B., and Bianca Blom. 2016. "Pathogens Use and Abuse MicroRNAs to Deceive the Immune System" International Journal of Molecular Sciences 17, no. 4: 538. https://doi.org/10.3390/ijms17040538