
Exploring the Interaction Natures in Plutonyl (VI) Complexes with Topological Analyses of Electron Density

Abstract
:
1. Introduction
2. Computational Details
3. Results and Discussion
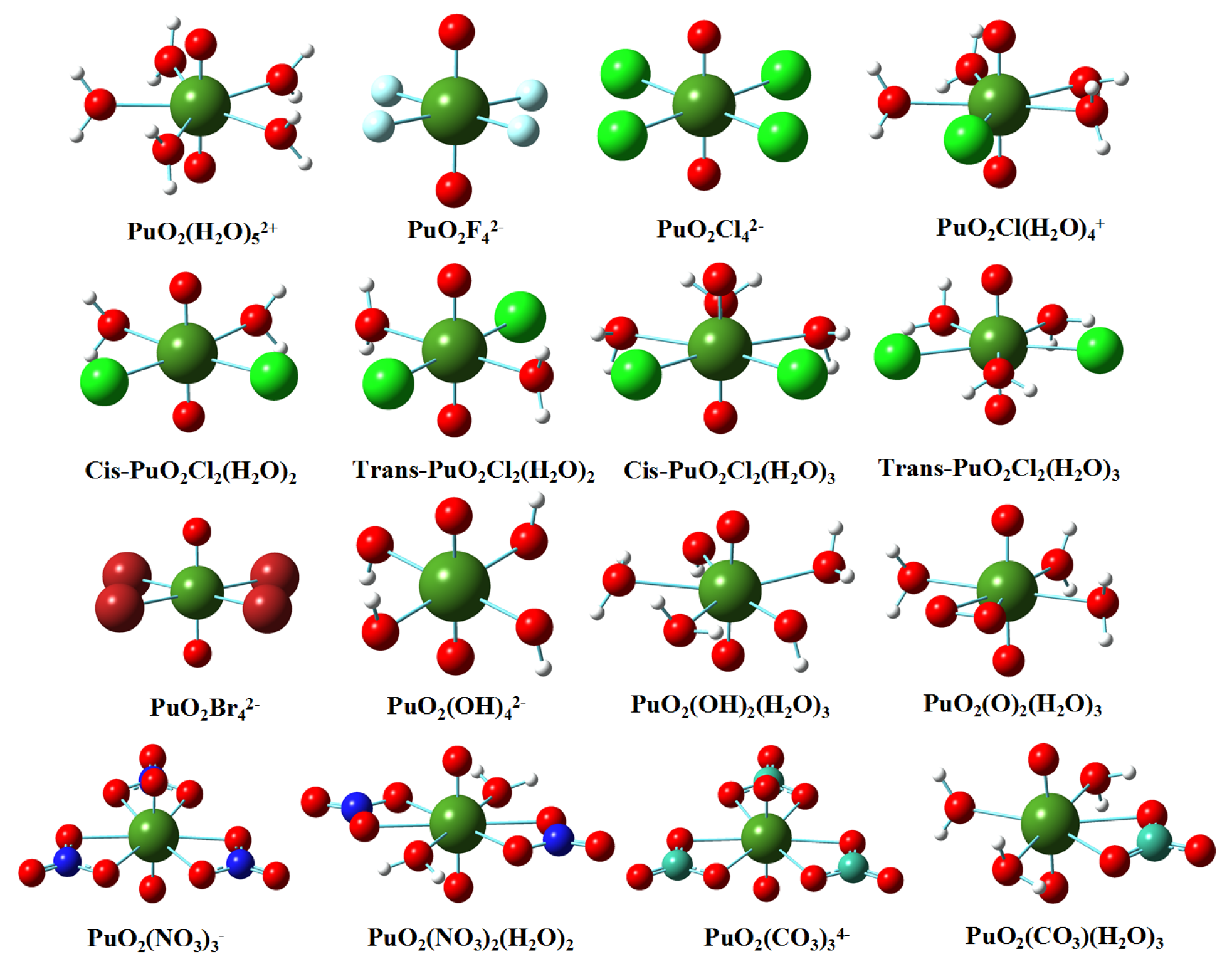
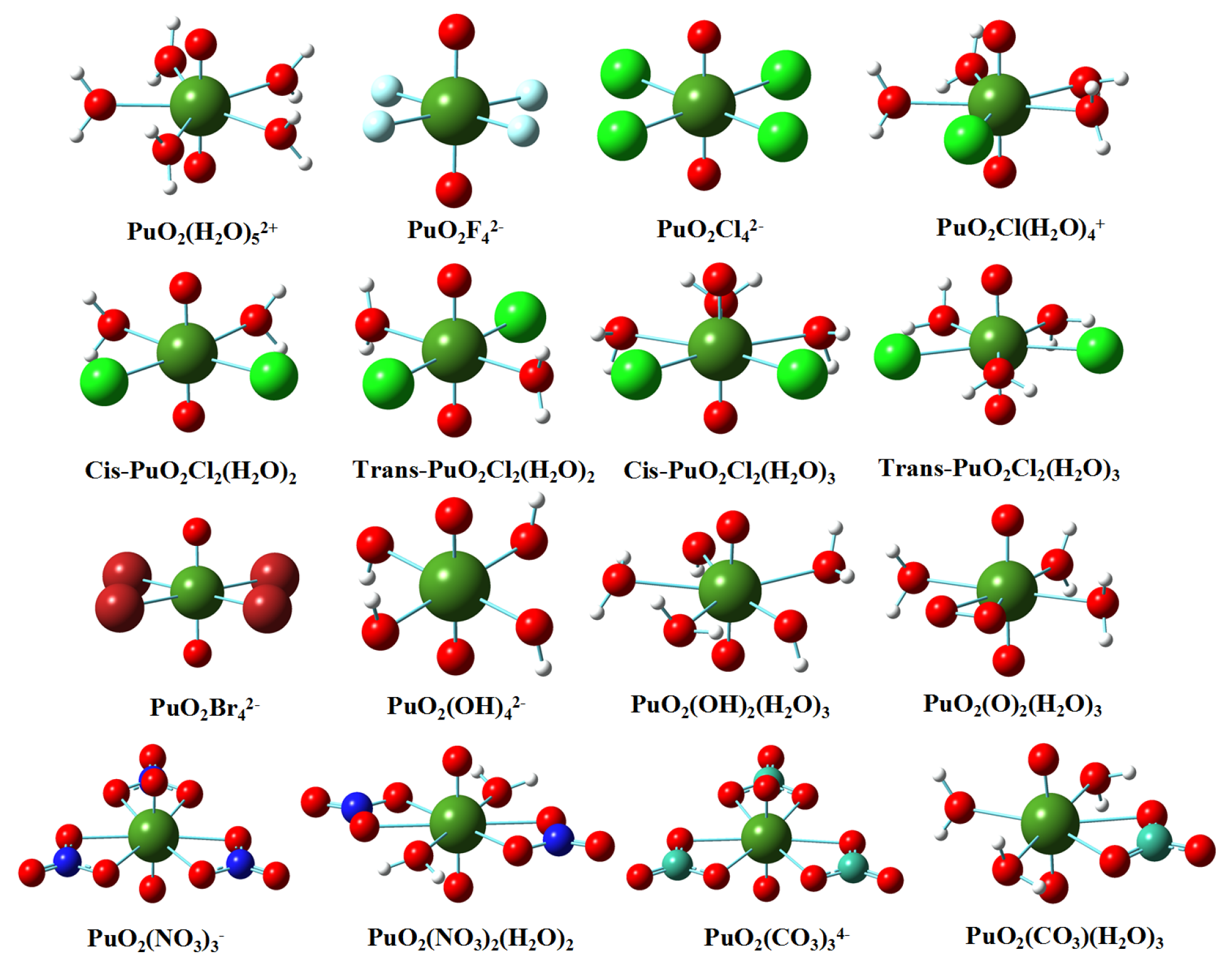
3.1. Structures in Gaseous and Solution Phases
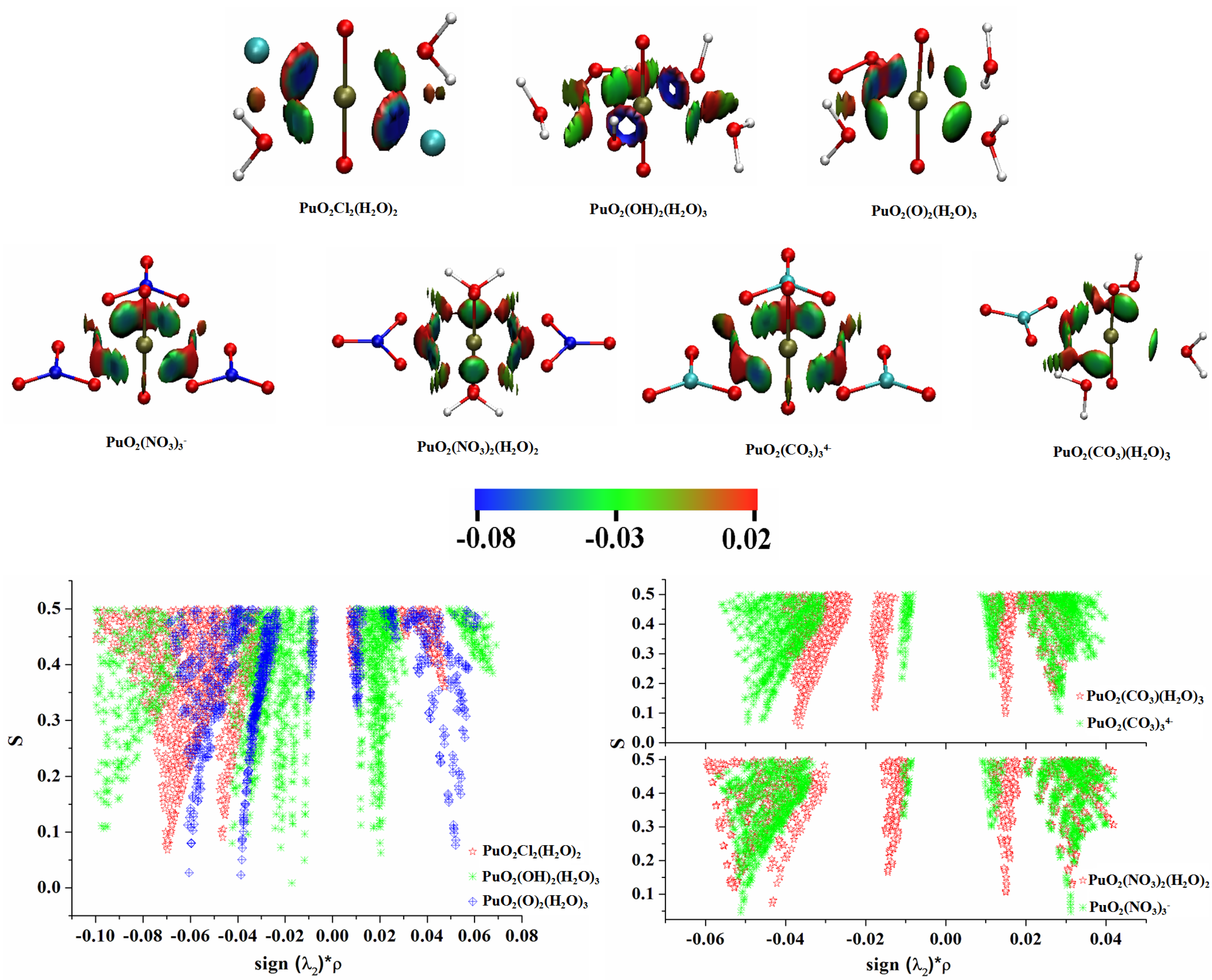
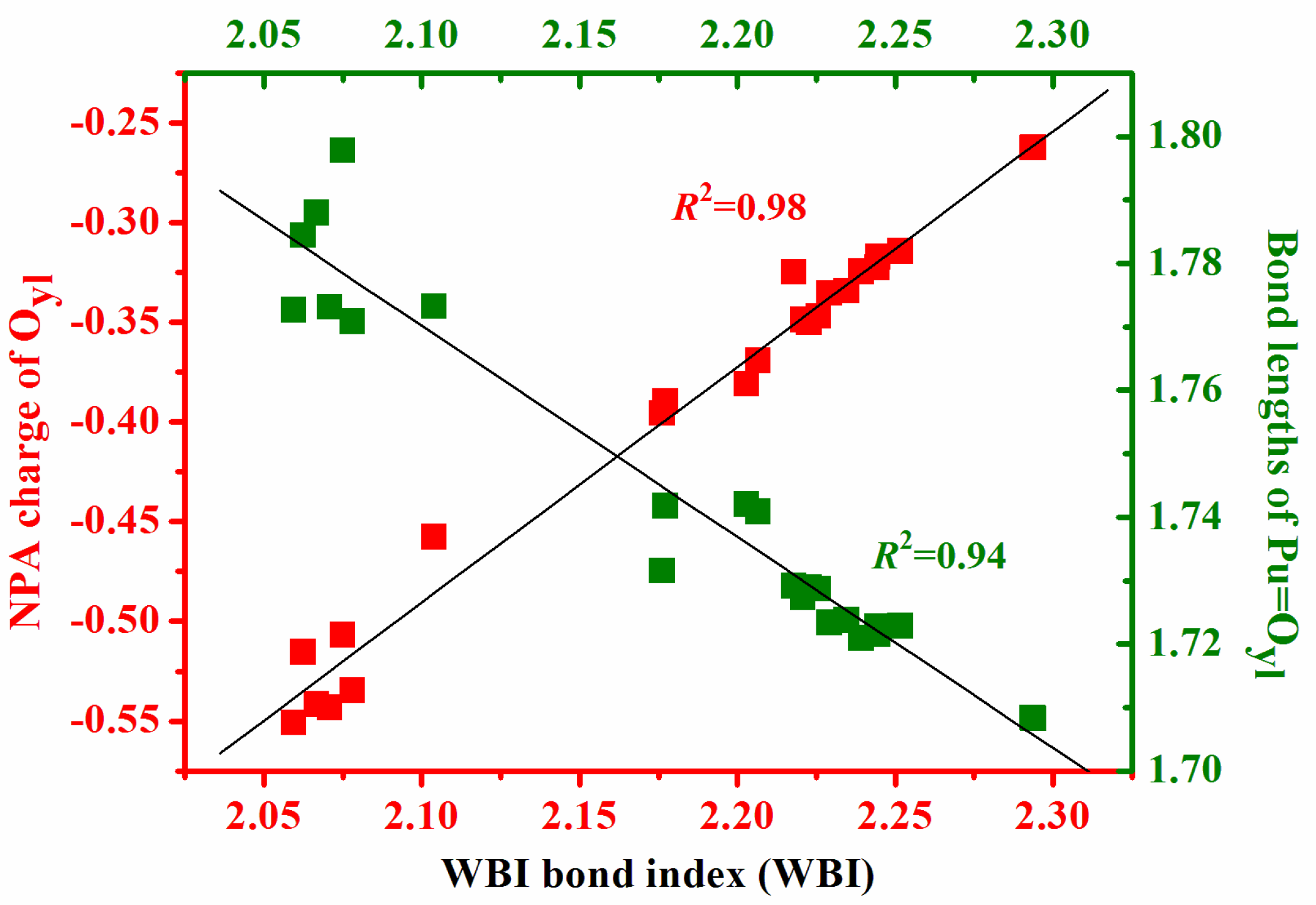
3.2. Natural Bonding Orbital Analyses
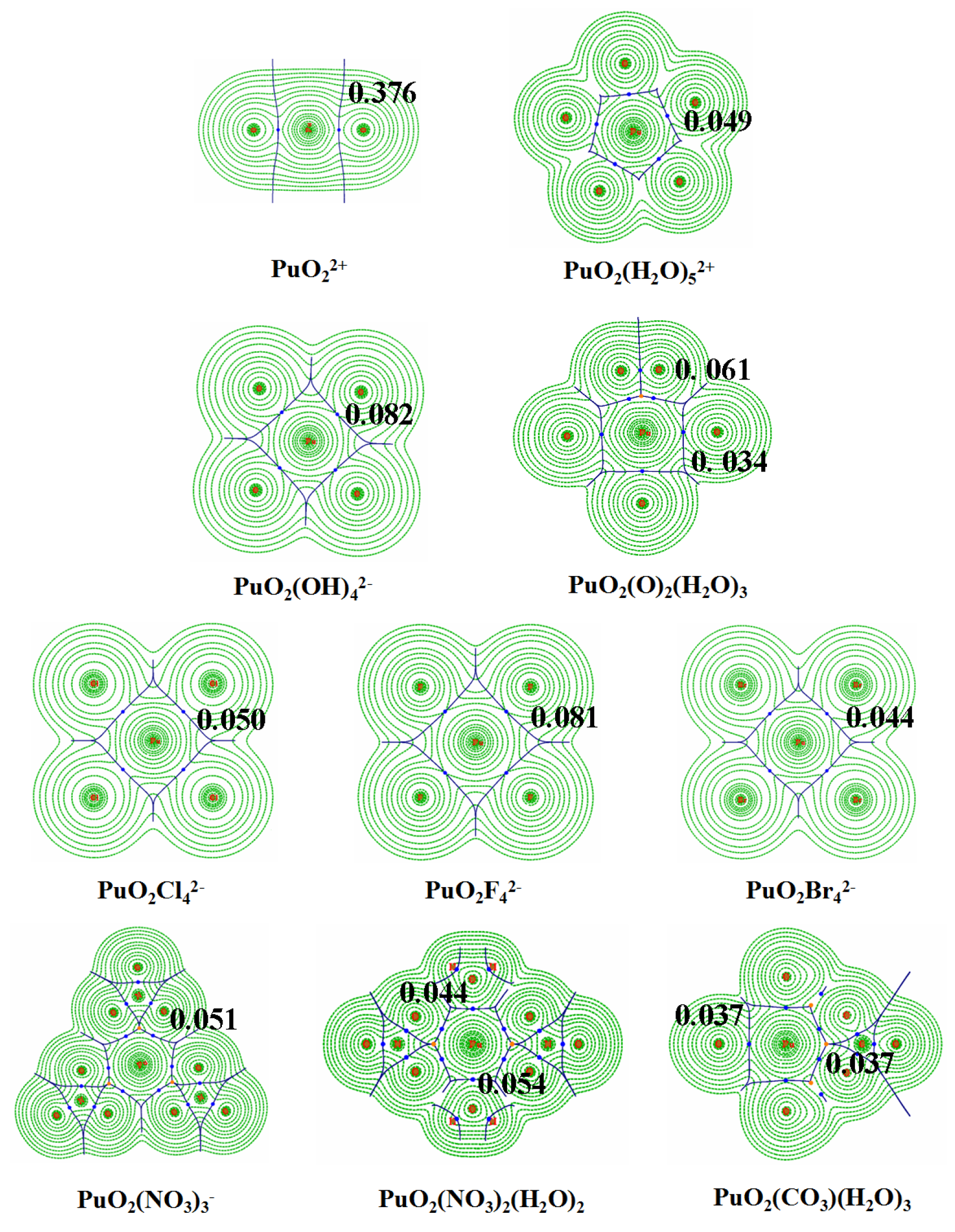
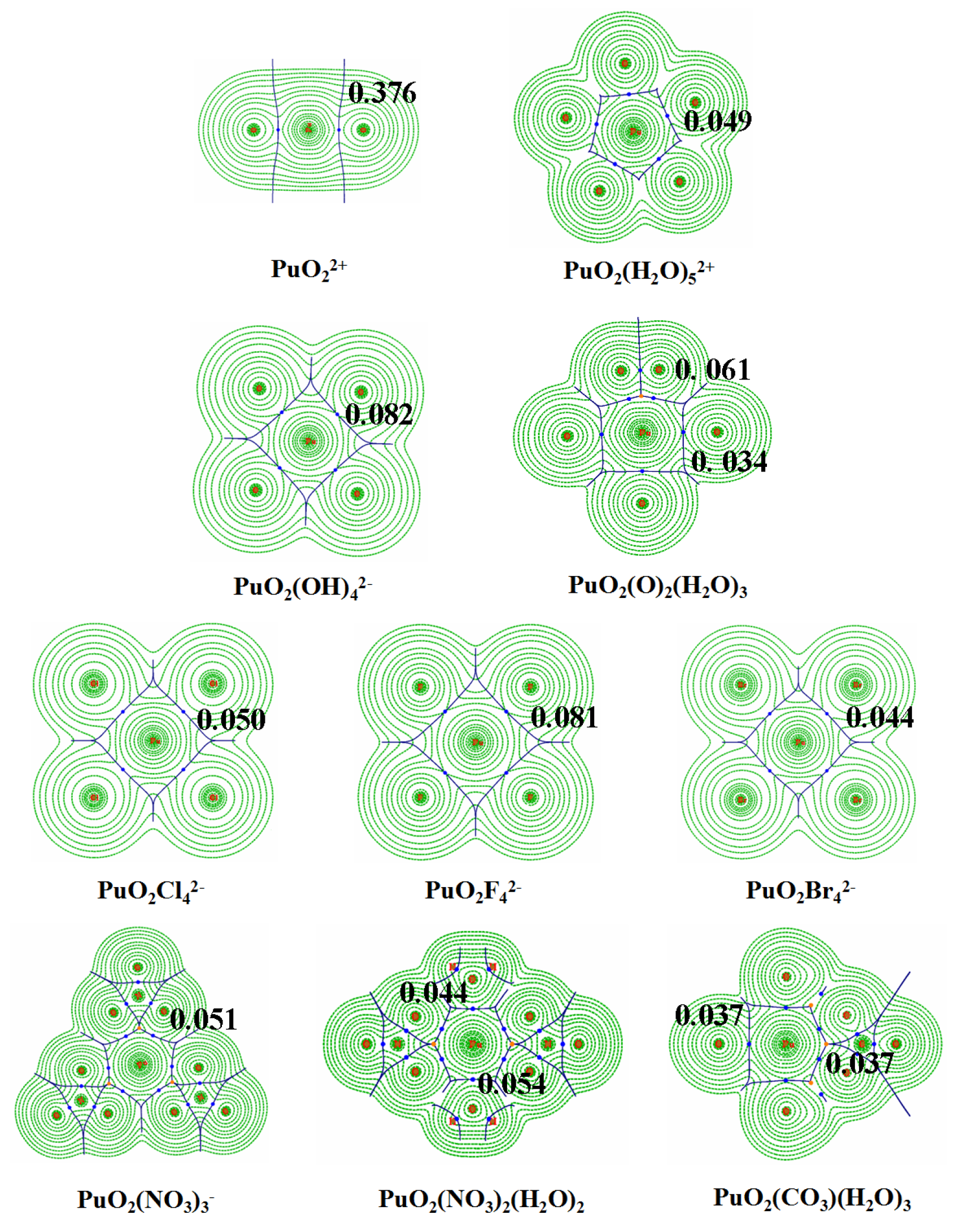
3.3. QTAIM Topological Analyses
3.4. Interaction Quantum Atom (IQA) Analyses
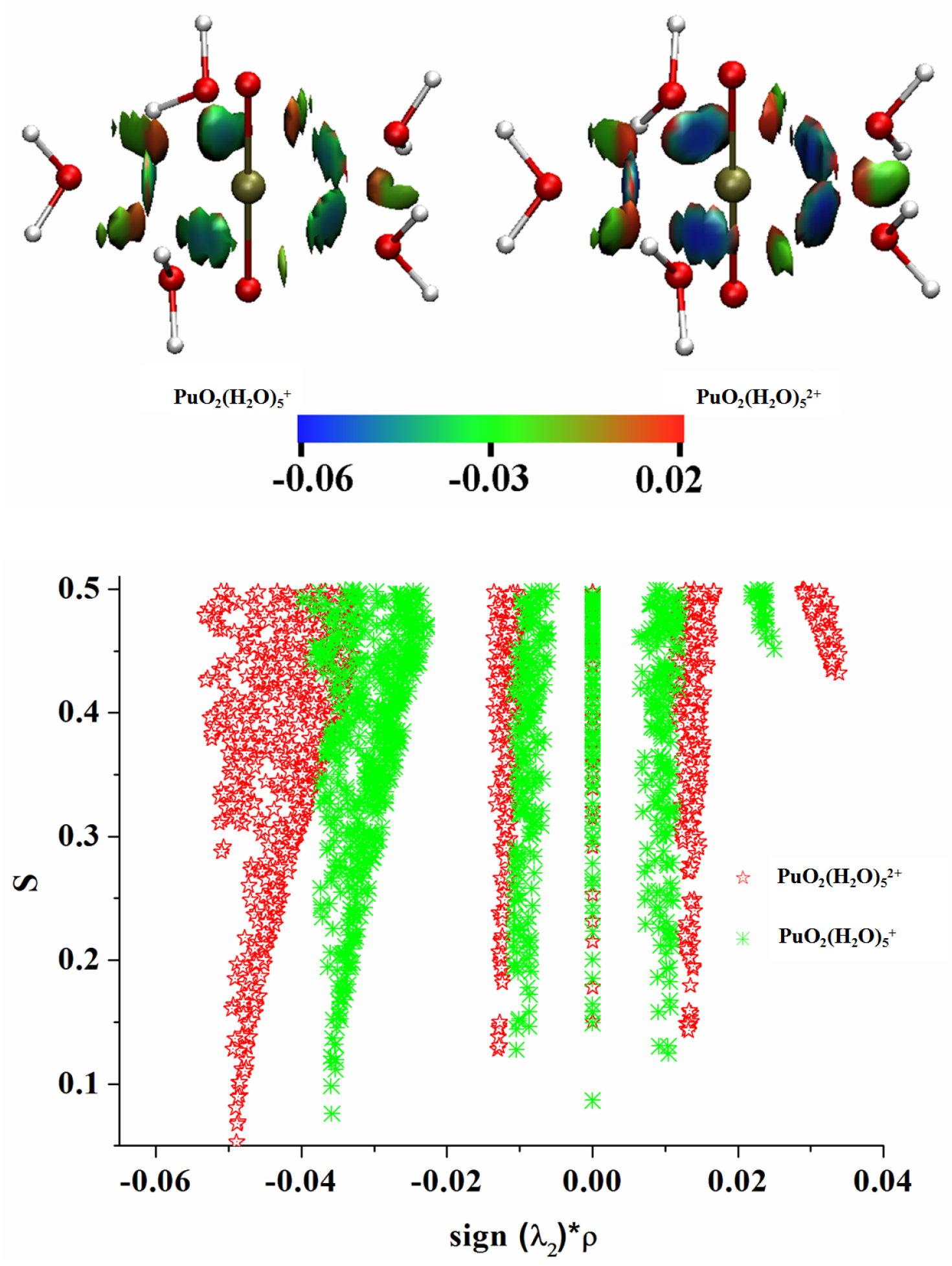
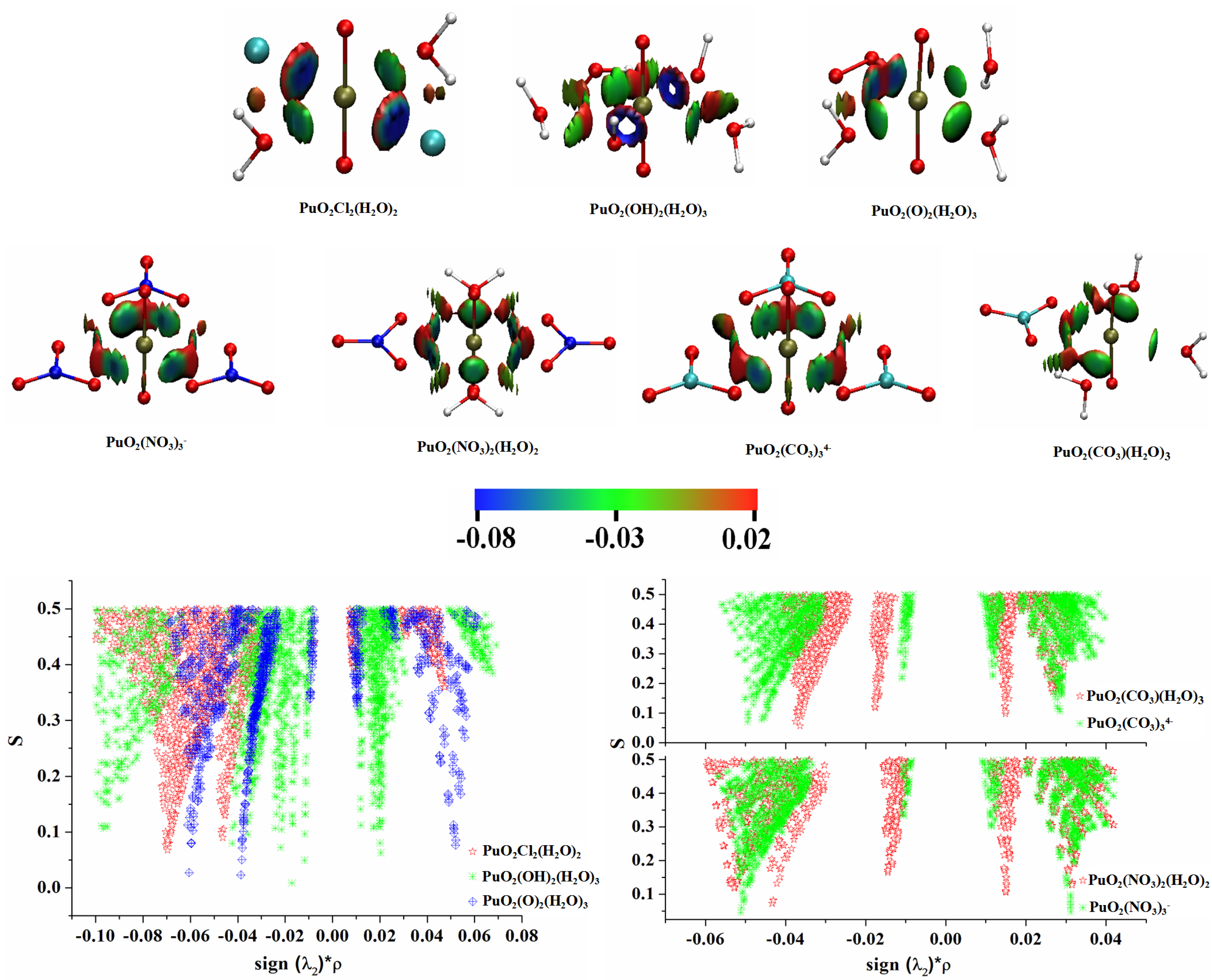
3.5. Electron Localization Function (ELF) Analyses
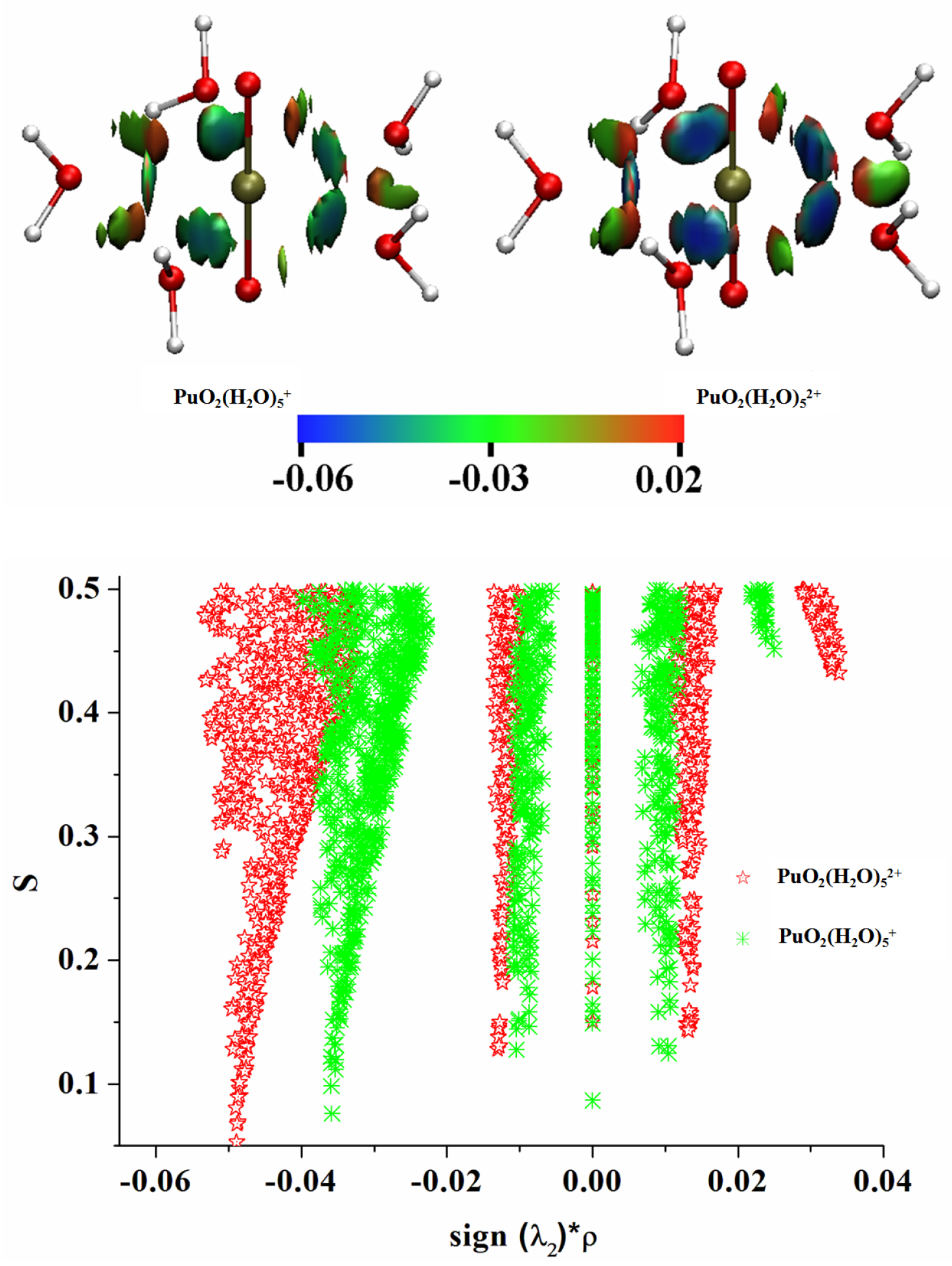
3.6. Noncovalent Interaction (NCI) Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Madic, C.; Begun, G.M.; Hobart, D.E.; Hahn, R.L. Raman spectroscopy of neptunyl and plutonyl Ions in aqueous solution: Hydrolysis of Np(V1) and Pu(V1) and disproportionation of Pu(V). Inorg. Chem. 1984, 23, 1914–1921. [Google Scholar] [CrossRef]
- Conradson, S.D.; Abney, K.D.; Begg, B.D.; Brady, E.D.; Clark, D.L.; den Auwer, C.; Ding, M.; Dorhout, P.K.; Espinosa-Faller, F.J.; Gordon, P.L.; et al. Higher order speciation effects on plutonium L3 X-ray absorption near edge spectra. Inorg. Chem. 2004, 43, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Runde, W.; Reilly, S.D.; Neu, M.P. Spectroscopic investigation of the formation of PuO2Cl+ and PuO2Cl2 in NaCl solutions and application for natural brine solutions. Geochim. Cosmochim. Acta 1999, 63, 3443–3449. [Google Scholar] [CrossRef]
- Gaunt, A.J.; May, I.; Neu, M.P.; Reilly, S.D.; Scott, B.L. Structural and spectroscopic characterization of Plutonyl(VI) Nitrate under acidic conditions. Inorg. Chem. 2011, 50, 4244–4246. [Google Scholar] [CrossRef] [PubMed]
- Correction note: Shamov, G.A.; Schreckenbach, G. Density functional studies of actinyl aquo complexes studied using small-core effective core potentials and a scalar four-component relativistic method. J. Phys. Chem. A 2006, 110, 12072. [Google Scholar] [CrossRef]
- Cao, Z.J.; Balasubramanian, K. Theoretical studies of UO2(H2O, NpO2(H2O, and PuO2(H2O complexes (n = 4–6) in aqueous solution and gas phase. J. Chem. Phys. 2005, 123, 114309. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.E.; Marston, J.B. Strong correlations in actinide redox reactions. J. Chem. Phys. 2011, 134, 064510. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Balasubramanian, K. Electronic structure and spectra of plutonyl complexes and their hydrated forms: PuO2CO3 and PuO2CO3·nH2O (n = 1, 2). Chem. Phys. Lett. 2004, 399, 67–72. [Google Scholar] [CrossRef]
- Austin, J.P.; Sundararajan, M.; Vincent, M.A.; Hillier, I.H. The geometric structures, vibrational frequencies and redox properties of the actinyl coordination complexes ([AnO2(L)n]m; An = U, Pu, Np; L = H2O, Cl−, , CH3, OH−) in aqueous solution, studied by density functional theory methods. Dalton Trans. 2009, 5902–5909. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, L.; Willetts, A.; Skylaris, C.K.; Handy, N.C.; Spencer, S.; Ioannou, A.G.; Simper, A.M. A relativistic density functional study on the uranium hexafluoride and plutonium hexafluoride monomer and dimer species. J. Am. Chem. Soc. 1998, 120, 11727–11731. [Google Scholar] [CrossRef]
- Hay, P.J.; Martin, R.L. Theoretical studies of the structures and vibrational frequencies of actinide compounds using relativistic effective core potentials with Hartree-Fock and density functional methods: UF6, NpF6, and PuF6. J. Chem. Phys. 1998, 109, 3875–3881. [Google Scholar] [CrossRef]
- Odoh, S.O.; Schreckenbach, G. Theoretical study of the structural properties of plutonium(IV) and (VI) complexes. J. Phys. Chem. A 2011, 115, 14110–14119. [Google Scholar] [CrossRef] [PubMed]
- Vallet, V.; Wahlgren, U.; Grenthe, I. Probing the nature of chemical bonding in uranyl(VI) complexes with quantum chemical methods. J. Phys. Chem. A 2012, 116, 12373–12380. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.W.B. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Kirker, I.; Kaltsoyannis, N. Does covalency really increase across the 5f series? A comparison of molecular orbital, natural population, spin and electron density analyses of AnCp3 (An = Th–Cm; Cp = η5-C5H5). Dalton Trans. 2011, 40, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting quantum atoms: A correlated energy decomposition scheme based on the quantum theory of atoms in molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W.T. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. ORCA, an Ab Initio, DFT and Semiempirical Electronic Structure Package, Version 3.0.0; University of Bonn: Bonn, Germany, 2009.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision B.2.; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Van Wuellen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392–399. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.Y.; Landis, C.R.; Neese, F. All-electron scalar relativistic basis sets for third-row transition metal atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-Electron scalar relativistic basis sets for the actinides. J. Chem. Theory Comput. 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Sinnecker, S.; Rajendran, A.; Klamt, A.; Diedenhofen, M.; Neese, F. Calculation of solvent shifts on electronic g-tensors with the conductor-like screening model (COSMO) and its self-consistent generalization to real solvents (direct COSMO-RS). J. Phys. Chem. A 2006, 110, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Dolg, M.; Stoll, H. Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. J. Chem. Phys. 2003, 118, 487. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core actinide pseudopotential basis sets. J. Mol. Struct. (Theochem.) 2004, 673, 203–209. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. J. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll. version 11.10.16. 2011. Available online: aim.tkgristmill.com (accessed on 10 November 2015).
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Basile, L.J.; Sullivan, J.C.; Ferraro, J.R.; Labonvil, P. The Raman scattering of uranyl and transuranium V, VI, and VII ions. Appl. Spectrosc. 1974, 28, 142–145. [Google Scholar] [CrossRef]
- Ismail, N.; Heully, J.L.; Saue, T.; Daudey, J.P.; Marsden, C.J. Theoretical studies of the actinides: Method calibration for the and ions. Chem. Phys. Lett. 1999, 300, 296–302. [Google Scholar] [CrossRef]
- La Macchia, G.; Infante, I.; Raab, J.; Gibson, J.K.; Gagliardi, L. A theoretical study of the ground state and lowest excited states of PuO0/+/+2 and PuO20/+/+2. Phys. Chem. Chem. Phys. 2008, 10, 7278–7283. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Farrugia, L.J.; Senn, H.M. Metal–metal and metal–ligand bonding at a QTAIM Catastrophe: A combined experimental and theoretical charge density study on the alkylidyne cluster Fe3(μ-H)(μ-COMe)(CO)10. J. Phys. Chem. A 2010, 114, 13418–13433. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E. Chemical bonds without bonding electron density—Does the difference electron-density analysis suffice for a description of the chemical bond. Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Mihaiv, V.P. Density functionals of chemical bonding. Int. J. Mol. Sci. 2008, 9, 1050–1095. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gas | Solution | Expt | |||
|---|---|---|---|---|---|---|
| Pu–Oyl | Pu–OH2 | Pu–Oyl | Pu–OH2 | Pu–Oyl | Pu–OH2 | |
| 1.711(1.673) | 1.716(1.723) | |||||
| PuO2(H2O | 1.751(1.703) | 2.451(2.467) | 1.763(1.716) | 2.402(2.449) | 1.74 | 2.41 |
| 1.748(1.714) | 1.771(1.785) | |||||
| PuO2(H2O | 1.808(1.766) | 2.539(2.591) | 1.832(1.779) | 2.497(2.597) | 1.81 | 2.47 |
| Species | Gas | Solution | ||||
|---|---|---|---|---|---|---|
| Pu–Oyl | Pu–L | Pu–OH2 | Pu–Oyl | Pu–L | Pu–OH2 | |
| PuO2 | 1.811(1.773) | 2.222(2.225) | 1.806(1.766) | 2.201(2.209) | ||
| PuO2 | 1.772(1.742) | 2.749(2.743) | 1.775(1.737) | 2.706(2.692) | ||
| PuO2Cl2(H2O)2 | ||||||
| Cis- | 1.768(1.729) | 2.612(2.582) | 2.445(2.513) | 1.768(1.732) | 2.671(2.664) | 2.345(2.476) |
| Trans- | 1.767(1.729) | 2.623(2.591) | 2.417(2.481) | 1.768(1.728) | 2.670(2.640) | 2.349(2.421) |
| PuO2Cl2(H2O)3 | ||||||
| Cis- | 1.767(1.725) | 2.665(2.619) | 2.574(2.624) | 1.771(1.729) | 2.738(2.680) | 2.458(2.556) |
| Trans- | 1.767(1.722) | 2.717(2.661) | 2.490(2.558) | 1.771(1.728) | 2.757(2.703) | 2.441(2.523) |
| Expt | 1.75 | 2.70 | 2.49 | |||
| PuO2Cl(H2O | 1.763(1.723) | 2.617(2.566) | 2.493(2.542) | 1.768(1.719) | 2.722(2.634) | 2.424(2.486) |
| Expt | 1.75 | 2.75 | 2.43 | |||
| PuO2 | 1.770(1.732) | 2.907(2.904) | 1.776(1.736) | 2.853(2.862) | ||
| PuO2(OH | 1.859(1.798) | 2.281(2.288) | 1.836(1.791) | 2.243(2.272) | ||
| PuO2(OH)2(H2O)3 | 1.836(1.742) | 2.267(2.199) | 2.548(2.611) | 1.800(1.755) | 2.193(2.201) | 2.523(2.588) |
| PuO2(O)2(H2O)3 | 1.827(1.788) | 2.398(2.379) | 2.526(2.595) | 1.838(1.796) | 2.444(2.411) | 2.471(2.563) |
| PuO2(NO3 | 1.762(1.729) | 2.500(2.493) | 1.761(1.737) | 2.493(2.485) | ||
| PuO2(NO3)2(H2O)2 | 1.767(1.722) | 2.483(2.474) | 2.466(2.524) | 1.770(1.730) | 2.512(2.490) | 2.434(2.505) |
| Expt | 1.727 | 2.432 | 2.497 | |||
| PuO2(CO3 | 1.815(1.784) | 2.558(2.544) | 1.809(1.780) | 2.467(2.469) | ||
| PuO2(CO3)(H2O)3 | 1.814(1.773) | 2.620(2.607) | 2.514(2.581) | 1.825(1.781) | 2.678(2.673) | 2.456(2.536) |
| Species | q | q | q | EC | EC | EC | WBI | WBI |
|---|---|---|---|---|---|---|---|---|
| (Pu) | (Oyl) | (L) | (Pu) | (Oyl) | (L) | (Pu=O) | (Pu–L) | |
| 2.04 | −0.52 | – | – | 2.05 | – | |||
| 2.50 | −0.25 | – | – | 2.24 | – | |||
| PuO2(H2O | 1.44 | −0.54 | −0.89 | 2.07 | (0.24) a | |||
| PuO2(H2O | 1.44 | −0.26 | −0.85 | 2.29 | (0.39) | |||
| PuO2 | 1.39 | −0.46 | −0.62 | 2.10 | 0.65 | |||
| PuO2 | 0.68 | −0.39 | −0.48 | 2.18 | 0.86 | |||
| PuO2Cl2(H2O)2 | ||||||||
| Cis- | 0.86 | −0.35 | −0.28 | 2.23 | 1.14/(0.35) | |||
| Trans- | 0.86 | −0.35 | −0.27 | 2.22 | 1.14/(0.35) | |||
| PuO2Cl2(H2O)3 | ||||||||
| Cis- | 0.81 | −0.34 | −0.3 | 2.23 | 1.08/(0.29) | |||
| Trans- | 0.85 | −0.34 | −0.36 | 2.23 | 1.01/(0.31) | |||
| PuO2Cl(H2O | 1.09 | −0.32 | −0.18 | 2.25 | 1.26/(0.32) | |||
| PuO2 | 0.57 | −0.40 | −0.44 | 2.18 | 0.89 | |||
| PuO2(OH | 1.07 | −0.51 | −0.94 | 2.08 | 0.79 | |||
| PuO2(OH)2(H2O)3 | 1.13 | −0.37 | −0.86 | 2.21 | 0.95/(0.26) | |||
| PuO2(O)2(H2O)3 | 1.31 | −0.54 | −0.31 | 2.07 | 0.40/(0.24) | |||
| PuO2(NO3 | 1.13 | −0.32 | −0.42 | 2.22 | 0.41 | |||
| PuO2(NO3)2(H2O)2 | 1.15 | −0.32 | −0.44 | 2.24 | 0.43/(0.32) | |||
| PuO2(CO3 | 1.22 | −0.52 | −0.73 | 2.06 | 0.41 | |||
| PuO2(CO3)(H2O)3 | 1.40 | −0.55 | −0.68 | 2.06 | 0.25/(0.25) |
| Pu-L BCPs | |||||
|---|---|---|---|---|---|
| Species | (Pu,L) | ||||
| 0.334 | 0.263 | −0.322 | 2.181 | 1.90 | |
| 0.376 | 0.349 | −0.393 | 2.374 | 1.88 | |
| PuO2(H2O | 0.035 | 0.150 | 0.002 | 0.204 | 0.95 |
| PuO2(H2O | 0.049 | 0.209 | 4.8 × | 0.299 | 0.99 |
| PuO2 | 0.081 | 0.363 | −0.005 | 0.578 | 1.05 |
| PuO2 | 0.050 | 0.113 | −0.008 | 0.577 | 1.21 |
| PuO2Cl2(H2O)2 | |||||
| Cis- | 0.071 (0.045) | 0.156 (0.184) | −0.016 (3.6 × ) | 0.822 (0.287) | 1.30 |
| Trans- | 0.070 (0.047) | 0.143 (0.204) | −0.016 (6.2 × ) | 0.823 (0.290) | 1.31 |
| PuO2Cl2(H2O)3 | |||||
| Cis- | 0.066 (0.036) | 0.143 (0.131) | −0.014 (3.2 × ) | 0.742 (0.230) | 1.28 |
| Trans- | 0.061 (0.045) | 0.132 (0.181) | −0.012 (3.8 × ) | 0.673 (0.266) | 1.26 |
| PuO2Cl(H2O | 0.074 (0.045) | 0.156 (0.180) | -0.018 (2.1 × ) | 0.927 (0.281) | 1.31 |
| PuO2 | 0.044 | 0.082 | −0.007 | 0.589 | 1.25 |
| PuO2(OH | 0.082 | 0.283 | −0.011 | 0.666 | 1.14 |
| PuO2(OH)2(H2O)3 | 0.105 (0.036) | 0.360 (0.136) | −0.022 (3.5 × ) | 0.827 (0.213) | 1.20 |
| PuO2(O)2(H2O)3 | 0.061 (0.034) | 0.241 (0.156) | −0.004 (0.002) | 0.387 (0.211) | 1.06 |
| PuO2(NO3 | 0.051 | 0.185 | −0.002 | 0.324 | 1.04 |
| PuO2(NO3)2(H2O)2 | 0.054 (0.044) | 0.188 (0.176) | −0.003 (6.0 | 0.349 (0.247) | 1.05 |
| PuO2(CO3 | 0.050 | 0.158 | −0.002 | 0.370 | 1.06 |
| PuO2(CO3)(H2O)3 | 0.037 (0.037) | 0.143 (0.155) | 1.7 × (8.1 × ) | 0.228 (0.240) | 0.99 |
| Complexes | Atoms | / | |||||||
|---|---|---|---|---|---|---|---|---|---|
| (a.u) | (a.u) | (a.u) | (a.u) | (kcal/mol) | (kcal/mol) | (kcal/mol) | |||
| PuO2(H2O | Pu–O(H2O) | −66.39 | −53.69 | 58.35 | 61.09 | −402.23 | −39.53 | −441.77 | 0.09 |
| PuO2 | Pu–F | −78.60 | −66.90 | 72.76 | 72.27 | −293.67 | −74.05 | −367.72 | 0.20 |
| PuO2 | Pu–Cl | −115.93 | −103.53 | 111.48 | 107.66 | −199.55 | −59.61 | −259.16 | 0.23 |
| PuO2 | Pu–Br | −220.93 | −201.90 | 216.87 | 205.69 | −176.96 | −56.48 | −233.43 | 0.24 |
| PuO2(OH | Pu–O(OH−) | −71.53 | −58.09 | 62.90 | 66.07 | −412.27 | −81.58 | −493.85 | 0.17 |
| PuO2(O)2(H2O)3 | Pu–O() | −63.33 | −56.27 | 60.51 | 58.90 | −123.62 | −46.44 | −170.06 | 0.27 |
| PuO2(NO3 | Pu–O() | −61.93 | −53.36 | 57.74 | 57.23 | −198.92 | −39.53 | −238.45 | 0.17 |
| Pu–N() | −37.68 | −39.63 | 42.89 | 34.82 | 247.87 | −1.88 | 245.98 | −0.008 | |
| PuO2(CO3 | Pu–O() | −64.34 | −51.98 | 56.21 | 59.50 | −384.04 | −38.91 | −422.94 | 0.09 |
| Pu–C() | −23.43 | −33.31 | 36.03 | 21.66 | 596.13 | −1.26 | 594.88 | −0.002 |
| Complexes | ||
|---|---|---|
| PuO2(H2O | 0.604 | 0.122 |
| PuO2 | 0.579 | 0.172 |
| PuO2 | 0.593 | 0.229 |
| PuO2 | 0.597 | 0.247 |
| PuO2(OH | 0.567 | 0.227 |
| PuO2(O)2(H2O)3 | 0.581 | 0.153 |
| PuO2(NO3 | 0.576 | 0.166 |
| PuO2(CO3 | 0.594 | 0.152 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.; Sun, X.; Jiang, G. Exploring the Interaction Natures in Plutonyl (VI) Complexes with Topological Analyses of Electron Density. Int. J. Mol. Sci. 2016, 17, 414. https://doi.org/10.3390/ijms17040414
Du J, Sun X, Jiang G. Exploring the Interaction Natures in Plutonyl (VI) Complexes with Topological Analyses of Electron Density. International Journal of Molecular Sciences. 2016; 17(4):414. https://doi.org/10.3390/ijms17040414
Chicago/Turabian StyleDu, Jiguang, Xiyuan Sun, and Gang Jiang. 2016. "Exploring the Interaction Natures in Plutonyl (VI) Complexes with Topological Analyses of Electron Density" International Journal of Molecular Sciences 17, no. 4: 414. https://doi.org/10.3390/ijms17040414