
Transcriptome and Gene Ontology (GO) Enrichment Analysis Reveals Genes Involved in Biotin Metabolism That Affect l-Lysine Production in Corynebacterium glutamicum


Abstract
:
1. Introduction
2. Results
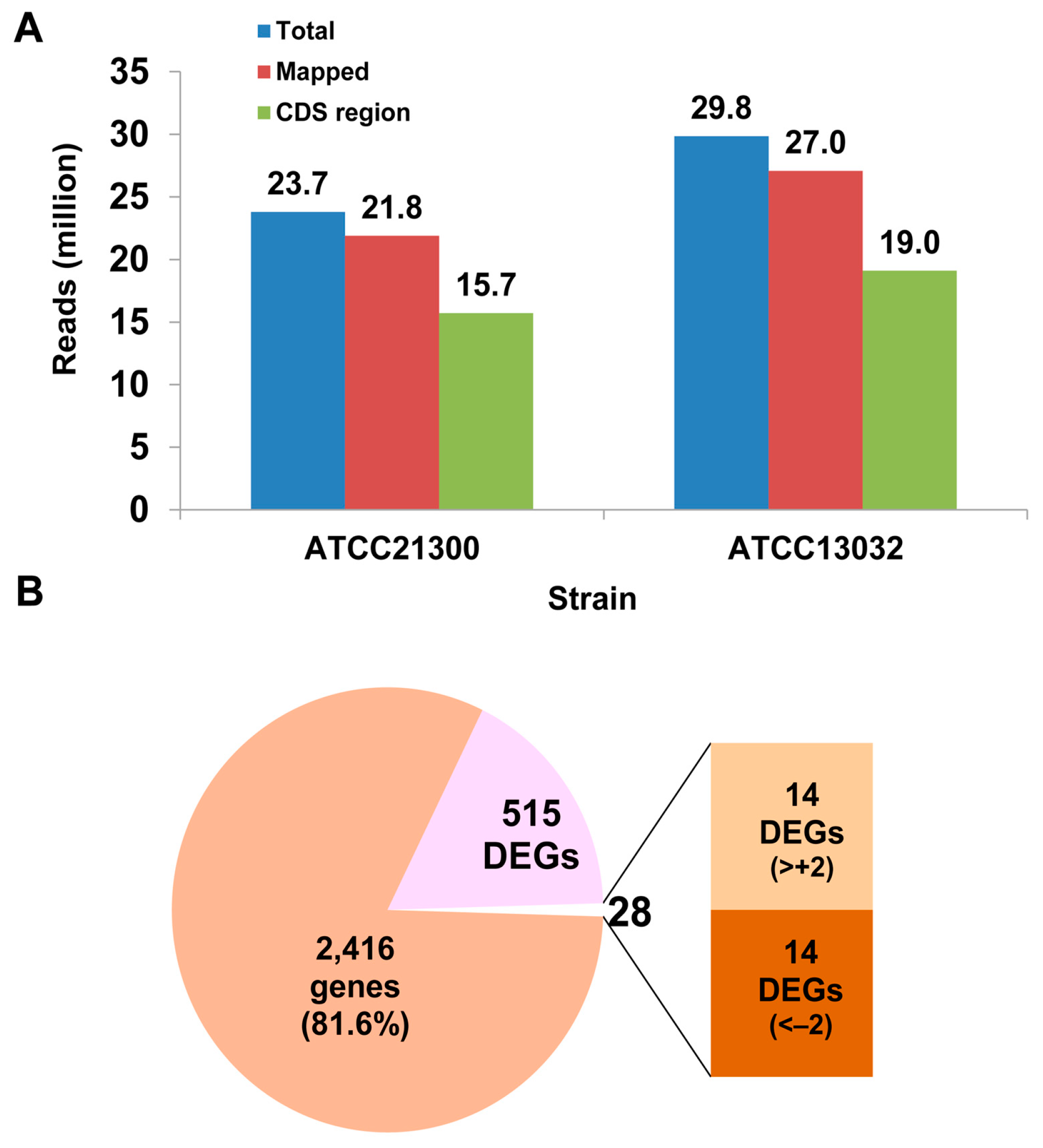
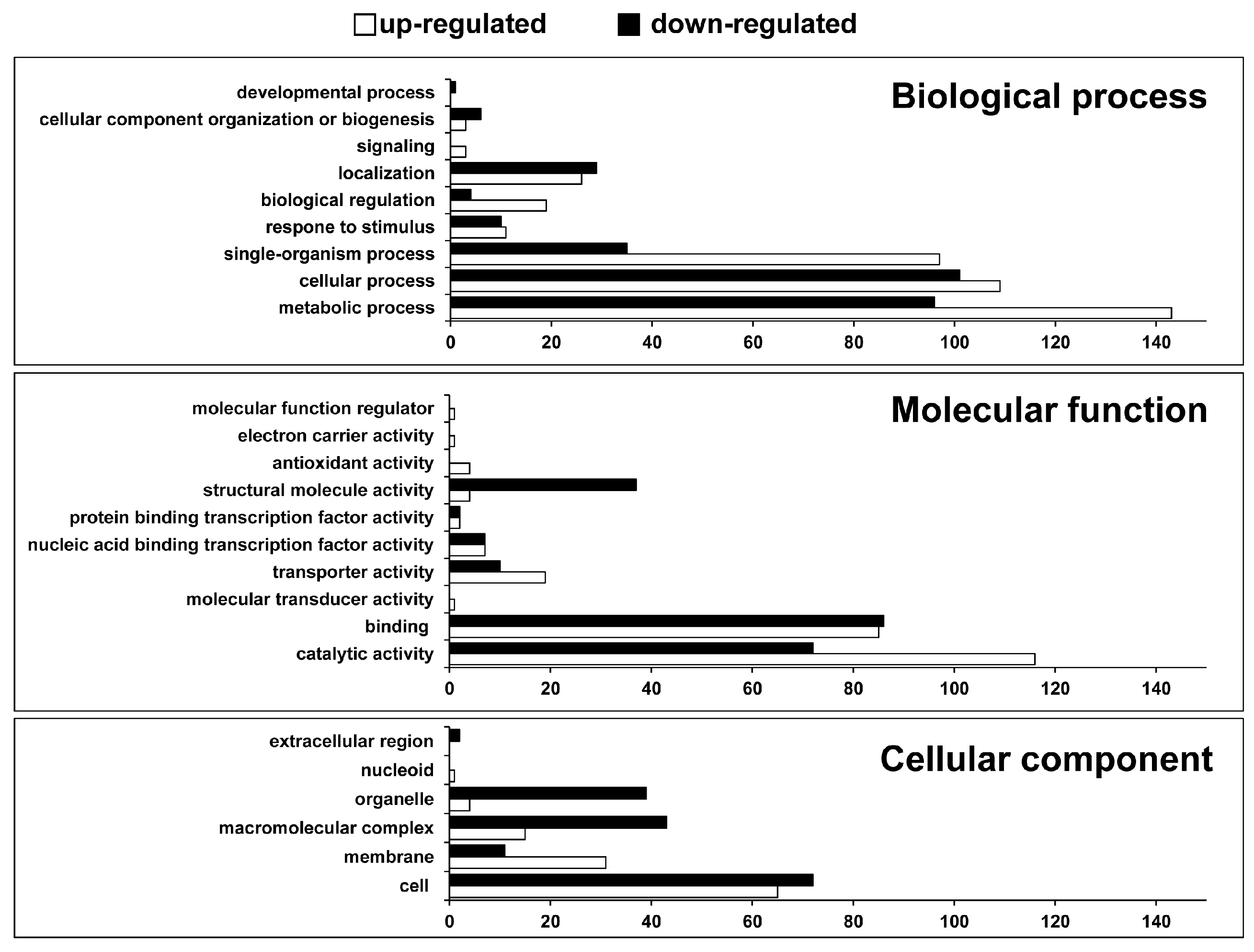
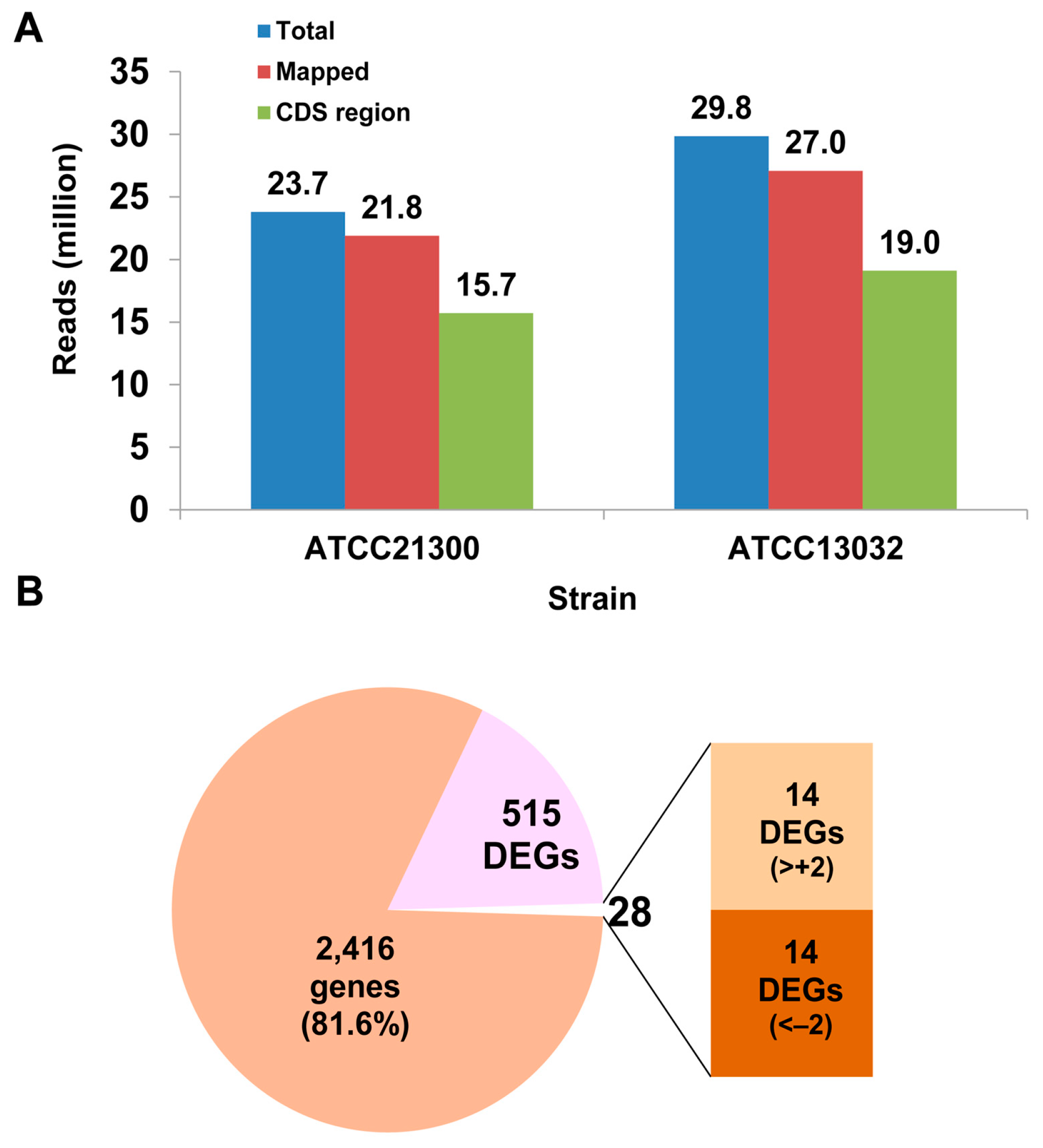
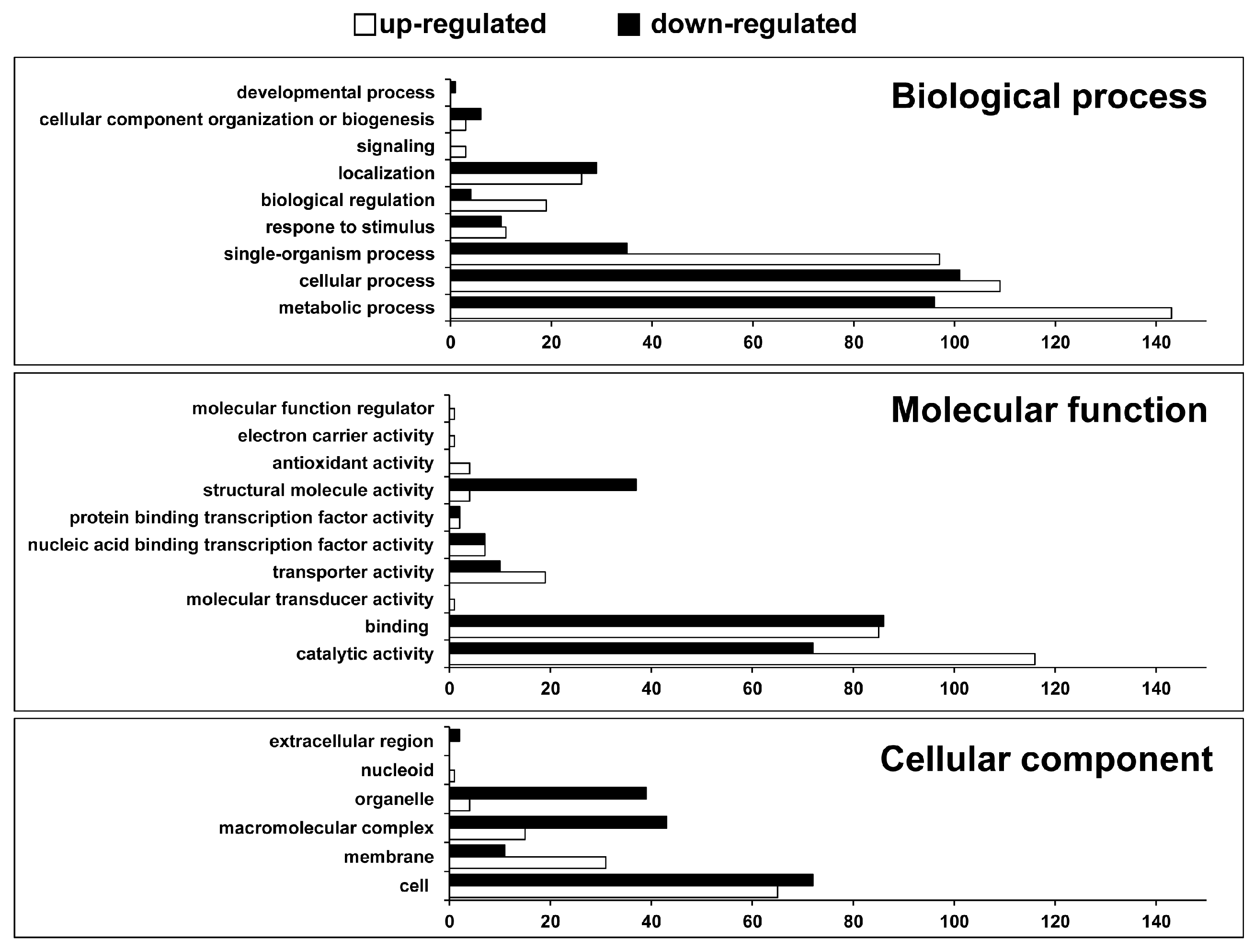
2.1. Identification of Differentially Expressed Genes and Gene Ontology (GO) Enrichment Analysis
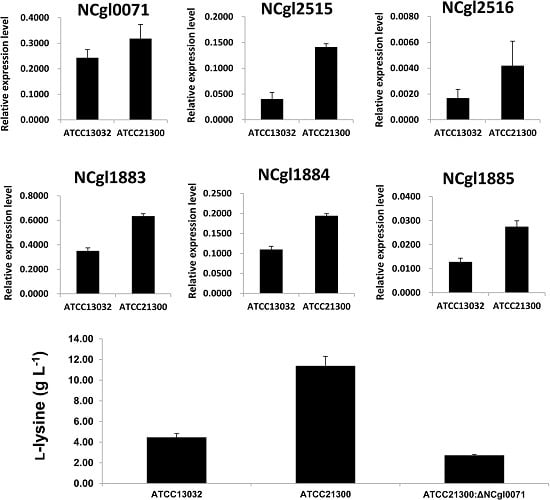
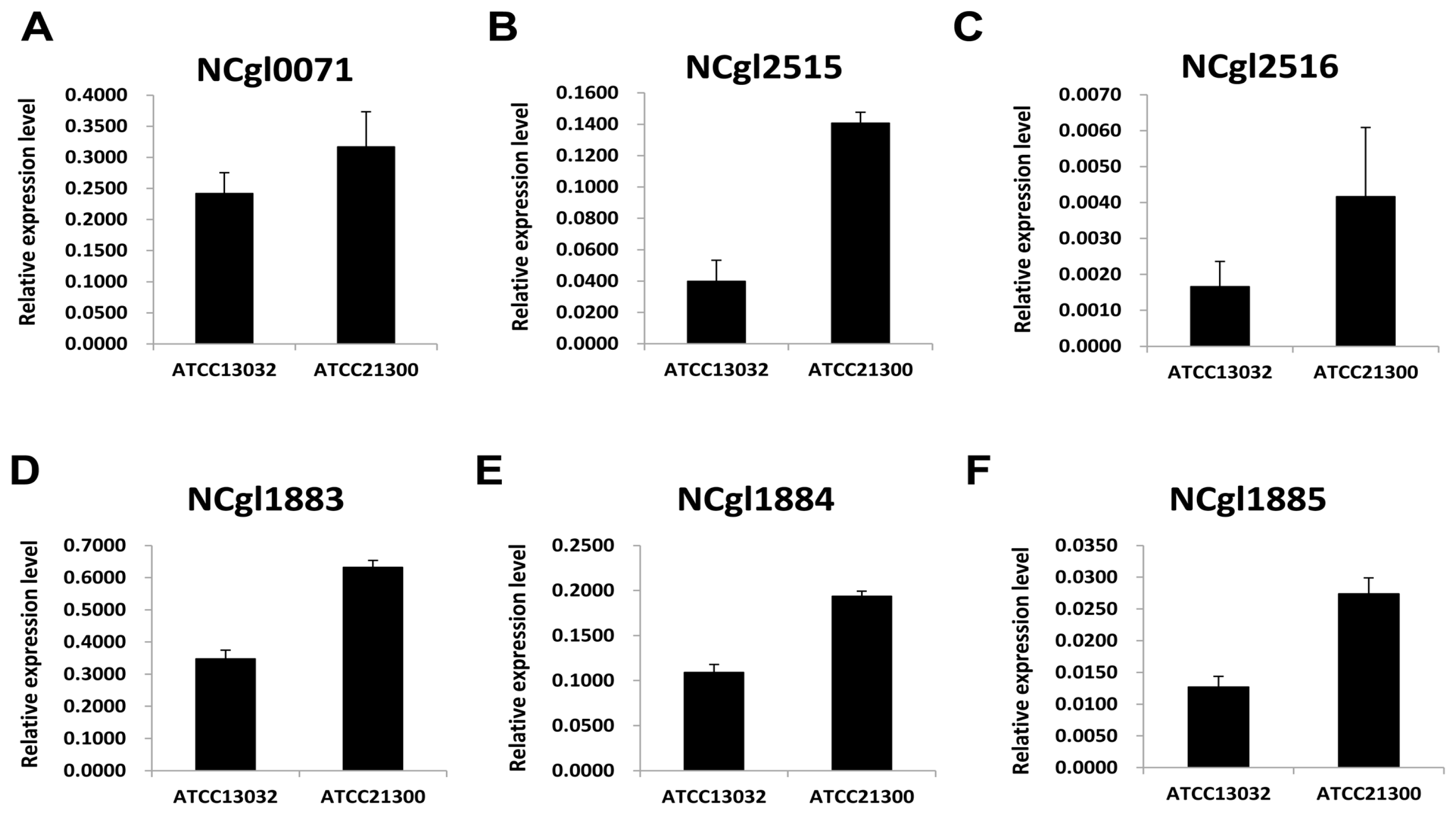
2.2. Expression Patterns of Genes Involved in Biotin Metabolism
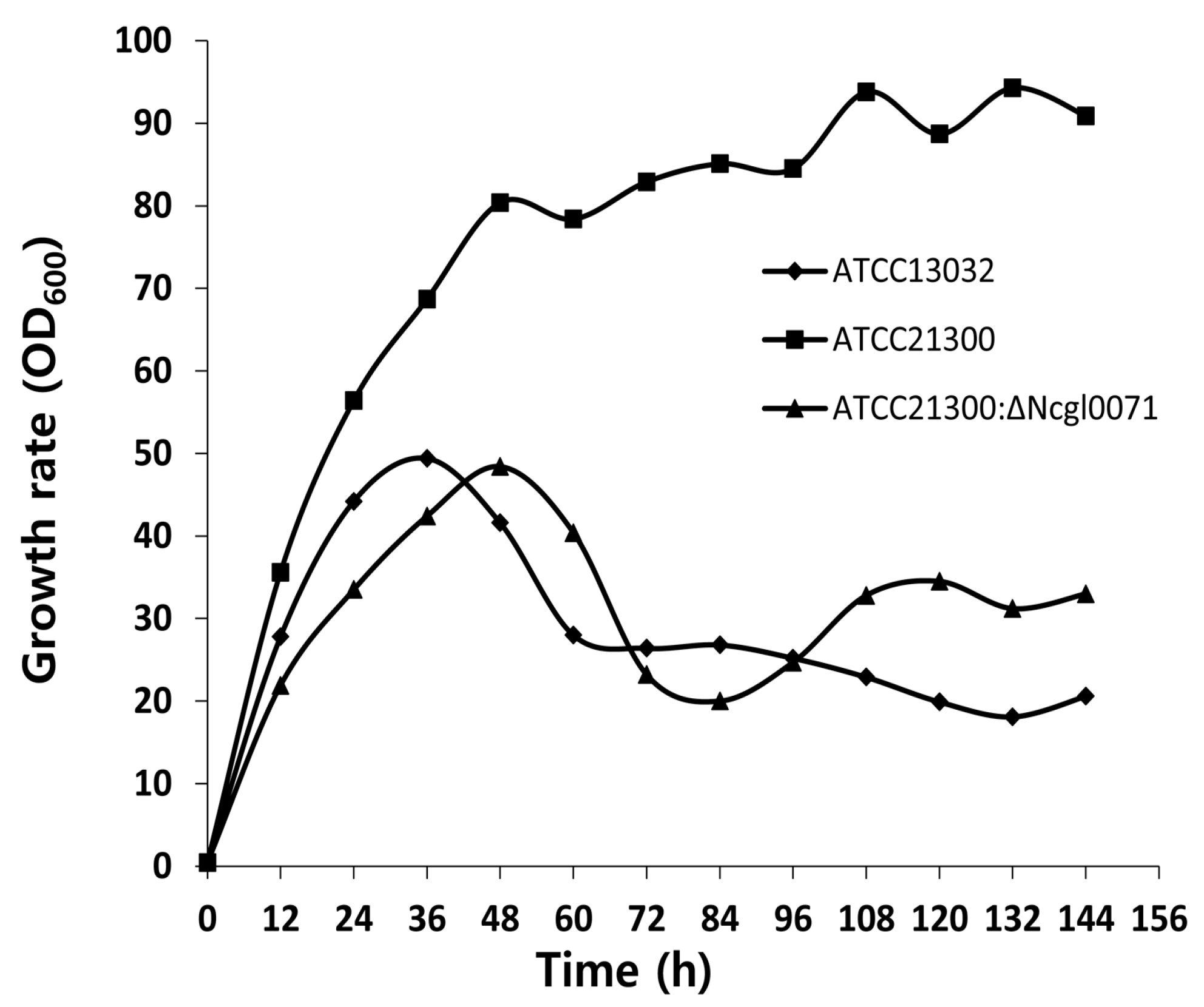
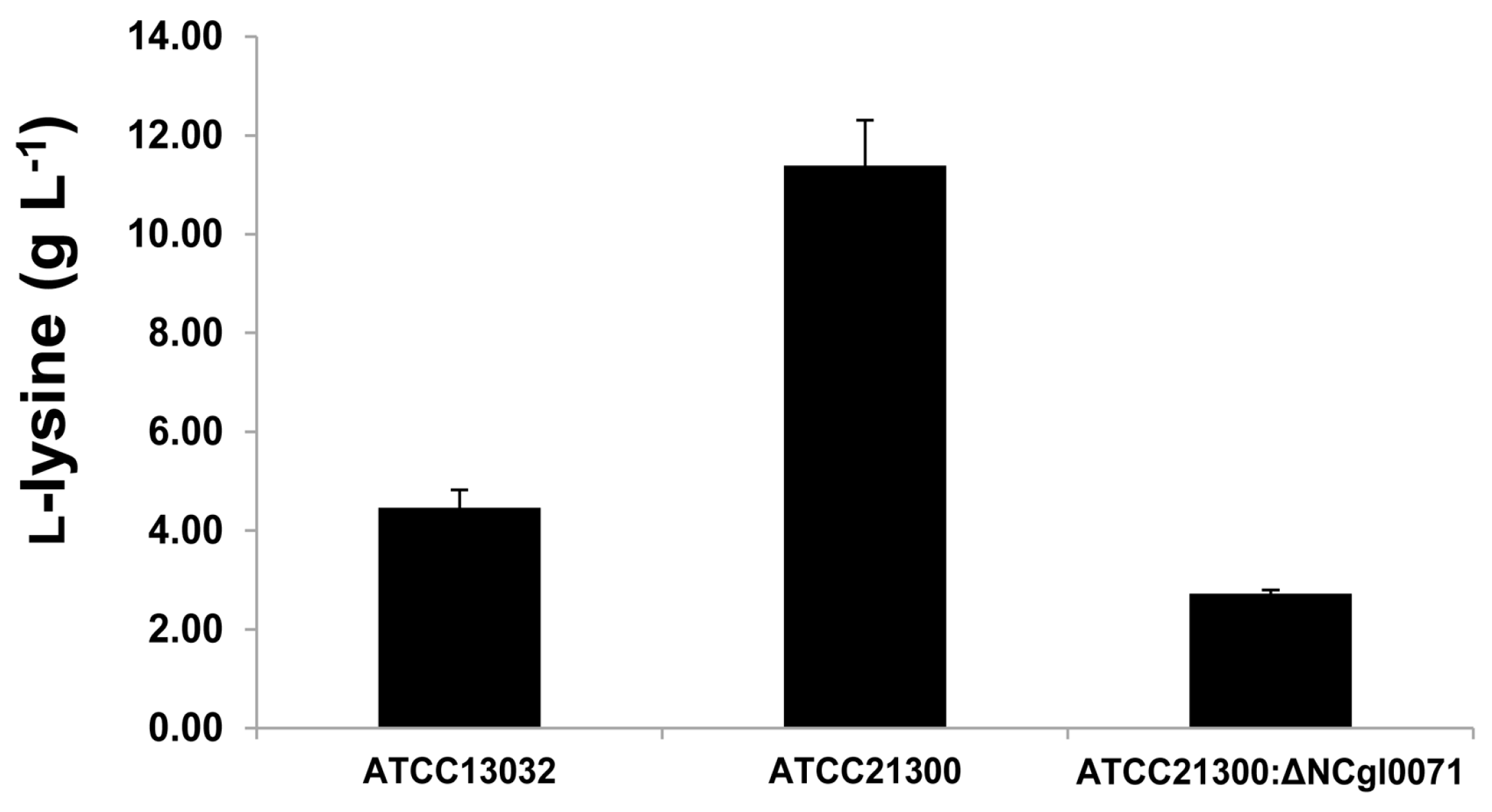
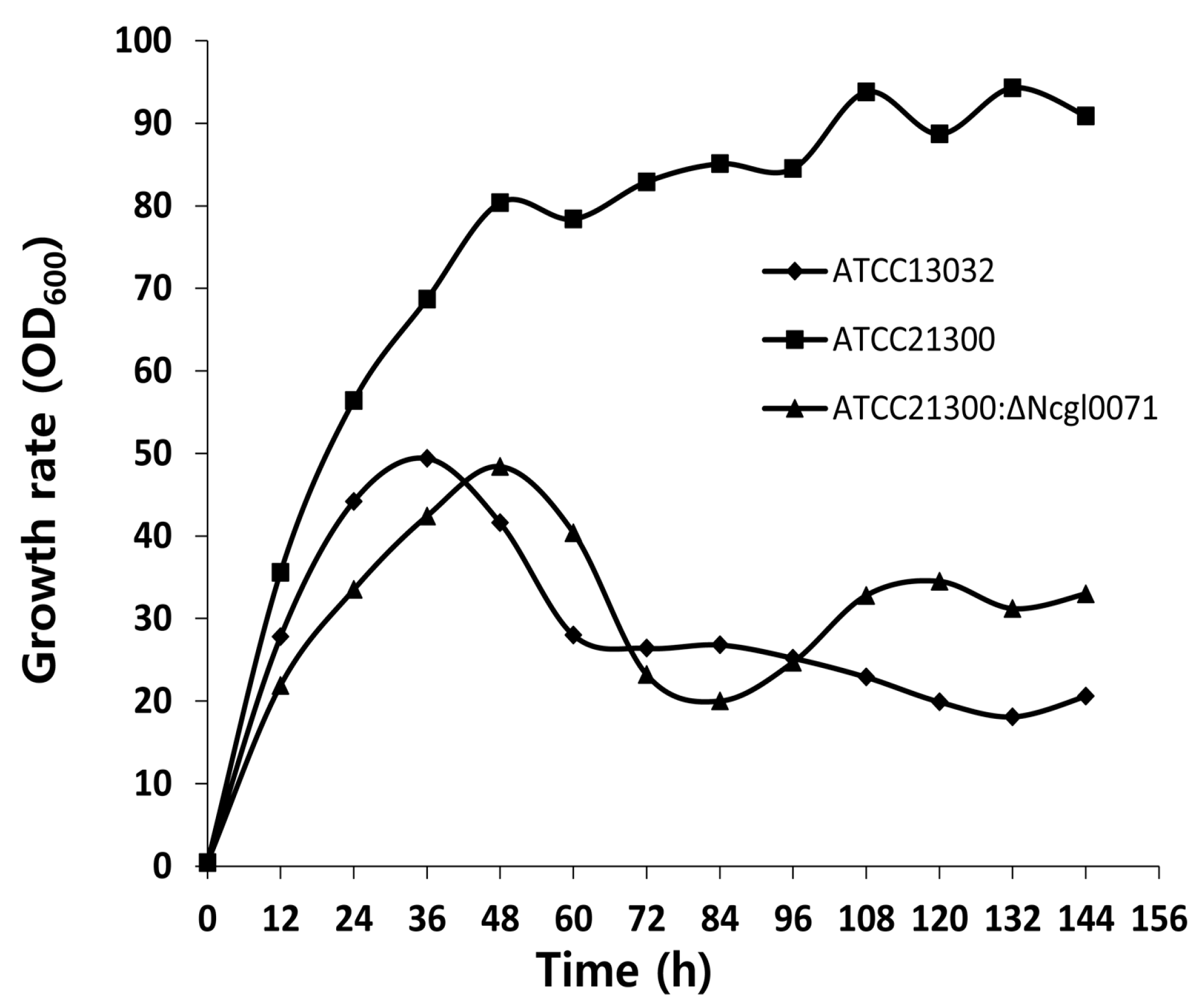
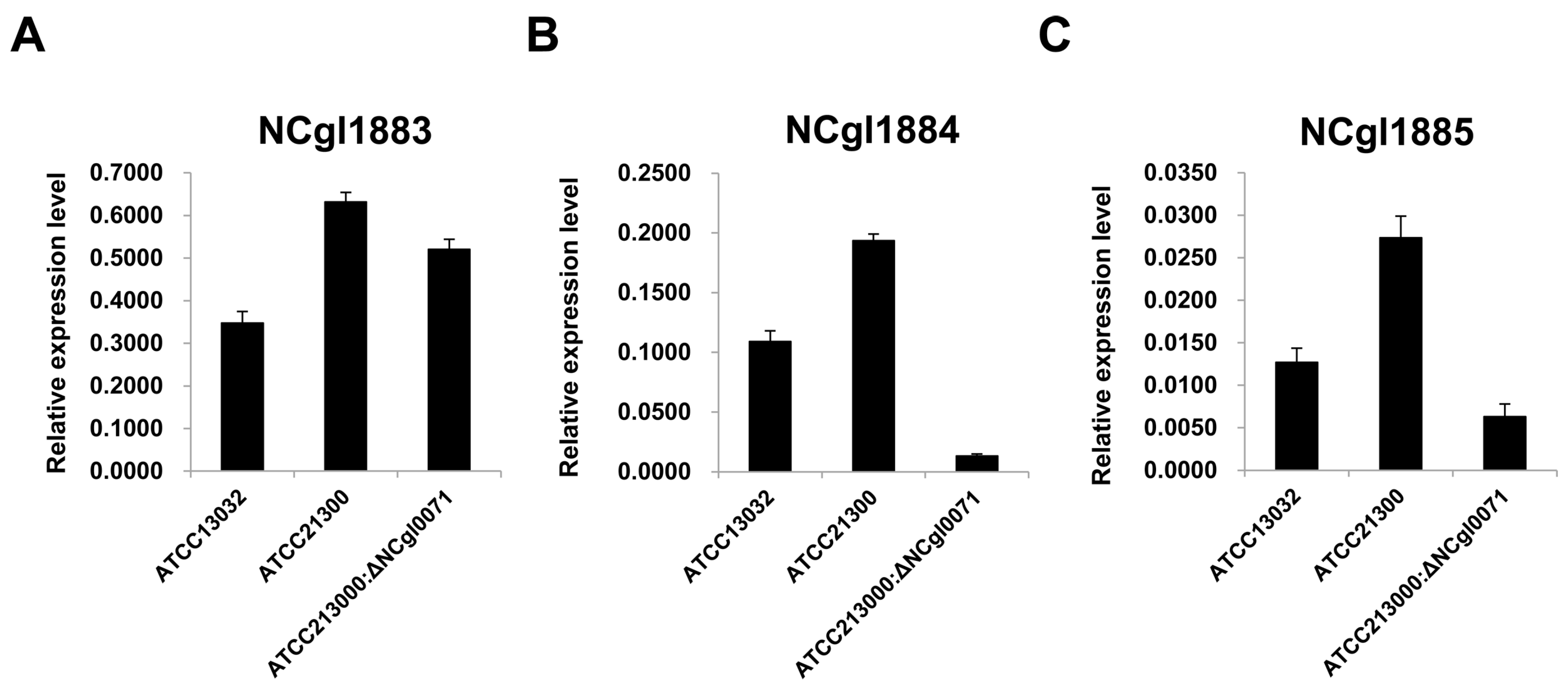
2.3. Effects of bioB Mutation on l-Lysine Production, Growth Rate, and Biotin Transport-Related Gene (NCgl1883, NCgl1884, and NCgl1885) Expression
3. Discussion
4. Materials and Methods
4.1. Bacterial Culture and Total RNA Isolation
4.2. Illumina Sequencing and Raw Data Analysis
4.3. Validation of Differentially Expressed Genes (DEGs) Using Quantitative Real Time PCR
4.4. Site-Specific Gene Disruption
4.5. High Performance Liquid Chromatography Analysis of l-Lysine Production
4.6. Growth Rate Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DEGs | differentially expressed genes |
| GO | gene ontology |
| NGS | next-generation sequencing |
| qRT-PCR | quantitative real-time RT-PCR |
| RPKM | read per kilobase per million mapped reads |
| TCA cycle | tricarboxylic acid cycle |
References
- Kelle, R.; Hermann, T.; Bathe, B. l-Lysine production. In Handbook of Corynebacterium glutamicum; Eggeling, L., Bott, M., Eds.; CRC Press: Florida, FL, USA, 2005; pp. 465–488. [Google Scholar]
- Eggeling, L.; Bott, M. A giant market and a powerful metabolism: l-lysine provided by Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2015, 99, 3387–3394. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, R.T. Industrial strain improvement: Mutagenesis and random screening procedures. Enzym. Microb. Technol. 1984, 6, 3–10. [Google Scholar] [CrossRef]
- Sahm, H.; Eggeling, L.; de Graaf, A.A. Pathway analysis and metabolic engineering in Corynebacterium glutamicum. J. Biol. Chem. 2000, 381, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Loos, A.; Glanemann, C.; Willis, L.B.; O'Brien, X.M.; Lessard, P.A.; Gerstmeir, R.; Guillouet, S.; Sinskey, A.J. Development and validation of Corynebacterium DNA microarrays. Appl. Environ. Microbiol. 2001, 67, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Muffler, A.; Bettermann, S.; Haushalter, M.; Hörlein, A.; Neveling, U.; Schramm, M.; Sorgenfrei, O. Genome-wide transcription profiling of Corynebacterium glutamicum after heat shock and during growth on acetate and glucose. J. Bacteriol. 2002, 98, 255–268. [Google Scholar] [CrossRef]
- Mizukami, T.; Hamu, A.; Ikeda, M.; Oka, T.; Katsumata, R. Cloning of the ATP phosphoribosyl transferase gene of Corynebacterium glutamicum and application of the gene to l-histidine production. Biosci. Biotechnol. Biochem. 1994, 58, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Reinscheid, D.J.; Kronemeyer, W.; Eggeling, L.; Eikmanns, B.J.; Sahm, H. Stable expression of hom-1-thrB in Corynebacterium glutamicum and its effect on the carbon flux to threonine and related amino acids. Appl. Environ. Microbiol. 1994, 60, 126–132. [Google Scholar] [PubMed]
- Krämer, R. Genetic and physiological approaches for the production of amino acids. J. Biotechnol. 1996, 45, 1–21. [Google Scholar] [CrossRef]
- Peters-Wendisch, P.G.; Schiel, B.; Wendisch, V.F.; Katsoulidis, E.; Möckel, B.; Sahm, H.; Eikmanns, B.J. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J. Mol. Microbiol. Biotechnol. 2001, 3, 295–300. [Google Scholar] [PubMed]
- Tauch, A.; Homann, I.; Mormann, S.; Rüberg, S.; Billault, A.; Bathe, B.; Brand, S.; Brockmann-Gretza, O.; Rückert, C.; Schischka, N.; et al. Strategy to sequence the genome of Corynebacterium glutamicum ATCC13032: Use of a cosmid and a bacterial artificial chromosome library. J. Biotechnol. 2002, 95, 25–38. [Google Scholar] [CrossRef]
- Ikeda, M.; Nakagawa, S. The Corynebacterium glutamicum genome: Features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 2003, 62, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, J.; Bathe, B.; Bartels, D.; Bischoff, N.; Bott, B.; Burkovski, A.; Dusch, N.; Eggeling, L.; Eikmanns, B.J.; Gaigalat, L.; et al. The complete Corynebacterium glutamicum ATCC13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J. Biotechnol. 2003, 104, 5–25. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, K.; Hohama, K.; Vertès, A.A.; Kobayashi, M.; Kurusu, Y.; Yukawa, H. Genomic organization of the biotin biosynthetic genes of coryneform bacteria: Cloning and sequencing of the bioA–bioD genes from Brevibacterium flavum. DNA Seq. 1993, 4, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Shiio, I. Microbial production of l-lysine III. Production by mutants resistant S-(2-aminoethyl)-l-cysteine. J. Gen. Appl. Microbiol. 1970, 16, 373–391. [Google Scholar] [CrossRef]
- Vrljic, M.; Sahm, H.; Eggeling, L. A new type of transporter with a new type of cellular function: l-Lysine export from Corynebacterium glutamicum. Mol. Microbiol. 1996, 22, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, J.; Hayashi, M.; Mitsuhashi, S.; Ikeda, M. Efficient 40 degrees C fermentation of l-lysine by a new Corynebacterium glutamicum mutant developed by genome breeding. Appl. Microbiol. Biotechnol. 2003, 62, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Moritz, B.; Striegel, K.; De Graff, A.A.; Sham, H. Kinetic properties of the glucose-6-phosphate and 6-phosphogluconate dehydrogenases from Corynebacterium glutamicum and their application for predicting pentose phosphate pathway flux in vivo. Eur. J. Biochem. 2000, 267, 3442–3452. [Google Scholar] [CrossRef] [PubMed]
- Koffas, M.A.G.; Jung, G.Y.; Aon, J.C.; Stephanopoulos, G. Effect of pyruvate carboxylase overexpression on the physiology of Corynebacterium glutamicum. Appl. Environ. Microbiol. 2002, 68, 5422–5428. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Udaka, S.; Shimono, M. Studies on the amino acid fermentation: Part I. Production of l-glutamic acid by various microorganisms. J. Gen. Appl. Microbiol. 1957, 3, 193–205. [Google Scholar] [CrossRef]
- Udaka, S. Screening method for microorganisms accumulating metabolites and its use in the isolation of Micrococcus glutamicus. J. Bacteriol. 1960, 79, 754–755. [Google Scholar] [PubMed]
- Sandmann, G.; Yukawa, H. Vitamin synthesis: Carotenoids, biotin, and pantothenate. In Handbook of Corynebacterium glutamicum; Eggeling, L., Bott, M., Eds.; CRC Press: Florida, FL, USA, 2005; pp. 397–415. [Google Scholar]
- Hatakeyama, K.; Kobayashi, M.; Yukawa, H. Analysis of biotin biosynthesis pathway in Coryneform bacteria: Brevibacterium flavum. Methods Enzymol. 1997, 279, 339–348. [Google Scholar] [PubMed]
- Entcheva, P.; Phillips, D.A.; Streit, W.R. Functional analysis of Sinorhizobium meliloti genes involved in biotin synthesis and transport. Appl. Environ. Microbiol. 2002, 68, 2834–2848. [Google Scholar] [CrossRef]
- Ko, Y.T.; Chipley, J.R. Role of biotin in the production of lysine by Brevibacterium lactofermentum. Microbios 1984, 40, 161–171. [Google Scholar] [PubMed]
- Brune, I.; Götker, S.; Schneider, J.; Rodionov, D.A.; Tauch, A. Negative transcriptional control of biotin metabolism genes by the TetR-type regulator BioQ in biotin-auxotrophic Corynebacterium glutamicum ATCC 13032. J. Biotechnol. 2012, 159, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Nam, J.Y.; Cho, J.Y.; Lee, C.S.; Park, Y.J. Next-generation sequencing-based transcriptome analysis of l-lysine-producing Corynebacterium glutamicum ATCC 21300 strain. J. Microbiol. 2013, 51, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Jahn, C.E.; Charkowski, A.O.; Willis, D.K. Evaluation of isolation methods and RNA integrity for bacterial RNA quantitation. J. Microbiol. Methods 2008, 75, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Tauch, A.; Jäger, W.; Kalinowski, J.; Thierbach, G.; Pühler, A. Small mobilizable muti-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: Selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 1994, 145, 69–73. [Google Scholar] [CrossRef]
- Yoon, K.H.; Cho, J.Y. Transcriptional analysis of the gum gene cluster from Xanthomonas oryzae pathovar oryzae. Biotechnol. Lett. 2007, 29, 95–103. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus ID | Gene Name | Product | RPKM * | log2 Fold Change | Signature (p-value < 0.001) | |
|---|---|---|---|---|---|---|
| ATCC21300 | ATCC13032 | |||||
| NCgl2515 | bioA | adenosylmethionine-8-amino-7-oxononanoate aminotransferase | 200.53 | 11.49 | 4.08 | TRUE |
| NCgl2516 | bioD | dithiobiotin synthetase | 264.71 | 52.29 | 2.30 | TRUE |
| NCgl0071 | bioB | biotin synthase | 1522.14 | 77.71 | 4.25 | TRUE |
| NCgl1883 | bioY homolog | hypothetical protein | 858.40 | 35.04 | 4.57 | TRUE |
| NCgl1884 | bioM homolog | ABC transporter ATPase | 751.37 | 60.68 | 3.59 | TRUE |
| NCgl1885 | - | ABC transporter permease | 322.68 | 61.30 | 2.36 | TRUE |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-I.; Kim, J.-H.; Park, Y.-J. Transcriptome and Gene Ontology (GO) Enrichment Analysis Reveals Genes Involved in Biotin Metabolism That Affect l-Lysine Production in Corynebacterium glutamicum. Int. J. Mol. Sci. 2016, 17, 353. https://doi.org/10.3390/ijms17030353
Kim H-I, Kim J-H, Park Y-J. Transcriptome and Gene Ontology (GO) Enrichment Analysis Reveals Genes Involved in Biotin Metabolism That Affect l-Lysine Production in Corynebacterium glutamicum. International Journal of Molecular Sciences. 2016; 17(3):353. https://doi.org/10.3390/ijms17030353
Chicago/Turabian StyleKim, Hong-Il, Jong-Hyeon Kim, and Young-Jin Park. 2016. "Transcriptome and Gene Ontology (GO) Enrichment Analysis Reveals Genes Involved in Biotin Metabolism That Affect l-Lysine Production in Corynebacterium glutamicum" International Journal of Molecular Sciences 17, no. 3: 353. https://doi.org/10.3390/ijms17030353