
Microglia-Mediated Neuroinflammation and Neurotrophic Factor-Induced Protection in the MPTP Mouse Model of Parkinson’s Disease-Lessons from Transgenic Mice


Abstract
:
1. Introduction
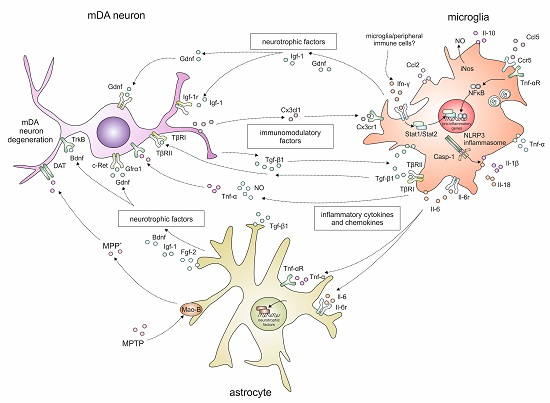
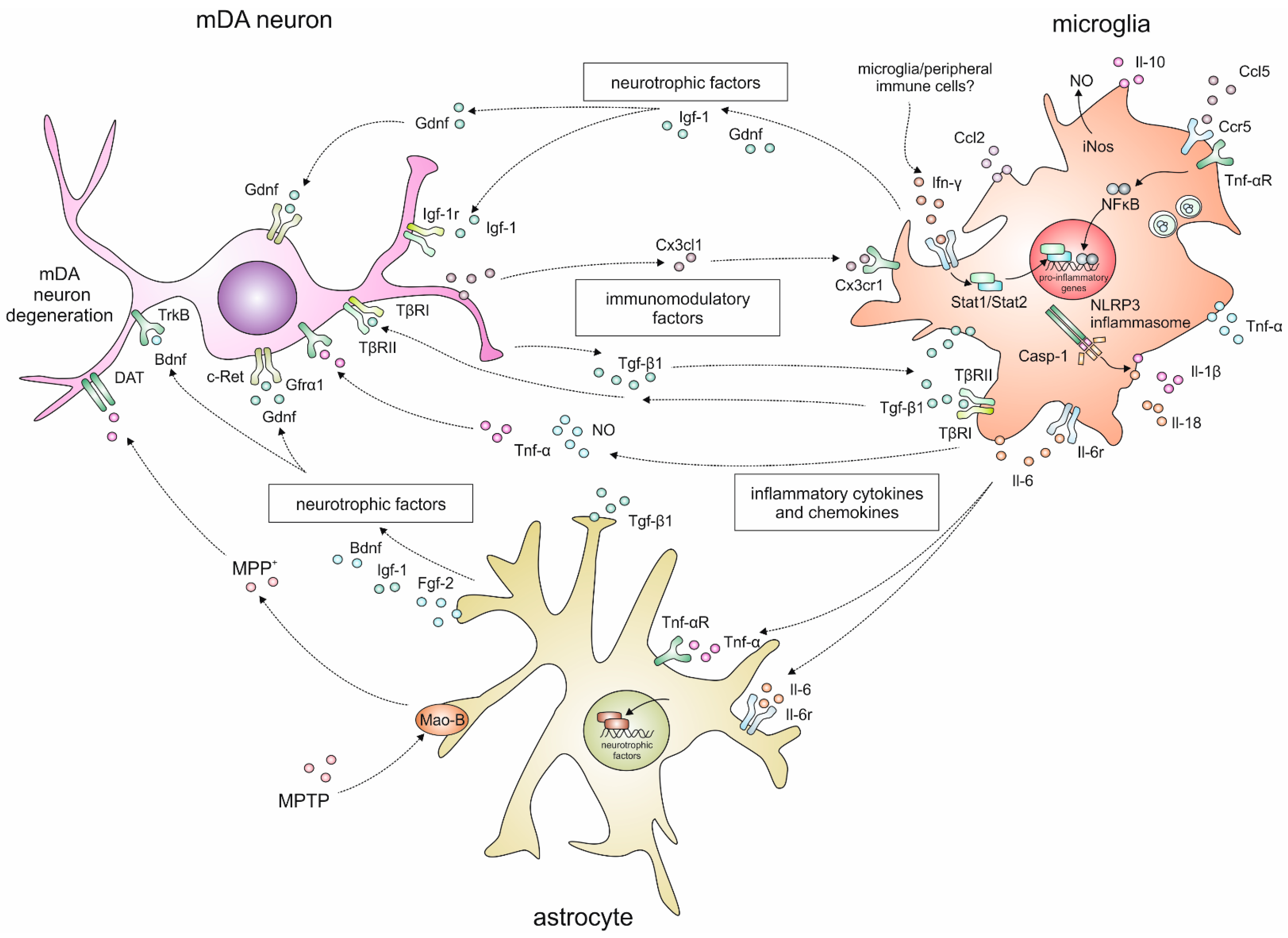
2. Neuroinflammatory Responses in the 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Mouse Model for Parkinson’s Disease
2.1. Temporal and Spatial Glia Reactions after MPTP Intoxication
2.2. Cytokine Signalling
2.3. Inflammasome Activation

{kind=link}
{kind=link}
| Gene | Gene Description | Receptor | Transgenic Modification | MPTP Administration and Dosage | Neuroinflammation | Neurodegeneration | Reference |
|---|---|---|---|---|---|---|---|
| Ccl2 (Mcp-1) | Chemokine (C-C motif) ligand 2 chemokine (C-C motif) receptor 2 | Ccr2 | Mcp-1/Ccl2 homozygous knockout mice (B6.129S4-Ccl2tm1Rol/J) | 4 × 20 mg/kg i.p., 2 h intervals | No differences in RNA expression of Tnf-α, Il-6, Cx3cr1, Tweak/Tnfsf12, Sdf-1/Cxcl12, Fn14/Tnfrsf12a and Cxcr4. | No difference in Th-ir mDA neuron numbers | [46] |
| Ccl2/Ccl3 (Mcp-1/Mip-1α) | Chemokine (C-C motif) ligand 2 Chemokine (C-C motif) receptor 2 | Ccr2 | Double knockout for CCL2 and CCR2 (Ccl2−/−Ccr2−/−) | 3 × 18.2 mg/kg s.c., 2 h intervals | Not described. | Slight reduction in dopamine and DOPAC levels | [60] |
| Ccl5 | Chemokine (C-C motif) receptor 5 | Ccr5 | Ccr5−/− | 4 × 15 mg/kg i.p., 1.5 h intervals | Microglia activation even under baseline conditions, increased Iba1 protein after MPTP. | Reduced Th-ir mDA neuron numbers and fibre density | [61] |
| Cx3cl1 (Fractalkine) | Chemokine (C-X-3-C motif) ligand 1 Chemokine (C-X3-C motif) receptor 1 | Cx3cr1 | Cx3cl1−/−, Cx3cr1−/−, Cx3cr1+/− | 4 × 10 mg/kg i.p., 1 h intervals | Cx3cr1−/− mice exhibit increased microglial activation in the SN after MPTP. | Increased loss of Nissl+ und Th-ir mDA neurons in Cx3cl1−/−and Cx3cr1−/− mice as compared to WT mice. | [59] |
| Cx3cl1 (Fractalkine) | chemokine (C-X-3-C motif) ligand 1 chemokine (C-X3-C motif) receptor 1 | Cx3cr1 | Cx3cl1−/− | 4 × 10 mg/kg i.p., 1 h intervals | Increased CD68+ and CD11b+ area in SNpc, increased Tnf-α and ll-1β concentrations in VM. | Reduced Th protein and Th-ir mDA neuron numbers. | [58] |
| Ifn-γ | Interferon-γ Interferon-γ receptor | Ifn-γr | Ifn-γ−/−, Ifn-γr−/− | 5 × 25 mg/kg i.p., 24 h intervals | No microglia activation. | Reduced loss of Th-ir mDA neurons and Th protein. | [56] |
| Il-18 (Part of the inflammasome) | Interleukin-18 | - | Il-18−/− | 4 × 10 mg/kg i.p., 2 h intervals | Normal microglia activation day 1-3, strongly reduced microglia activation on day 7. | No loss of Th-ir mDA neurons. | [84] |
| Il-1α | Interleukin-1 α | Il-1r | Il-1rI−/− | 1 × 40 mg/kg s.c. | Higher Igf-1 mRNA expression. | Reduced DAT on and higher striatal serotonin transporter density. | [62] |
| Il-1α/β (Part of the inflammasome) | Interleukin-1 α Interleukin-1 β | - | Il-1α/β−/− | 4 × 20 mg/kg i.p., 2 h intervals | Decreased percentage of CD68+ microglia in olfactory bulb and CPu. | Not described. | [83] |
| Il-1β (Part of the inflammasome) | Interleukin-1 β | - | Dominant negative inhibitor of interleukin-1β convertase enzyme (ICE) | 4 × 15 mg/kg, 2 h intervals | Not described. | No reduction in dopamine, DOPAC or HVA after MPTP, no reduction in Th-ir cell count. | [78] |
| Il-6 | Interleukin-6 | - | Il-6−/− | 30 mg/kg | No difference in microglial response. | Increased loss of striatal dopamine levels and loss of Th-ir mDA neurons. | [63] |
| Il-6 | Interleukin-6 | - | Il-6−/− | 30 mg/kg, s.c. | Normal astrogliosis but compromised microgliosis at day 7; time-dependent decrease in iNos expression. | - | [64] |
| Nlrp3 | NLR family, Pyrin Domain Containing 3 | - | Nlrp3−/− | 5 × 20 mg/kg, i.p., 2 h intervals | Reduced Il-1β and Il-18 production; impaired caspase-1 activation. | Resistant to MPTP-induced loss of Th-ir mDA neurons. | [81] |
| Tnf-α | Tumour necrosis factor-α receptor | Tnf-αr1 and Tnf-αr2 | Tnf-αr1−/−, Tnf-αr2−/− Tnf-αr1−/−Tnf-αr2−/− B6;129S-Tnf-ar1tm1 Imx, Tnf-br1tm1 Imx/J | 1 × 12.5 mg/kg, s.c. | Attenuated microglial activation in the CPu of Tnf-αr1−/−Tnf-αr2−/− mice. | Reduced Th-ir mDA loss, but exacerbated neuronal damage in hippocampus. | [69] |
| Tnf-α | Tumour necrosis factor-α | - | Tnf-a−/− | Acute: 4 × 20 mg/kg i.p., 2 h intervals Sub-acute: 4 × 15 mg/kg i.p., 24 h intervals | Not described. | Reduced losses of striatal dopamine and metabolites, and of striatal Th-ir fibre density; no difference in Th-ir and DA transporter immunoreactivity in the SN; lower mortality (10%). | [66] |
| Tnf-α | Tumour necrosis factor-α | - | Tnf-a−/− (B6;129S6-TnftmlGkl/J) | 4 × 10 mg/kg i.p., 1 h intervals | Decreased number of activated microglia, reduced pro-inflammatory cytokines (Tnf-α and Il-1β). | Better BBB integrity, no change in Th-ir mDA neuron numbers. | [67] |
| Tnf-α | Tumour necrosis factor-α receptor | Tnf-αr1 and Tnf-αr2 | Tnf-αr1−/−, Tnf-αr2−/−, Tnf-αr1−/−Tnf-αr2−/− | 1 × 12.5 mg/kg b.w., s.c. | No significant upregulation of Gfap in CPu of Tnf-αr1−/−Tnf-αr2−/− mice. | No striatal Th and dopamine loss in Tnf-αr1−/−Tnf-αr2−/−, still neuronal damage in hippocampus. | [68] |
| Tnf-α | Tumour necrosis factor α | Tnf-αr1 and Tnf-αr2 | Tnf-αr1−/−, Tnf-αr2−/−, Tnf-αr1−/−Tnf-αr2−/− | 4 × 15 mg/kg b.w., i.p., 2 h intervals. | Not described. | No difference in Th-ir mDA neuron numbers; slight reduction of dopamine in Tnf-αr1−/−Tnf-αr2−/−. | [70] |
| Tnf-α | Tumour necrosis factor α | Tnf-αr1 and Tnf-αr2 | Tnf-αr1−/−/Tnf-αr2−/− | 8 × 15 mg/kg b.w., i.p., 24 h intervals. | Not described. | No difference in dopamine levels and DAT-ir neurons. | [71] |
| Tnf-α Ifn-γ | Tumour necrosis factor-α; Interferon-γ | Ifn-γ−/− (B6.129S7-Ifngtm1Ts/J), Tnf-α−/− (B6.129S-Tnf. tm1Gkl/J) | 1 × 20 mg/kg b.w., i.p. | Ifn-γ−/−:no microgliosis but morphological change in microglia. Tnf-α−/−: only slight activation of microglia. Tnf-α−/−, Ifn-γ−/−: no astrocyte activation. | No change in Th-ir mDA neuron numbers after 24 h. | [57] |
3. Growth Factors and Neurotrophic Factors
4. Concluding Remarks
| Gene | Gene Description | Transgenic Mouse/Exogenous Treatment | Transgenic Modification | MPTP Administration and Dosage | Neuroinflammation | Neurodegeneration | Reference | |
|---|---|---|---|---|---|---|---|---|
| Bdnf | Brain-derived neurotrophic factor | Bdnf+/− mice | Haploinsufficient | Yes, 4 × 20 mg/kg b.w., i.p. at 2 h intervals | Not described. | Bdnf+/− mice do not show differences compared to WT mice. | [91] | |
| Cdnf | Cerebral dopamine neurotrophic factor | Pre and post treatment with Cdnf 5 μg/μL (bilateral striatal injections) before and after MPTP | None | Yes, 4 × 15 mg/kg b.w, i.p. for pre-treatment and 20mg/kg b.w, i.p. for post-treatment respectively | Not described. | Exogenous Cdnf proves neuroprotective and neurorestorative for the NS system. | [135] | |
| Egf | Epidermal growth factor | Infusion of Egf 5 μg/week | None | Yes, 7 × 30 mg/kg b.w., i.p. | Not described. | Partial restoration of dopamine and DOPAC. | [112] | |
| aFgf or Fgf-1 | acidic Fgf or Fibroblast growth factor-1 | Stereotactic injections of aFgf 0.5 μg/μL | None | Yes, 4 × 20 mg/kg b.w., i.p. | Not described. | Dopamine concentration and striatal fibre density partially recovered in young mice (8 w) but not in old mice (12 m). | [113] | |
| bFgf or Fgf-2 | Fibroblast growth factor-2 | Fgf-2−/− | Complete knockout | Yes, 3 × 20 mg/kg b.w., i.p. | Not described. | No significant differences in the NS system between WT and Fgf-2−/− mice. | [119] | |
| Fgf-2 | Fibroblast growth factor-2 | Gelfoam containing 4 μg Fgf-2 or cytochrome-c | None | Yes, 3 × 20 mg/kg b.w., i.p. | Co-staining of Fgf-2 with presumed microglial cells. | Not described. | [118] | |
| Fgf-2 | Fibroblast growth factor-2 | Gel foam containing 4 μg Fgf-2 or cytochrome-c | None | Yes, 3 × 20 mg/kg b.w., i.p. | Not described. | Moderates reduction of striatal dopamine and reverses losses of Th-ir mDA neurons. | [114] | |
| Fgf-2 | Fibroblast growth factor-2 | Gel foam containing 4 μg Fgf-2 or cytochrome-c | None | Yes, 3 × 20 mg/kg b.w., i.p. | No excessive reactive astrocytosis after Fgf-2 application. | Striatal dopamine content was checked to asses MPTP effect. | [117] | |
| Fgf-2 | Fibroblast growth factor-2 | Intraventricular infusion of hrFGF-22 μg/24 h | None | Yes, 40 mg/kg b.w., s.c | hrFGF-2 induces increase in astroglial reaction and numbers in SN and CPu. | hrFGF-2 treatment reduced MPTP-induced losses of mDA neurons and fibres. Locomotor activity was fully recovered after hrFGF-2 treatment. | [115] | |
| G-Csf | Granulocyte-colony stimulating factor | 8x250 μg/kg, b.w., s.c. | None | Yes, 5 × 30 mg/kg b.w., i.p. | G-csf treatment after MPTP reduced microglial burden in the CPu. | G-csf treatment after MPTP improved rotarod performance. | [129] | |
| Gdnf | Glial derived neurotrophic factor | Transplants of foetal neural tissues (Gdnf−/− or Gdnf-/+ or Gdnf+/−) into MPTP treated adult WT mice | Gdnf−/− or Gdnf+/− foetal neural tissues transplanted into ventral CPu of MPTP lesioned WT mice | Yes, 4 × 30 mg/kg/day | Not described. | Gdnf−/− grafts pre-incubated with Gdnf show increased Th-ir mDA neuron numbers. | [98] | |
| Gdnf | Glial derived neurotrophic factor | Unilateral stereotactic injections of Gdnf 5 g/μL | None | Yes, 4 × 20 mg/kg b.w., i.p. | Not described. | Gdnf administration induces recovery of the NS system in both young and aged mice. | [99] | |
| Gdnf | Glial derived neurotrophic factor | Intracerebral injections of Gdnf (5 μg/μL) before and after MPTP | None | Yes, 2 × 40 mg/kg b.w., s.c. | Not described. | Protection of mDA neurons; recovery of Th fibres and dopamine in the CPu. Motor behaviour increased above normal levels. | [100] | |
| Gdnf | Glial derived neurotrophic factor | Intrastriatal injections of Gdnf 2 μg/μL | None | Yes, 7 × 35 mg/kg, b.w., s.c. | Not described. | Increased locomotor activity, striatal dopamine and metabolite levels, Increase in Th-ir mDA neurons. | [101] | |
| Gdnf | Glial derived neurotrophic factor | Gdnf lentiviral construct in a macrophage-specific synthetic promoter | None | Yes,15 mg/kg b.w., free base MPTP on day 1, 25 mg/kg b.w., on day 2, and 30 mg/kg b.w., on days 3–7, s.c. | Putative neuroprotective effects of Gdnf expressing macrophage/microglia on Th-ir mDA neurons. | Macrophage-mediated Gdnf treatment ameliorated MPTP-induced degeneration of mDA neurons and Th fibre terminals, stimulated axon regeneration, and reversed hypoactivity in the open field test. | [103] | |
| Gdnf | Glial derived neurotrophic factor | Gel foam containing 1 μg/μL Gdnf or cytochrome-c | None | Yes, 3 × 20 mg/kg b.w., i.p. | Not described. | Exogenous Gdnf reduced loss of striatal DA fibres. | [102] | |
| Gfrα1 | Gdnf receptor (Glial cell line derived neurotrophic factor family receptor α 1) | Gfrα1+/− | Haploinsufficient; substitution with phosphoglycerate kinase | Yes, 4 × 20 mg/kg b.w., i.p. | Higher CD45-ir, microglial response in the SN. | Increase in Th-ir mDA neuron death. | [106] | |
| Gm-Csf | Granulocyte-macrophage-colony stimulating factor | 5x50 μg/kg i.p. | None | Yes, 4 × 16 mg/kg b.w., s.c. | Gm-csf pre-treatment altered microglial morphology (reduced microgliosis) and Treg induction. | Neuroprotection of Th-ir mDA neurons and striatal fibres by adoptive transfer of Gm-Csf-induced Treg to MPTP mice. | [132] | |
| Igf-1r | Insulin-like growth factor-1 receptor | Igf-1r+/− | Haploinsufficient | Yes, 2 × 30 mg/kg b.w., i.p. at 3 h intervals | Increase in neuroinflammatory responses (particularly microglia numbers in SNpc and SNr). | Increase in Th-ir mDA neuron death. | [121] | |
| Ngf | Nerve growth factor | Stereotactic injections of 0.4 μg/μL Ngf into the right ventricle | None | Yes, 5 × 30 mg/kg b.w., i.p. | Not described. | Restoration of dopamine and HVA levels after Ngf injections. | [93] | |
| Ntn | Neurturin | Ntn lentiviral construct in a microglia specific synthetic promoter | None | Yes, 15 mg/kg b.w., free base MPTP on day 1, 25 mg/kg b.w., on day 2, and 30 mg/kg b.w., on days 3–7, s.c. | Neuroprotective effects of Ntn expressing microglia on mDA neurons. | Reduction in MPTP-induced degeneration of mDA neurons in the SN and fibre terminals in the CPu. | [104] | |
| Pdgf | Platelet derived growth factor | Pdgf-bb delivery (36 ng/day) for 2 w into the right lateral ventricle via osmotic pumps | None | For cell proliferation: 4 × 15 mg/kg b.w., i.p. For neurorestoration 1 × 40 mg/kg b.w., s.c. | Not described. | Pgdf-bb administration lead to an increase in striatal Th expression and DAT sites. | [122] | |
| Ret | Gdnf receptor (rearranged during transfection) | Dat-Retlx/lx | Conditional knockout | Yes, 5 × 30 mg/kg b.w., i.p. | No difference in MPTP-induced astrogliosis in CPu. | Ret deficiency does not increase MPTP vulnerability in the SN, but is essential for regeneration in the CPu. | [105] | |
| Tgf-β2 | Transforming growth factor-β 2 | Tgf- β2+/− | Haploinsufficient | Yes, 40 + 20 mg/kg b.w., s.c. | Not described. | Marginally reduced Th-ir mDA neurons. Reduced striatal dopamine turnover. | [110] | |
| Tgf-β | Transforming growth factor-β | Gel foam containing anti Tgf-β pan mAB or isotype control mouse IgG (5 μg) | None | Yes, 3 × 20 mg/kg b.w., i.p. | Not described. | Simultaneous application of Gdnf and Tgf-β neutralising antibodies abolished the neuroprotective effects of Gdnf. | [102] | |
| Tgf-β1 | Transforming growth factor-β 1 | AAV- mediated Tgf-β1 overexpression | None | Yes, 30 mg/kg b.w., s.c. | Not described. | Overexpression of Tgf-β1 in the NS aggravates the Parkinsonian state caused by MPTP injury in adult mice. | [111] | |
| TrkB | Bdnf receptor (Tyrosine receptor kinase B) | TrkB hypomorphic mutant, fBneo/fBneo (expresses TrkB at 1/4-1/3 of the normal amount) | Conditional knockout | Yes, 5 × 25 mg/kg b.w., i.p. | Increased reactive astrogliosis in the CPu of mutant mice. | Mutant mice exhibit selective and late neurodegeneration. mDA neurons show enhanced vulnerability to MPTP. | [92] | |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abdullah, R.; Basak, I.; Patil, K.S.; Alves, G.; Larsen, J.P.; Møller, S.G. Parkinson’s disease and age: The obvious but largely unexplored link. Exp. Gerontol. 2015, 68, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Neuropathology of sporadic Parkinson’s disease: Evaluation and changes of concepts. Mov. Disord. 2012, 27, 8–30. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Postuma, R.B.; Bloem, B.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.M.; Hardy, J.; Lang, A.E.; et al. Time to redefine PD? Introductory statement of the MDS task force on the definition of Parkinson’s disease. Mov. Disord. 2014, 29, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.G.; Postuma, R. Premotor and nonmotor features of Parkinson’s disease. Curr. Opin. Neurol. 2014, 27, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Buchman, A.S.; Shulman, J.M.; Nag, S.; Leurgans, S.E.; Arnold, S.E.; Morris, M.C.; Schneider, J.A.; Bennett, D.A. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 2012, 71, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.-S. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 2007, 35, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Naini, A.B.; Jakowec, M.; Petzinger, G.; Miller, R.; Akram, M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A technical review of its utility and safety. J. Neurochem. 2001, 76, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, W.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Nicklas, W.J.; Youngster, S.K.; Kindt, M.V.; Heikkila, R.E. MPTP, MPP+ and mitochondrial function. Life Sci. 1987, 40, 721–729. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M. MPTP: A review of its mechanisms of neurotoxicity. Clin. Neurosci. Res. 2001, 1, 407–418. [Google Scholar] [CrossRef]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [PubMed]
- Javitch, J.A.; Snyder, S.H. Uptake of MPP+ by dopamine neurons explains selectivity of parkinsonism-inducing neurotoxin, MPTP. Eur. J. Pharmacol. 1984, 106, 455–456. [Google Scholar] [CrossRef]
- Mayer, R.A.; Kindt, M.V.; Heikkila, R.E. Prevention of the nigrostriatal toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by inhibitors of 3,4-dihydroxyphenylethylamine transport. J. Neurochem. 1986, 47, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Del Zompo, M.; Piccardi, M.P.; Ruiu, S.; Quartu, M.; Gessa, G.L.; Vaccari, A. Selective MPP+ uptake into synaptic dopamine vesicles: Possible involvement in MPTP neurotoxicity. Br. J. Pharmacol. 1993, 109, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Klaidman, L.K.; Adams, J.D., Jr.; Leung, A.C.; Sam Kim, S.; Cadenas, E. Redox cycling of MPP+: Evidence for a new mechanism involving hydride transfer with xanthine oxidase, aldehyde dehydrogenase, and lipoamide dehydrogenase. Free Radic. Biol. Med. 1993, 15, 169–179. [Google Scholar] [CrossRef]
- Mizuno, Y.; Sone, N.; Saitoh, T. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J. Neurochem. 1987, 48, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, Z.L.; Sotgiu, A.; Sharp, D.E.; Hadjiconstantinou, M.; Neff, N.H. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and free radicals in vitro. Biochem. Pharmacol. 1988, 37, 4573–4574. [Google Scholar] [CrossRef]
- Hasegawa, E.; Takeshige, K.; Oishi, T.; Murai, Y.; Minakami, S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem. Biophys. Res. Commun. 1990, 170, 1049–1055. [Google Scholar] [CrossRef]
- Boyce, S.; Kelly, E.; Reavill, C.; Jenner, P.; Marsden, C.D. Repeated administration of N-methyl-4-phenyl 1,2,5,6-tetrahydropyridine to rats is not toxic to striatal dopamine neurones. Biochem. Pharmacol. 1984, 33, 1747–1752. [Google Scholar] [CrossRef]
- Chiueh, C.C.; Markey, S.P.; Burns, R.S.; Johannessen, J.N.; Pert, A.; Kopin, I.J. Neurochemical and behavioral effects of systemic and intranigral administration of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the rat. Eur. J. Pharmacol. 1984, 100, 189–194. [Google Scholar] [CrossRef]
- Sedelis, M.; Hofele, K.; Auburger, G.W.; Morgan, S.; Huston, J.P.; Schwarting, R.K. MPTP susceptibility in the mouse: Behavioral, neurochemical, and histological analysis of gender and strain differences. Behav. Genet. 2000, 30, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Sedelis, M.; Hofele, K.; Auburger, G.W.; Morgan, S.; Huston, J.P.; Schwarting, R.K. Evidence for resistance to MPTP in C57BL/6 x BALB/c F1 hybrids as compared with their progenitor strains. Neuroreport 2000, 11, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Ferger, B. Neurochemical findings in the MPTP model of Parkinson’s disease. J. Neural Transm. 2001, 108, 1263–1282. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Jakowec, M.; Burke, R.E.; Przedborski, S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegener. J. Neurodegener. Disord. Neuroprot. Neuroregener. 1995, 4, 257–269. [Google Scholar] [CrossRef]
- Tatton, N.A.; Kish, S.J. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience 1997, 77, 1037–1048. [Google Scholar] [CrossRef]
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. α-Synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Imbert, C.; Deloire, X.; Bioulac, B.; Gross, C.E. A chronic MPTP model reproducing the slow evolution of Parkinson’s disease: Evolution of motor symptoms in the monkey. Brain Res. 1997, 766, 107–112. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Glial reactions in Parkinson’s disease. Mov. Disord. 2008, 23, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kohutnicka, M.; Lewandowska, E.; Kurkowska-Jastrzebska, I.; Członkowski, A.; Członkowska, A. Microglial and astrocytic involvement in a murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Immunopharmacology 1998, 39, 167–180. [Google Scholar] [CrossRef]
- Członkowska, A.; Kohutnicka, M.; Kurkowska-Jastrzebska, I.; Członkowski, A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 1996, 5, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.-K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [PubMed]
- Ciesielska, A.; Joniec, I.; Przybyłkowski, A.; Gromadzka, G.; Kurkowska-Jastrzebska, I.; Członkowska, A.; Członkowski, A. Dynamics of expression of the mRNA for cytokines and inducible nitric synthase in a murine model of the Parkinson’s disease. Acta Neurobiol. Exp. 2003, 63, 117–126. [Google Scholar]
- Luchtman, D.W.; Shao, D.; Song, C. Behavior, neurotransmitters and inflammation in three regimens of the MPTP mouse model of Parkinson’s disease. Physiol. Behav. 2009, 98, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Lofrumento, D.D.; Saponaro, C.; Cianciulli, A.; de Nuccio, F.; Mitolo, V.; Nicolardi, G.; Panaro, M.A. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation 2011, 18, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Shimoda, T.; Uno, K.; Tateishi, N.; Furuya, S.; Yagi, K.; Suzuki, K.; Fujita, S. The effects of MPTP on the activation of microglia/astrocytes and cytokine/chemokine levels in different mice strains. J. Neuroimmunol. 2008, 204, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Pattarini, R.; Smeyne, R.J.; Morgan, J.I. Temporal mRNA profiles of inflammatory mediators in the murine 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrimidine model of Parkinson’s disease. Neuroscience 2007, 145, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Shinagawa, R.; Yamada, M.; Mori, T.; Tateishi, N.; Fujita, S. Long-lasting reactive changes observed in microglia in the striatal and substantia nigral of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 2007, 1138, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Ros, C.M.; Annese, V.; Carrillo-de Sauvage, M.A.; Ros-Bernal, F.; Gómez, A.; Yuste, J.E.; Campuzano, C.M.; de Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci. Rep. 2012, 2, 809. [Google Scholar] [CrossRef] [PubMed]
- Kurkowska-Jastrzebska, I.; Wrońska, A.; Kohutnicka, M.; Członkowski, A.; Członkowska, A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication in mouse. Exp. Neurol. 1999, 156, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Banerjee, R.; Liu, J.; Gendelman, H.E.; Mosley, R.L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J. Leukoc. Biol. 2007, 82, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J. Cytokines: Past, present, and future. Int. J. Hematol. 2001, 74, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Historical review of cytokines. Eur. J. Immunol. 2007, 37, S34–S45. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef] [PubMed]
- Owens, T.; Khorooshi, R.; Wlodarczyk, A.; Asgari, N. Interferons in the central nervous system: A few instruments play many tunes. Glia 2014, 62, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of interferon-γ in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 2007, 27, 3328–3337. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Ros, C.M.; Annese, V.; Gómez, A.; Ros-Bernal, F.; Aguado-Llera, D.; Martínez-Pagán, M.E.; de Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2012, 3, e142. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Nash, K.R.; Grimmig, B.A.; Ranjit, S.; Small, B.; Bickford, P.C.; Gemma, C. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 2012, 32, 14592–14601. [Google Scholar] [CrossRef] [PubMed]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Kalkonde, Y.V.; Morgan, W.W.; Sigala, J.; Maffi, S.K.; Condello, C.; Kuziel, W.; Ahuja, S.S.; Ahuja, S.K. Chemokines in the MPTP model of Parkinson’s disease: Absence of CCL2 and its receptor CCR2 does not protect against striatal neurodegeneration. Brain Res. 2007, 1128, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-Y.; Lee, M.K.; Hong, J.T. Lack of CCR5 modifies glial phenotypes and population of the nigral dopaminergic neurons, but not MPTP-induced dopaminergic neurodegeneration. Neurobiol. Dis. 2013, 49, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Hébert, G.; Mingam, R.; Arsaut, J.; Dantzer, R.; Demotes-Mainard, J. A role of IL-1 in MPTP-induced changes in striatal dopaminergic and serotoninergic transporter binding: Clues from interleukin-1 type I receptor-deficient mice. Mol. Brain Res. 2005, 136, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Bolin, L.M.; Strycharska-Orczyk, I.; Murray, R.; Langston, J.W.; di Monte, D. Increased vulnerability of dopaminergic neurons in MPTP-lesioned interleukin-6 deficient mice. J. Neurochem. 2002, 83, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Bolin, L.M. Compromised reactive microgliosis in MPTP-lesioned IL-6 KO mice. Brain Res. 2003, 985, 89–97. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-α (TNF-α) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Ling, Z.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-α knockout and minocycline treatment attenuates blood brain barrier leakage in MPTP-treated mice. Neurobiol. Dis. 2007, 26, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-α. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Rousselet, E.; Callebert, J.; Parain, K.; Joubert, C.; Hunot, S.; Hartmann, A.; Jacque, C.; Perez-Diaz, F.; Cohen-Salmon, C.; Launay, J.-M.; et al. Role of TNF-α Receptors in Mice Intoxicated with the Parkinsonian Toxin MPTP. Exp. Neurol. 2002, 177, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Leng, A.; Mura, A.; Feldon, J.; Ferger, B. Tumor necrosis factor-α receptor ablation in a chronic MPTP mouse model of Parkinson’s disease. Neurosci. Lett. 2005, 375, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Cervia, D.; Sugama, S.; Conti, B. Interleukin 18 in the CNS. J. Neuroinflamm. 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, S.M.; Tyrrell, P.J.; Rothwell, N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 2005, 5, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Klevenyi, P.; Andreassen, O.; Ferrante, R.J.; Schleicher, J.R.; Friedlander, R.M.; Beal, M.F. Transgenic mice expressing a dominant negative mutant interleukin-1β converting enzyme show resistance to MPTP neurotoxicity. Neuroreport 1999, 10, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhai, Y.-Q.; Xu, L.-L.; Qiao, C.; Sun, X.-L.; Ding, J.-H.; Lu, M.; Hu, G. Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice. Exp. Neurol. 2014, 251, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Vroon, A.; Drukarch, B.; Bol, J.G.J.M.; Cras, P.; Brevé, J.J.P.; Allan, S.M.; Relton, J.K.; Hoogland, P.V.J.M.; van Dam, A.-M. Neuroinflammation in Parkinson’s patients and MPTP-treated mice is not restricted to the nigrostriatal system: Microgliosis and differential expression of interleukin-1 receptors in the olfactory bulb. Exp. Gerontol. 2007, 42, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Sugama, S.; Wirz, S.A.; Barr, A.M.; Conti, B.; Bartfai, T.; Shibasaki, T. Interleukin-18 null mice show diminished microglial activation and reduced dopaminergic neuron loss following acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment. Neuroscience 2004, 128, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Aron, L.; Klein, R. Repairing the parkinsonian brain with neurotrophic factors. Trends Neurosci. 2011, 34, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, S.V.; O’Keeffe, G.W.; Sullivan, A.M. Neurotrophic factors: From neurodevelopmental regulators to novel therapies for Parkinson’s disease. Neural Regen. Res. 2014, 9, 1708–1711. [Google Scholar] [PubMed]
- Weissmiller, A.M.; Wu, C. Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl. Neurodegener. 2012, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. The neurotrophin family of neurotrophic factors: An overview. Methods Mol. Biol. Clifton NJ 2012, 846, 1–12. [Google Scholar]
- Chen, L.-W.; Hu, H.-J.; Liu, H.-L.; Yung, K.K.L.; Chan, Y.S. Identification of brain-derived neurotrophic factor in nestin-expressing astroglial cells in the neostriatum of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. Neuroscience 2004, 126, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, K.M.; Jiao, Y.; Pagala, V.; Smeyne, R.J. Exercise does not protect against MPTP-induced neurotoxicity in BDNF haploinsufficient mice. PLoS ONE 2012, 7, e43250. [Google Scholar] [CrossRef] [PubMed]
- Baydyuk, M.; Nguyen, M.T.; Xu, B. Chronic deprivation of TrkB signaling leads to selective late-onset nigrostriatal dopaminergic degeneration. Exp. Neurol. 2011, 228, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Rios, C.; Sotelo, J. Ventricular injection of nerve growth factor increases dopamine content in the striata of MPTP-treated mice. Neurochem. Res. 1992, 17, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-W.; Zhang, J.-P.; Kwok-Yan Shum, D.; Chan, Y.-S. Localization of nerve growth factor, neurotrophin-3, and glial cell line-derived neurotrophic factor in nestin-expressing reactive astrocytes in the caudate-putamen of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/Bl mice. J. Comp. Neurol. 2006, 497, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Airavaara, M.; Voutilainen, M.H.; Wang, Y.; Hoffer, B. Neurorestoration. Parkinsonism Relat. Disord. 2012, 18, S143–S146. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Garcia de Yebenes, J.; Yebenes, J.; Mena, M.A. Neurotrophic factors in neurodegenerative disorders: Model of Parkinson’s disease. Neurotox. Res. 2000, 2, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Granholm, A.C.; Reyland, M.; Albeck, D.; Sanders, L.; Gerhardt, G.; Hoernig, G.; Shen, L.; Westphal, H.; Hoffer, B. Glial cell line-derived neurotrophic factor is essential for postnatal survival of midbrain dopamine neurons. J. Neurosci. 2000, 20, 3182–3190. [Google Scholar] [PubMed]
- Date, I.; Aoi, M.; Tomita, S.; Collins, F.; Ohmoto, T. GDNF administration induces recovery of the nigrostriatal dopaminergic system both in young and aged parkinsonian mice. Neuroreport 1998, 9, 2365–2369. [Google Scholar] [CrossRef] [PubMed]
- Tomac, A.; Lindqvist, E.; Lin, L.F.; Ogren, S.O.; Young, D.; Hoffer, B.J.; Olson, L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 1995, 373, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.C.; Ni, D.R.; Wu, M.C.; Kuo, J.S.; Chia, L.G. Glial cell line-derived neurotrophic factor protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in C57BL/6 mice. Neurosci. Lett. 1998, 252, 87–90. [Google Scholar] [CrossRef]
- Schober, A.; Peterziel, H.; von Bartheld, C.S.; Simon, H.; Krieglstein, K.; Unsicker, K. GDNF applied to the MPTP-lesioned nigrostriatal system requires TGF-β for its neuroprotective action. Neurobiol. Dis. 2007, 25, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Biju, K.C.; Zhou, Q.; Li, G.; Imam, S.Z.; Roberts, J.L.; Morgan, W.W.; Clark, R.A.; Li, S. Macrophage-mediated GDNF delivery protects against dopaminergic neurodegeneration: A therapeutic strategy for Parkinson’s disease. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Biju, K.C.; Santacruz, R.A.; Chen, C.; Zhou, Q.; Yao, J.; Rohrabaugh, S.L.; Clark, R.A.; Roberts, J.L.; Phillips, K.A.; Imam, S.Z.; et al. Bone marrow-derived microglia-based neurturin delivery protects against dopaminergic neurodegeneration in a mouse model of Parkinson’s disease. Neurosci. Lett. 2013, 535, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Kowsky, S.; Pöppelmeyer, C.; Kramer, E.R.; Falkenburger, B.H.; Kruse, A.; Klein, R.; Schulz, J.B. RET signaling does not modulate MPTP toxicity but is required for regeneration of dopaminergic axon terminals. Proc. Natl. Acad. Sci. USA 2007, 104, 20049–20054. [Google Scholar] [CrossRef] [PubMed]
- Boger, H.A.; Middaugh, L.D.; Zaman, V.; Hoffer, B.; Granholm, A.-C. Differential effects of the dopamine neurotoxin MPTP in animals with a partial deletion of the GDNF receptor, GFR α1, gene. Brain Res. 2008, 1241, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Krieglstein, K.; Suter-Crazzolara, C.; Unsicker, K. Development of mesencephalic dopaminergic neurons and the transforming growth factor-β superfamily. J. Neural Transm. Suppl. 1995, 46, 209–216. [Google Scholar] [PubMed]
- Krieglstein, K.; Henheik, P.; Farkas, L.; Jaszai, J.; Galter, D.; Krohn, K.; Unsicker, K. Glial cell line-derived neurotrophic factor requires transforming growth factor-β for exerting its full neurotrophic potential on peripheral and CNS neurons. J. Neurosci. 1998, 18, 9822–9834. [Google Scholar] [PubMed]
- Schober, A.; Hertel, R.; Arumäe, U.; Farkas, L.; Jaszai, J.; Krieglstein, K.; Saarma, M.; Unsicker, K. Glial cell line-derived neurotrophic factor rescues target-deprived sympathetic spinal cord neurons but requires transforming growth factor-β as cofactor in vivo. J. Neurosci. 1999, 19, 2008–2015. [Google Scholar] [PubMed]
- Andrews, Z.B.; Zhao, H.; Frugier, T.; Meguro, R.; Grattan, D.R.; Koishi, K.; McLennan, I.S. Transforming growth factor β2 haploinsufficient mice develop age-related nigrostriatal dopamine deficits. Neurobiol. Dis. 2006, 21, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Capelo, A.; Colin, P.; Guibert, B.; Biguet, N.F.; Mallet, J. Transforming growth factor β1 overexpression in the nigrostriatal system increases the dopaminergic deficit of MPTP mice. Mol. Cell. Neurosci. 2003, 23, 614–625. [Google Scholar] [CrossRef]
- Hadjiconstantinou, M.; Fitkin, J.G.; Dalia, A.; Neff, N.H. Epidermal growth factor enhances striatal dopaminergic parameters in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mouse. J. Neurochem. 1991, 57, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Date, I.; Notter, M.F.; Felten, S.Y.; Felten, D.L. MPTP-treated young mice but not aging mice show partial recovery of the nigrostriatal dopaminergic system by stereotaxic injection of acidic fibroblast growth factor (aFGF). Brain Res. 1990, 526, 156–160. [Google Scholar] [CrossRef]
- Otto, D.; Unsicker, K. Basic FGF reverses chemical and morphological deficits in the nigrostriatal system of MPTP-treated mice. J. Neurosci. 1990, 10, 1912–1921. [Google Scholar] [PubMed]
- Chadi, G.; Møller, A.; Rosén, L.; Janson, A.M.; Agnati, L.A.; Goldstein, M.; Ogren, S.O.; Pettersson, R.F.; Fuxe, K. Protective actions of human recombinant basic fibroblast growth factor on MPTP-lesioned nigrostriatal dopamine neurons after intraventricular infusion. Exp. Brain Res. 1993, 97, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Otto, D.; Unsicker, K. FGF-2 modulates dopamine and dopamine-related striatal transmitter systems in the intact and MPTP-lesioned mouse. Eur. J. Neurosci. 1993, 5, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Unsicker, K. FGF-2 in the MPTP model of Parkinson’s disease: Effects on astroglial cells. Glia 1994, 11, 47–56. [Google Scholar]
- Wirth, S.B.; Rufer, M.; Unsicker, K. Early effects of FGF-2 on glial cells in the MPTP-lesioned striatum. Exp. Neurol. 1996, 137, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zechel, S.; Jarosik, J.; Kiprianova, I.; Schober, A.; Unsicker, K.; von Bohlen und Halbach, O. FGF-2 deficiency does not alter vulnerability of the dopaminergic nigrostriatal system towards MPTP intoxication in mice. Eur. J. Neurosci. 2006, 23, 1671–1675. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.C.; Gluckman, P.D.; Feldman, E.L.; Werther, G.A. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 2005, 26, 916–943. [Google Scholar] [CrossRef] [PubMed]
- Nadjar, A.; Berton, O.; Guo, S.; Leneuve, P.; Dovero, S.; Diguet, E.; Tison, F.; Zhao, B.; Holzenberger, M.; Bezard, E. IGF-1 signaling reduces neuro-inflammatory response and sensitivity of neurons to MPTP. Neurobiol. Aging 2009, 30, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Zachrisson, O.; Zhao, M.; Andersson, A.; Dannaeus, K.; Häggblad, J.; Isacson, R.; Nielsen, E.; Patrone, C.; Rönnholm, H.; Wikstrom, L.; et al. Restorative effects of platelet derived growth factor-BB in rodent models of Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 49–63. [Google Scholar] [PubMed]
- Nikkhah, G.; Odin, P.; Smits, A.; Tingström, A.; Othberg, A.; Brundin, P.; Funa, K.; Lindvall, O. Platelet-derived growth factor promotes survival of rat and human mesencephalic dopaminergic neurons in culture. Exp. Brain Res. 1993, 92, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Othberg, A.; Odin, P.; Ballagi, A.; Ahgren, A.; Funa, K.; Lindvall, O. Specific effects of platelet derived growth factor (PDGF) on fetal rat and human dopaminergic neurons in vitro. Exp. Brain Res. 1995, 105, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Root, R.K.; Dale, D.C. Granulocyte Colony-Stimulating Factor and Granulocyte-Macrophage Colony-Stimulating Factor: Comparisons and Potential for Use in the Treatment of Infections in Nonneutropenic Patients. J. Infect. Dis. 1999, 179, S342–S352. [Google Scholar] [CrossRef] [PubMed]
- Schäbitz, W.-R.; Kollmar, R.; Schwaninger, M.; Juettler, E.; Bardutzky, J.; Schölzke, M.N.; Sommer, C.; Schwab, S. Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke J. Cereb. Circ. 2003, 34, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Krüger, C.; Steigleder, T.; Weber, D.; Pitzer, C.; Laage, R.; Aronowski, J.; Maurer, M.H.; Gassler, N.; Mier, W.; et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Investig. 2005, 115, 2083–2098. [Google Scholar] [CrossRef] [PubMed]
- Meuer, K.; Pitzer, C.; Teismann, P.; Krüger, C.; Göricke, B.; Laage, R.; Lingor, P.; Peters, K.; Schlachetzki, J.C.M.; Kobayashi, K.; et al. Granulocyte-colony stimulating factor is neuroprotective in a model of Parkinson’s disease. J. Neurochem. 2006, 97, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Sava, V.; Rowe, A.; Li, K.; Cao, C.; Mori, T.; Sanchez-Ramos, J. Granulocyte-colony stimulating factor (G-CSF) enhances recovery in mouse model of Parkinson’s disease. Neurosci. Lett. 2011, 487, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-Q.; Arai, H.; Ren, Y.-R.; Oizumi, H.; Zhang, N.; Seike, S.; Furuya, T.; Yasuda, T.; Mizuno, Y.; Mochizuki, H. Recombinant human granulocyte colony-stimulating factor protects against MPTP-induced dopaminergic cell death in mice by altering Bcl-2/Bax expression levels. J. Neurochem. 2006, 99, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Choi, B.H.; Huang, X.; Snyder, B.J.; Bukhari, S.; Kong, T.-H.; Park, H.; Park, H.C.; Park, S.R.; Ha, Y. Granulocyte-macrophage colony-stimulating factor promotes survival of dopaminergic neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced murine Parkinson’s disease model. Eur. J. Neurosci. 2009, 29, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Kosloski, L.M.; Kosmacek, E.A.; Olson, K.E.; Mosley, R.L.; Gendelman, H.E. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J. Neuroimmunol. 2013, 265, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, P.; Saarma, M. Novel CDNF/MANF family of neurotrophic factors. Dev. Neurobiol. 2010, 70, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, P.; Voutilainen, M.H.; Laurén, J.; Peränen, J.; Leppänen, V.-M.; Andressoo, J.-O.; Lindahl, M.; Janhunen, S.; Kalkkinen, N.; Timmusk, T.; et al. Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 2007, 448, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Airavaara, M.; Harvey, B.K.; Voutilainen, M.H.; Shen, H.; Chou, J.; Lindholm, P.; Lindahl, M.; Tuominen, R.K.; Saarma, M.; Hoffer, B.; et al. CDNF protects the nigrostriatal dopamine system and promotes recovery after MPTP treatment in mice. Cell Transplant. 2012, 21, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, V.; Zöller, T.; Attaai, A.; Spittau, B. Microglia-Mediated Neuroinflammation and Neurotrophic Factor-Induced Protection in the MPTP Mouse Model of Parkinson’s Disease-Lessons from Transgenic Mice. Int. J. Mol. Sci. 2016, 17, 151. https://doi.org/10.3390/ijms17020151
Machado V, Zöller T, Attaai A, Spittau B. Microglia-Mediated Neuroinflammation and Neurotrophic Factor-Induced Protection in the MPTP Mouse Model of Parkinson’s Disease-Lessons from Transgenic Mice. International Journal of Molecular Sciences. 2016; 17(2):151. https://doi.org/10.3390/ijms17020151
Chicago/Turabian StyleMachado, Venissa, Tanja Zöller, Abdelraheim Attaai, and Björn Spittau. 2016. "Microglia-Mediated Neuroinflammation and Neurotrophic Factor-Induced Protection in the MPTP Mouse Model of Parkinson’s Disease-Lessons from Transgenic Mice" International Journal of Molecular Sciences 17, no. 2: 151. https://doi.org/10.3390/ijms17020151