
Apoptosis in Cellular Society: Communication between Apoptotic Cells and Their Neighbors

Abstract
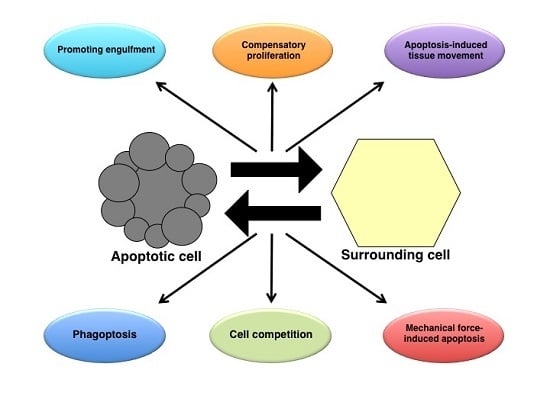
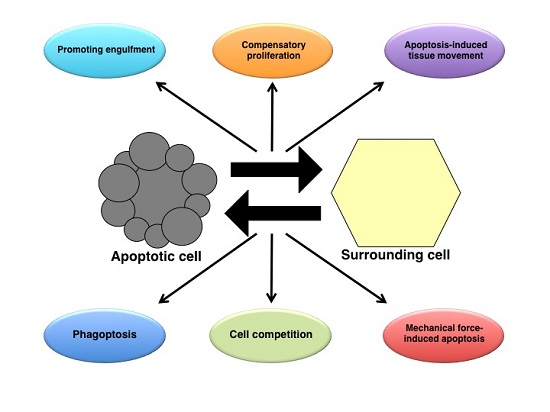
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
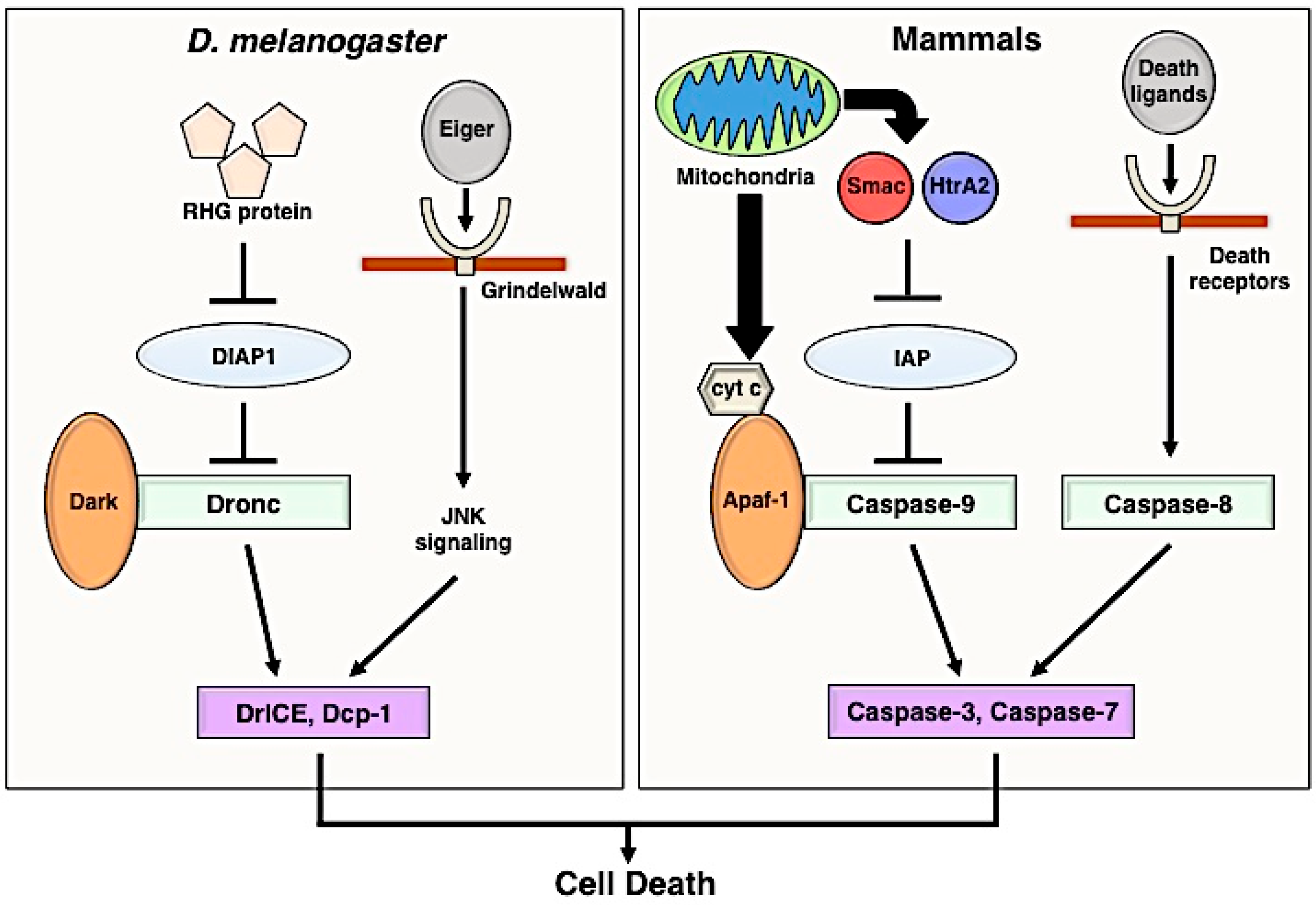
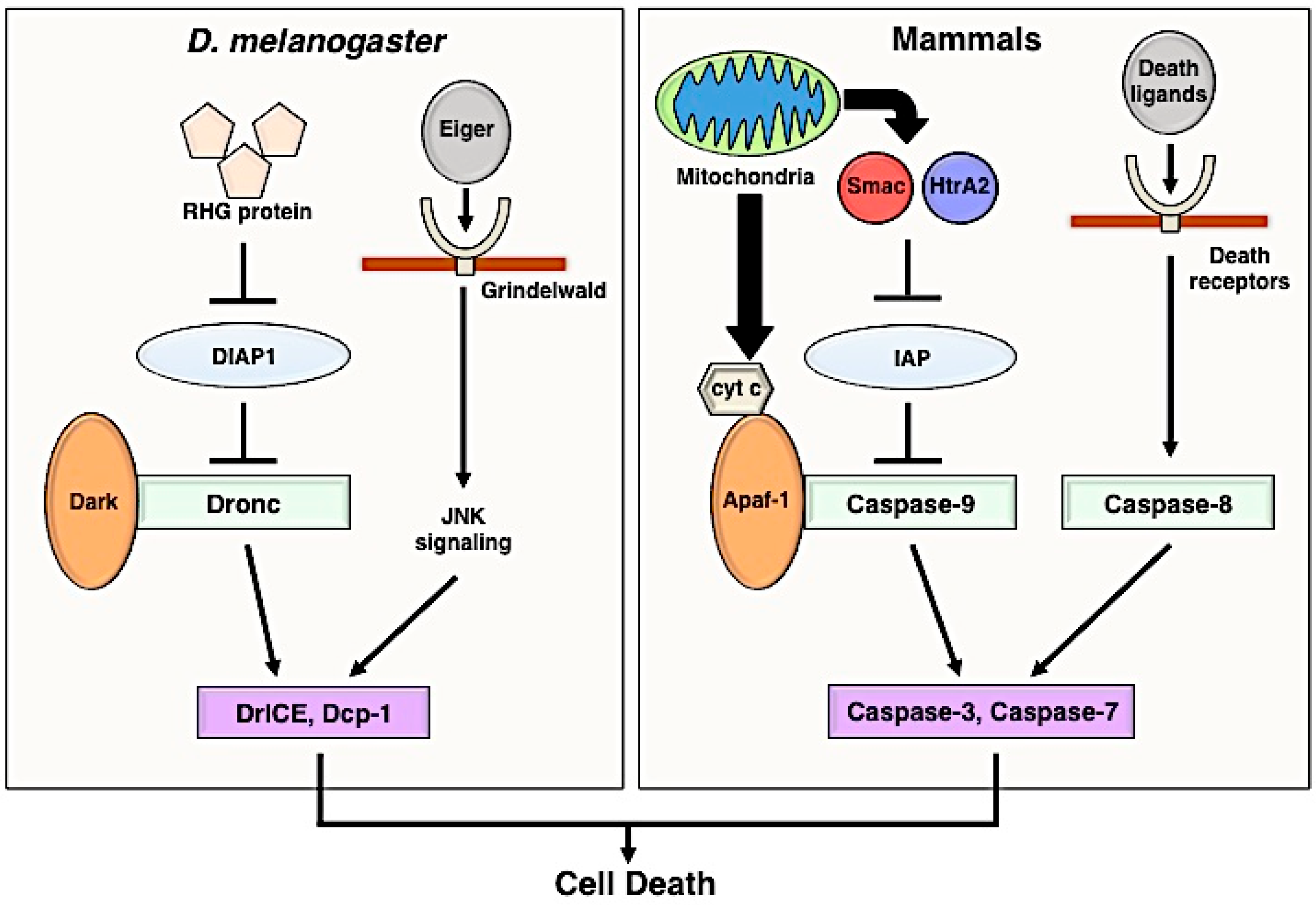
2. Apoptosis Signaling Pathway
3. Engulfment and Apoptosis
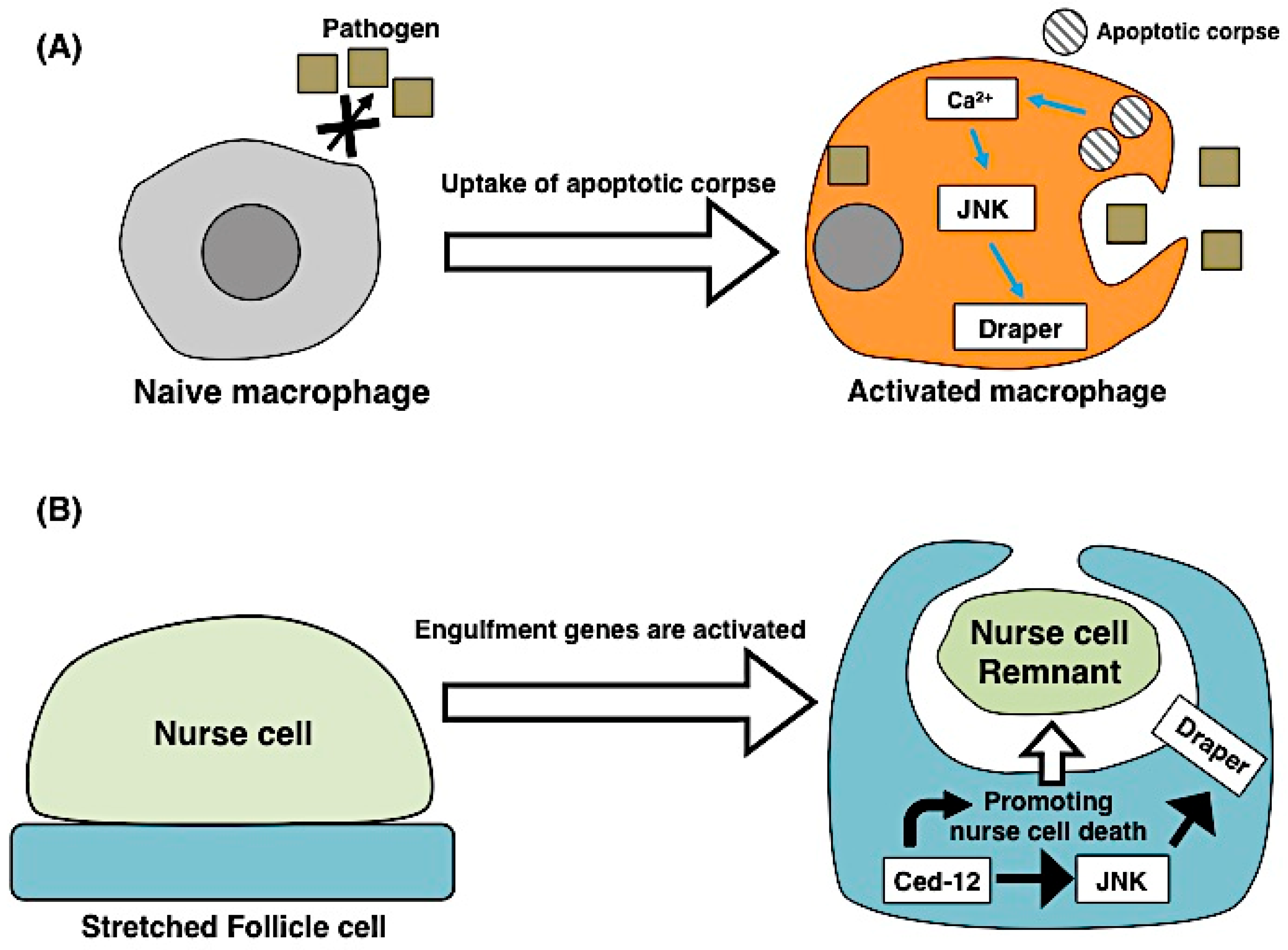
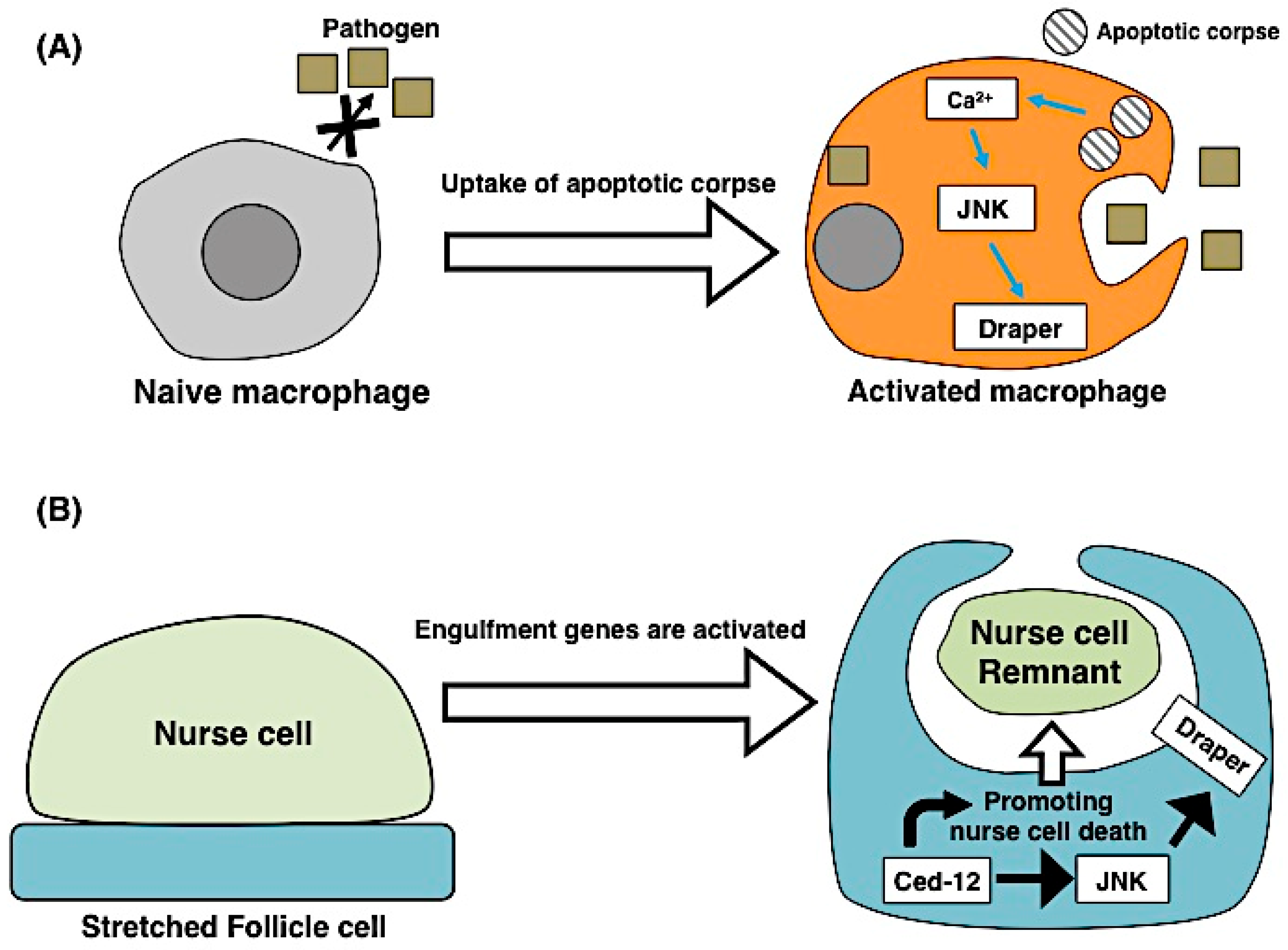
3.1. Apoptosis Induces Engulfment
3.2. Engulfing Cells Contribute to Apoptosis
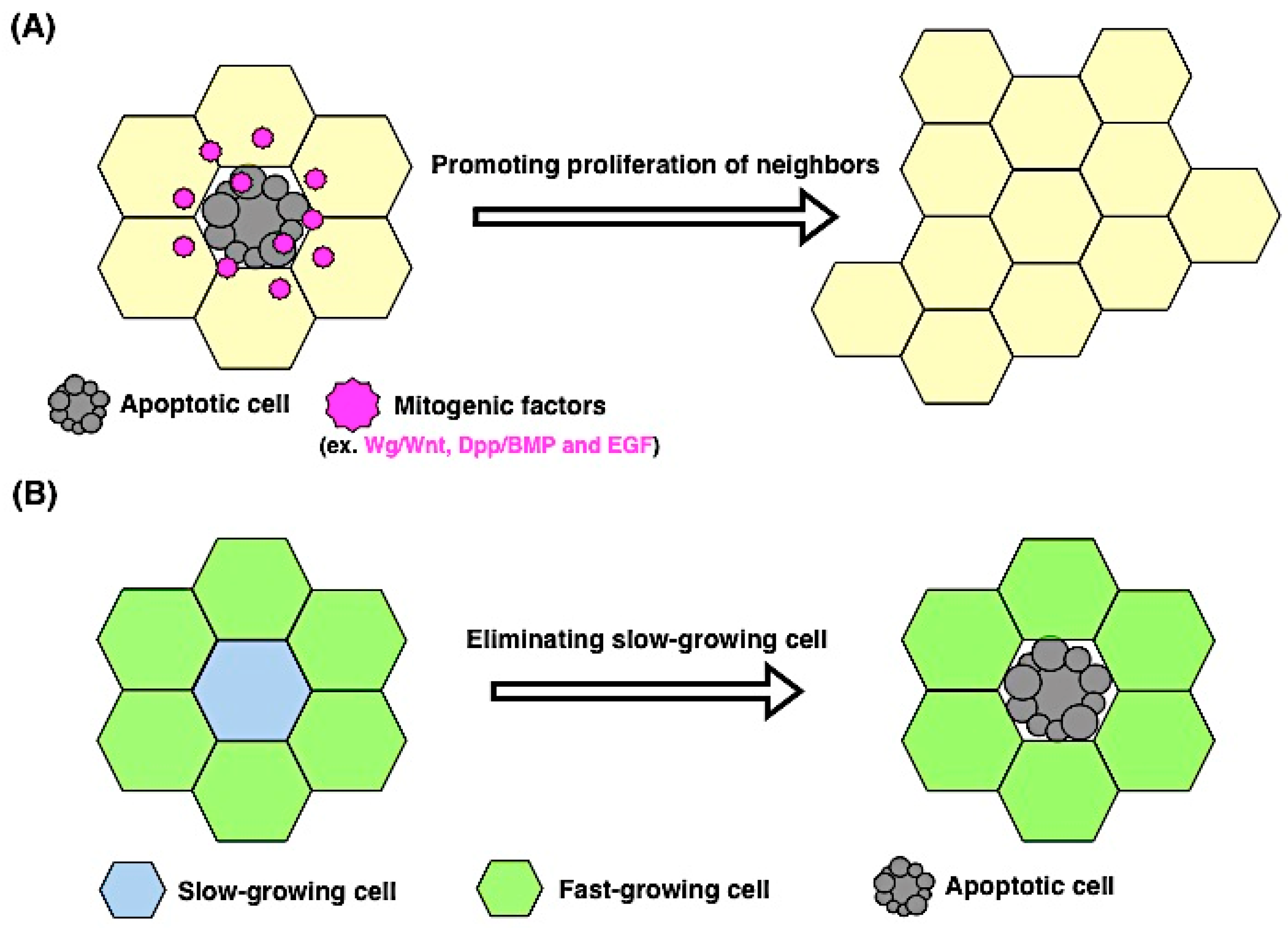
4. Proliferation and Apoptosis
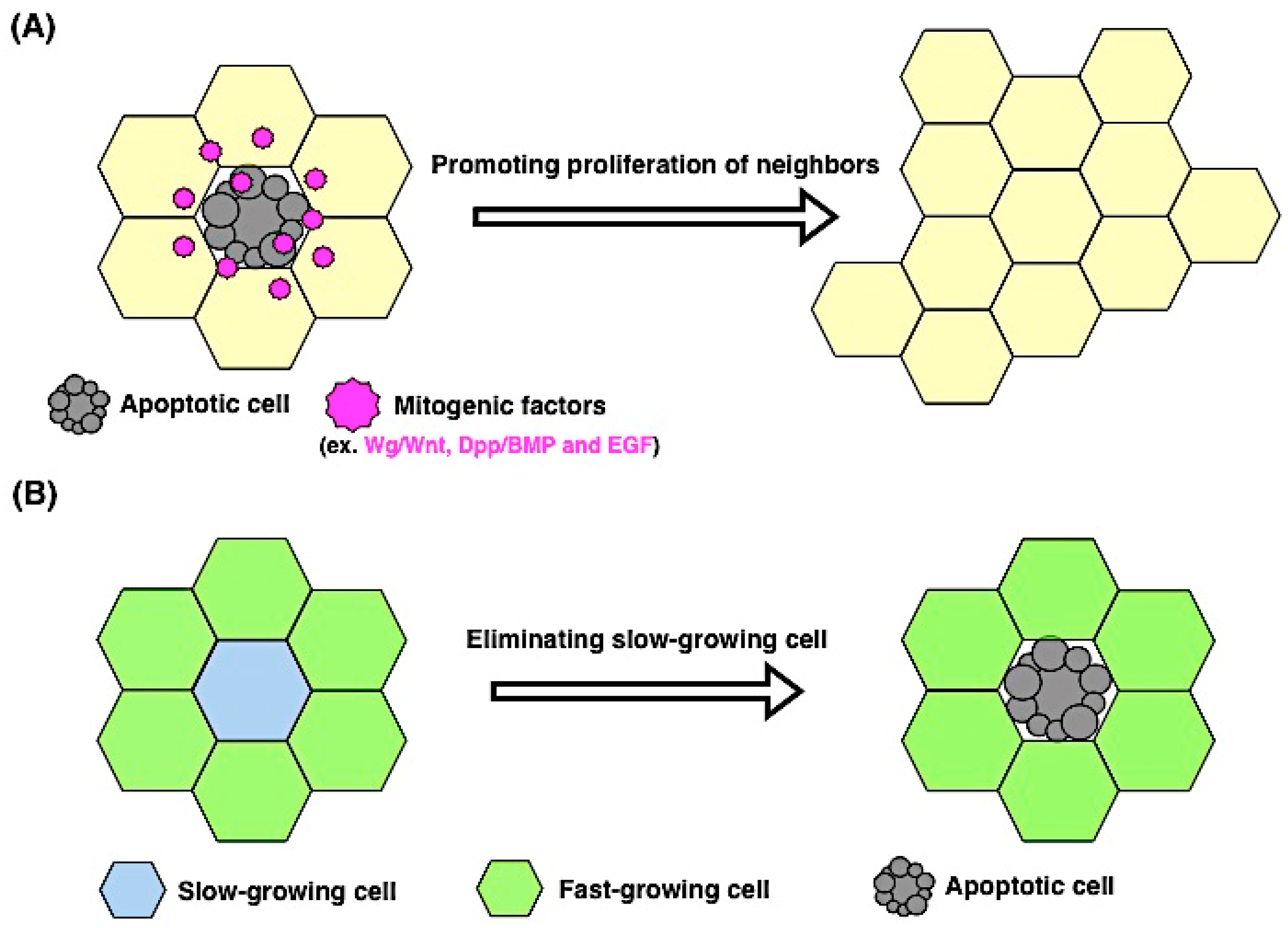
4.1. Apoptosis Induces Proliferation
4.2. Proliferating Cells Contribute to Apoptosis
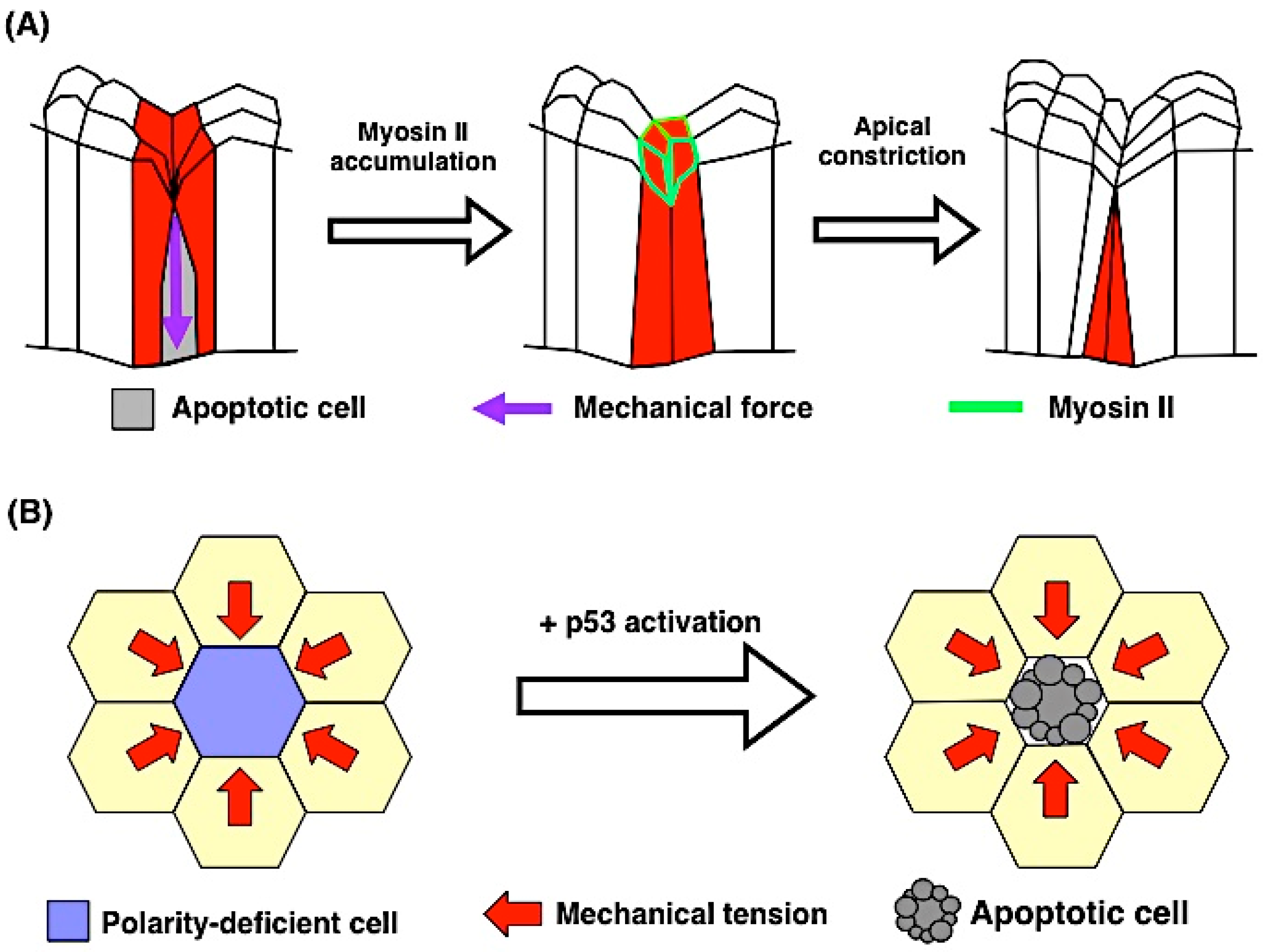
5. Force and Apoptosis
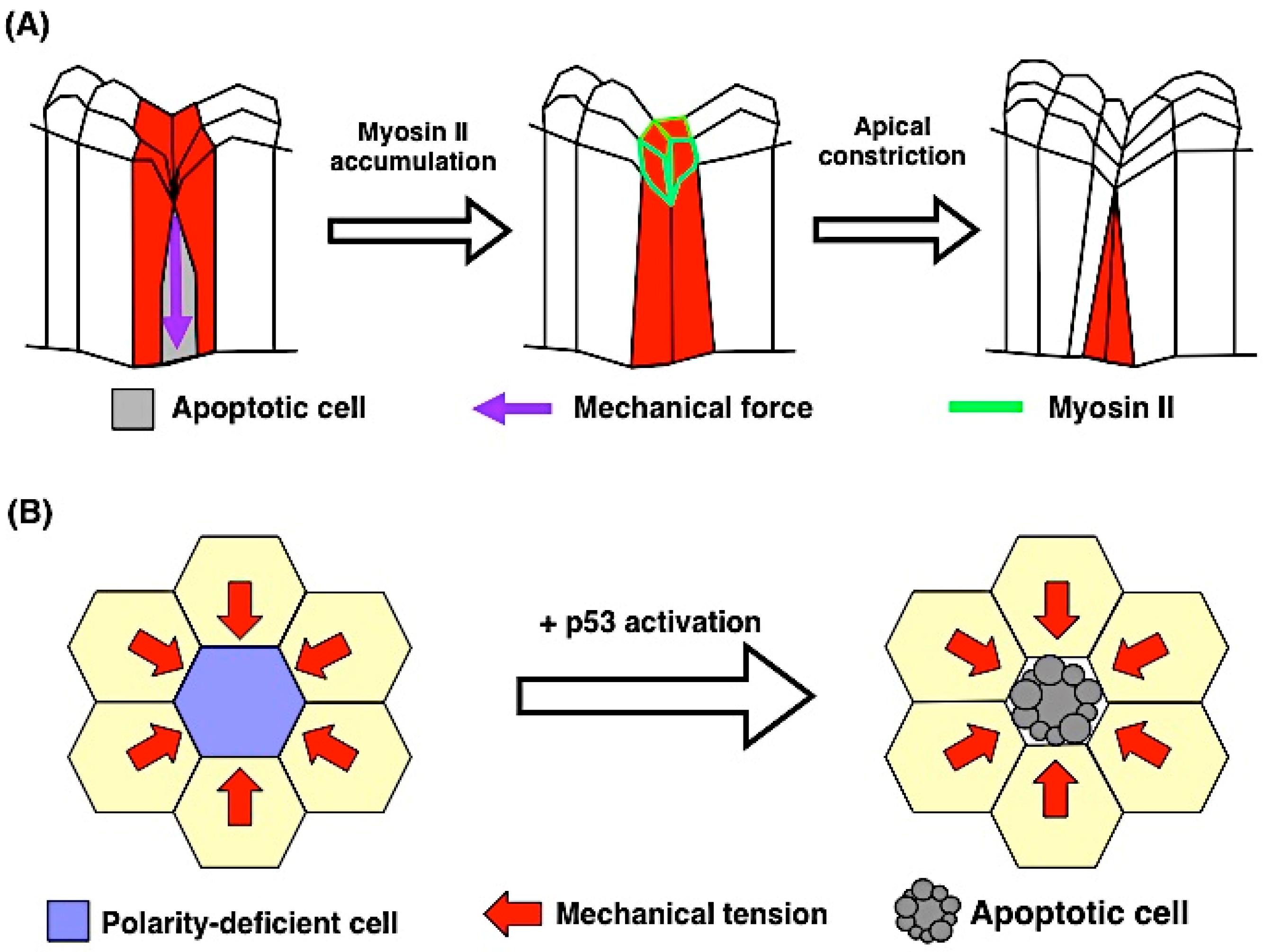
5.1. Apoptosis Induces Mechanical Force
5.2. Mechanical Force Contributes to Apoptosis
6. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eroglu, M.; Derry, W.B. Your neighbours matter—Non-autonomous control of apoptosis in development and disease. Cell Death Differ. 2016, 23, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Garijo, A.; Steller, H. Spreading the word: Non-autonomous effects of apoptosis during development, regeneration and disease. Development 2015, 142, 3253–3262. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a Mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K.; Takio, K.; Takahashi, R. A Serine Protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef]
- Schile, A.J.; García-Fernández, M.; Steller, H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008, 22, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Holley, C.; Olson, M.R.; Colon-Ramos, D.A.; Kornbluth, S. Reaper eliminates IAP proteins through stimulated IAP degradation and ganeralized translational inhibition. Nat. Cell Biol. 2002, 4, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Lazebnik, Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999, 13, 3179–3184. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Grether, M.; Abrams, J.; Young, L.; Farrell, K.; Steller, H. Genetic control of programmed cell death in Drosophila. Science 1994, 264, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; McCall, K.; Agapite, J.; Hartwieg, E.; Steller, H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000, 19, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Silke, J.; Leevers, S.J.; Evan, G.I. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 2000, 19, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, Y.; Willecke, R.; Chen, Z.; Ding, T.; Bergmann, A. The effector caspases drICE and dcp-1 have partially overlapping functions in the apoptotic pathway in Drosophila. Cell Death Differ. 2006, 13, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Igaki, T.; Kanda, H.; Yamamoto-Goto, Y.; Kanuka, H.; Kuranaga, E.; Aigaki, T.; Miura, M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002, 21, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.S.; Colombani, J.; Palmerini, V.; Chakrabandhu, K.; Boone, E.; Röthlisberger, M.; Toggweiler, J.; Basler, K.; Mapelli, M.; Hueber, A.-O.; et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature 2015, 522, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Jäättelä, M.; Tschopp, J. Caspase-independent cell death in T lymphocytes. Nat. Immunol. 2003, 4, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Dixit, V.M. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 2010, 17, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Hanayama, R.; Kawane, K. Autoimmunity and the Clearance of Dead Cells. Cell 2010, 140, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Truman, L.A.; Ford, C.A.; Pasikowska, M.; Pound, J.D.; Wilkinson, S.J.; Dumitriu, I.E.; Melville, L.; Melrose, L.A.; Ogden, C.A.; Nibbs, R.; et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 2008, 112, 5026–5036. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Krahling, S.; Callahan, M.K.; Williamson, P.; Schlegel, R.A. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ. 1999, 6, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Miwa, M.; Miwa, K.; Hanayama, R.; Nagase, H.; Nagata, S.; Tanaka, M. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J. Exp. Med. 2004, 200, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Weavers, H.; Evans, I.R.; Martin, P.; Wood, W. Corpse Engulfment Generates a Molecular Memory that Primes the Macrophage Inflammatory Response. Cell 2016, 165, 1658–1671. [Google Scholar] [CrossRef] [PubMed]
- Stramer, B.; Wood, W.; Galko, M.J.; Redd, M.J.; Jacinto, A.; Parkhurst, S.M.; Martin, P. Live imaging of wound inflammation in Drosophila embryos reveals key roles for small GTPases during in vivo cell migration. J. Cell Biol. 2005, 168, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Reddien, P.W.; Cameron, S.; Horvitz, H.R. Phagocytosis promotes programmed cell death in C. elegans. Nature 2001, 412, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Hoeppner, D.J.; Hengartner, M.O.; Schnabel, R. Engulfment genes cooperate with CED-3 to promote cell death in Caenorhabditis elegans. Nature 2001, 412, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Lambie, E.J.; Bindu, S.; Mikeladze-Dvali, T.; Conradt, B. Engulfment pathways promote programmed cell death by enhancing the unequal segregation of apoptotic potential. Nat. Commun. 2015, 6, 10126. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, H.L.; Hovitz, H.R. Both the apoptotic suicide pathway and phagocytosis are required for a programmed cell death in Caenorhabditis elegans. BMC Biol. 2016, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Stravopodis, D.J.; Papassideri, I.; Robert-Nicoud, M.; Margaritis, L.H. Stage-specific apoptotic patterns during Drosophila oogenesis. Eur. J. Cell Biol. 2000, 79, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.I.; Timmons, A.K.; Klein, A.P.; Pritchett, T.L.; Welch, E.; Meehan, T.L.; Li, C.; McCall, K. Draper acts through the JNK pathway to control synchronous engulfment of dying germline cells by follicular epithelial cells. Development 2012, 139, 4029–4039. [Google Scholar] [CrossRef] [PubMed]
- Timmons, A.K.; Mondragon, A.A.; Schenkel, C.E.; Yalonetskaya, A.; Taylor, J.D.; Moynihan, K.E.; Etchegaray, J.I.; Meehan, T.L.; McCall, K. Phagocytosis genes nonautonomously promote developmental cell death in the Drosophila ovary. Proc. Natl. Acad. Sci. USA 2016, 113, E1246–E1255. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Hornik, T.C.; Vilalta, A.; Brown, G.C. Activated microglia cause reversible apoptosis of pheochromocytoma cells, inducing their cell death by phagocytosis. J. Cell Sci. 2016, 129, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Bergmann, A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev. Cell 2008, 14, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D.; Bergmann, A. The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008797. [Google Scholar] [CrossRef] [PubMed]
- Haynie, J.L.; Bryant, P.J. The effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc of Drosophila melanogaster. Wilhelm Roux’s Arch. Dev. Biol. 1977, 183, 85–100. [Google Scholar] [CrossRef]
- Pérez-Garijo, A.; Martín, F.A.; Morata, G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 2004, 131, 5591–5598. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.R.; Guo, M.; Hay, B.A. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr. Biol. 2004, 14, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D.; Gorenc, T.; Steller, H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 2004, 7, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, S.; Hernandez, J.; Yenigun, V.B.; Hertlein, G.; Fogarty, C.E.; Lindblad, J.L.; Bergmann, A. Genetic models of apoptosis-induced proliferation decipher activation of JNK and identify a requirement of EGFR signaling for tissue regenerative responses in Drosophila. PLoS Genet. 2014, 10, e1004131. [Google Scholar] [CrossRef] [PubMed]
- Wells, B.S.; Yoshida, E.; Johnston, L.A. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr. Biol. 2006, 16, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Senoo-Matsuda, N.; Hiromi, Y.; Miura, M. DRONC coordinates cell death and compensatory proliferation. Mol. Cell. Biol. 2006, 26, 7258–7268. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, C.E.; Diwanji, N.; Lindblad, J.L.; Tare, M.; Amcheslavsky, A.; Makhijani, K.; Brückner, K.; Fan, Y.; Bergmann, A. Extracellular reactive oxygen species drive apoptosis-induced proliferation via Drosophila macrophages. Curr. Biol. 2016, 26, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Gauron, C.; Rampon, C.; Bouzaffour, M.; Ipendey, E.; Teillon, J.; Volovitch, M.; Vriz, S. Sustained production of ROS triggers compensatory proliferation and is required for regeneration to proceed. Sci. Rep. 2013, 3, 2084. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Patel, P.H.; Kohlmaier, A.; Grenley, M.O.; McEwen, D.G.; Edgar, B.A. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 2009, 137, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, A.; Steller, H. Apoptosis, stem cells, and tissue regeneration. Sci. Signal. 2010, 3, re8. [Google Scholar] [CrossRef] [PubMed]
- Chera, S.; Ghila, L.; Dobretz, K.; Wenger, Y.; Bauer, C.; Buzgariu, W.; Martinou, J.-C.; Galliot, B. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev. Cell 2009, 17, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.W.; Christen, B.; Slack, J.M.W. Molecular pathways needed for regeneration of spinal cord and muscle in a vertebrate. Dev. Cell 2003, 5, 429–439. [Google Scholar] [CrossRef]
- Ho, D.M.; Whitman, M. TGF-β signaling is required for multiple processes during Xenopus tail regeneration. Dev. Biol. 2008, 315, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Slack, J.M.W. Requirement for Wnt and FGF signaling in Xenopus tadpole tail regeneration. Dev. Biol. 2008, 316, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.-Y. Apoptotic cells activate the "phoenix rising" pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.T.; et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Morata, G.; Ripoll, P. Minutes: mutants of Drosophila autonomously affecting cell division rate. Dev. Biol. 1975, 42, 211–221. [Google Scholar] [CrossRef]
- Simpson, P.; Morata, G. Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev. Biol. 1981, 85, 299–308. [Google Scholar] [CrossRef]
- Moreno, E.; Basler, K.; Morata, G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 2002, 416, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Baker, N.E. Engulfment is required for cell competition. Cell 2007, 129, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Prober, D.A.; Edgar, B.A.; Eisenman, R.N.; Gallant, P. Drosophila MYC regulates cellular growth during development. Cell 1999, 98, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Basler, K. dMyc transforms cells into super-competitors. Cell 2004, 117, 117–129. [Google Scholar] [CrossRef]
- De la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila MYC regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef]
- Rhiner, C.; López-Gay, J.M.; Soldini, D.; Casas-Tinto, S.; Martín, F.A.; Lombardía, L.; Moreno, E. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila. Dev. Cell 2010, 18, 985–998. [Google Scholar] [CrossRef] [PubMed]
- De la Cova, C.; Senoo-Matsuda, N.; Ziosi, M.; Wu, D.C.; Bellosta, P.; Quinzii, C.M.; Johnston, L.A. Supercompetitor status of Drosophila Myc cells requires p53 as a fitness sensor to reprogram metabolism and promote viability. Cell Metab. 2014, 19, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Clavería, C.; Giovinazzo, G.; Sierra, R.; Torres, M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature 2013, 500, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Di-Gregorio, A.; George, N.; Pozzi, S.; Sánchez, J.M.; Pernaute, B.; Rodríguez, T.A. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev. Cell 2013, 26, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Villa del Campo, C.; Clavería, C.; Sierra, R.; Torres, M. Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep. 2014, 8, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, M.M.; Madhavan, K. Morphogenesis of the epidermis of adult abdomen of Drosophila. J. Embryol. Exp. Morphol. 1980, 60, 1–31. [Google Scholar] [PubMed]
- Ninov, N.; Chiarelli, D.A.; Martín-Blanco, E. Extrinsic and intrinsic mechanisms directing epithelial cell sheet replacement during Drosophila metamorphosis. Development 2007, 134, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.-I.; Kuranaga, E.; Sugimura, K.; Miyawaki, A.; Miura, M. Nonautonomous apoptosis is triggered by local cell cycle progression during epithelial replacement in Drosophila. Mol. Cell. Biol. 2011, 31, 2499–2512. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, M.; Gradilla, A.-C.; Seijo, I.; Andrés, G.; Rodríguez-Navas, C.; González-Méndez, L.; Guerrero, I. Cytonemes are required for the establishment of a normal Hedgehog morphogen gradient in Drosophila epithelia. Nat. Cell Biol. 2013, 15, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, T.B.; Roy, S. Cytonemes as specialized signaling filopodia. Development 2014, 141, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Kiehart, D.P.; Galbraith, C.G.; Edwards, K.A.; Rickoll, W.L.; Montague, R.A. Multiple forces contribute to cell sheet morphogenesis for dorsal closure in Drosophila. J. Cell Biol. 2000, 149, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Toyama, Y.; Peralta, X.G.; Wells, A.R.; Kiehart, D.P.; Edwards, G.S. Apoptotic force and tissue dynamics during Drosophila embryogenesis. Science 2008, 321, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Muliyil, S.; Krishnakumar, P.; Narasimha, M. Spatial, temporal and molecular hierarchies in the link between death, delamination and dorsal closure. Development 2011, 138, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Gleichauf, R. Anatomie und variabilitat des geschlechtapparates von Drosopihla melanogaster. Z. Wiss. Zool. 1936, 148, 1–66. (In Germany) [Google Scholar]
- Adám, G.; Perrimon, N.; Noselli, S. The retinoic-like juvenile hormone controls the looping of left-right asymmetric organs in Drosophila. Development 2003, 130, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.K.; Lengyel, J.A. Embryonic head involution and rotation of male terminalia require the Drosophila locus head involution defective. Genetics 1991, 129, 783–789. [Google Scholar] [PubMed]
- Suzanne, M.; Petzoldt, A.G.; Spéder, P.; Coutelis, J.-B.; Steller, H.; Noselli, S. Coupling of apoptosis and L/R patterning controls stepwise organ looping. Curr. Biol. 2010, 20, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Kuranaga, E.; Matsunuma, T.; Kanuka, H.; Takemoto, K.; Koto, A.; Kimura, K.; Miura, M. Apoptosis controls the speed of looping morphogenesis in Drosophila male terminalia. Development 2011, 138, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Monier, B.; Gettings, M.; Gay, G.; Mangeat, T.; Schott, S.; Guarner, A.; Suzanne, M. Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature 2015, 518, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T. The mechanism of Drosophila leg development along the proximodistal axis. Dev. Growth Differ. 2004, 46, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Manjón, C.; Sánchez-Herrero, E.; Suzanne, M. Sharp boundaries of Dpp signalling trigger local cell death required for Drosophila leg morphogenesis. Nat. Cell Biol. 2007, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Weil, M.; Jacobson, M.D.; Raff, M.C. Is programmed cell death required for neural tube closure? Curr. Biol. 1997, 7, 281–284. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shinotsuka, N.; Nonomura, K.; Takemoto, K.; Kuida, K.; Yosida, H.; Miura, M. Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. J. Cell Biol. 2011, 195, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Raff, M.C.; Cramer, L.P. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr. Biol. 2001, 11, 1847–1857. [Google Scholar] [CrossRef]
- Gu, Y.; Forostyan, T.; Sabbadini, R.; Rosenblatt, J. Epithelial cell extrusion requires the sphingosine-1-phosphate receptor 2 pathway. J. Cell Biol. 2011, 193, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Marinari, E.; Mehonic, A.; Curran, S.; Gale, J.; Duke, T.; Baum, B. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature 2012, 484, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Levayer, R.; Dupont, C.; Moreno, E. Tissue Crowding Induces Caspase-Dependent Competition for Space. Curr. Biol. 2016, 26, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhoffer, G.T.; Loftus, P.D.; Yoshigi, M.; Otsuna, H.; Chien, C.-B.; Morcos, P.A.; Rosenblatt, J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Shea, J.; Slattum, G.; Firpo, M.A.; Alexander, M.; Mulvihill, S.J.; Golubovskaya, V.M.; Rosenblatt, J. Defective apical extrusion signaling contributes to aggressive tumor hallmarks. Elife 2015, 4, e04069. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, L.; Goschorska, M.; Kozyrska, K.; Duclos, G.; Kucinski, I.; Chessel, A.; Hampton-O’Neil, L.; Bradshaw, C.R.; Allen, G.E.; Rawlins, E.L.; et al. Mechanical cell competition kills cells via induction of lethal p53 levels. Nat. Commun. 2016, 7, 11373. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.; Wisniewska, K.A.; Lawrenson, K.; Garcia-Miranda, P.; Tada, M.; Kajita, M.; Mano, H.; Ishikawa, S.; Ikegawa, M.; Shimada, T.; et al. Loss of Scribble causes cell competition in mammalian cells. J. Cell Sci. 2012, 125, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, A.; Kuranaga, E.; Tonoki, A.; Misaki, K.; Yonemura, S.; Kanuka, H.; Miura, M. Homeostatic epithelial renewal in the gut is required for dampening a fatal systemic wound response in Drosophila. Cell Rep. 2013, 3, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Kashio, S.; Obata, F.; Zhang, L.; Katsuyama, T.; Chihara, T.; Miura, M. Tissue nonautonomous effects of fat body methionine metabolism on imaginal disc repair in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter-Hufford, A.E.; Lee, C.S.; Kinchen, J.M.; Sokolowski, J.D.; Arandjelovic, S.; Call, J.A.; Klibanov, A.L.; Yan, Z.; Mandell, J.W.; Ravichandran, K.S. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 2013, 497, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Garlena, R.A.; Lennox, A.L.; Baker, L.R.; Parsons, T.E.; Weinberg, S.M.; Stronach, B.E. The receptor tyrosine kinase Pvr promotes tissue closure by coordinating corpse removal and epidermal zippering. Development 2015, 142, 3403–3415. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawamoto, Y.; Nakajima, Y.-i.; Kuranaga, E. Apoptosis in Cellular Society: Communication between Apoptotic Cells and Their Neighbors. Int. J. Mol. Sci. 2016, 17, 2144. https://doi.org/10.3390/ijms17122144
Kawamoto Y, Nakajima Y-i, Kuranaga E. Apoptosis in Cellular Society: Communication between Apoptotic Cells and Their Neighbors. International Journal of Molecular Sciences. 2016; 17(12):2144. https://doi.org/10.3390/ijms17122144
Chicago/Turabian StyleKawamoto, Yuhei, Yu-ichiro Nakajima, and Erina Kuranaga. 2016. "Apoptosis in Cellular Society: Communication between Apoptotic Cells and Their Neighbors" International Journal of Molecular Sciences 17, no. 12: 2144. https://doi.org/10.3390/ijms17122144