
Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis

 ,
,
Abstract
:1. Introduction
2. Types of Heterogeneity
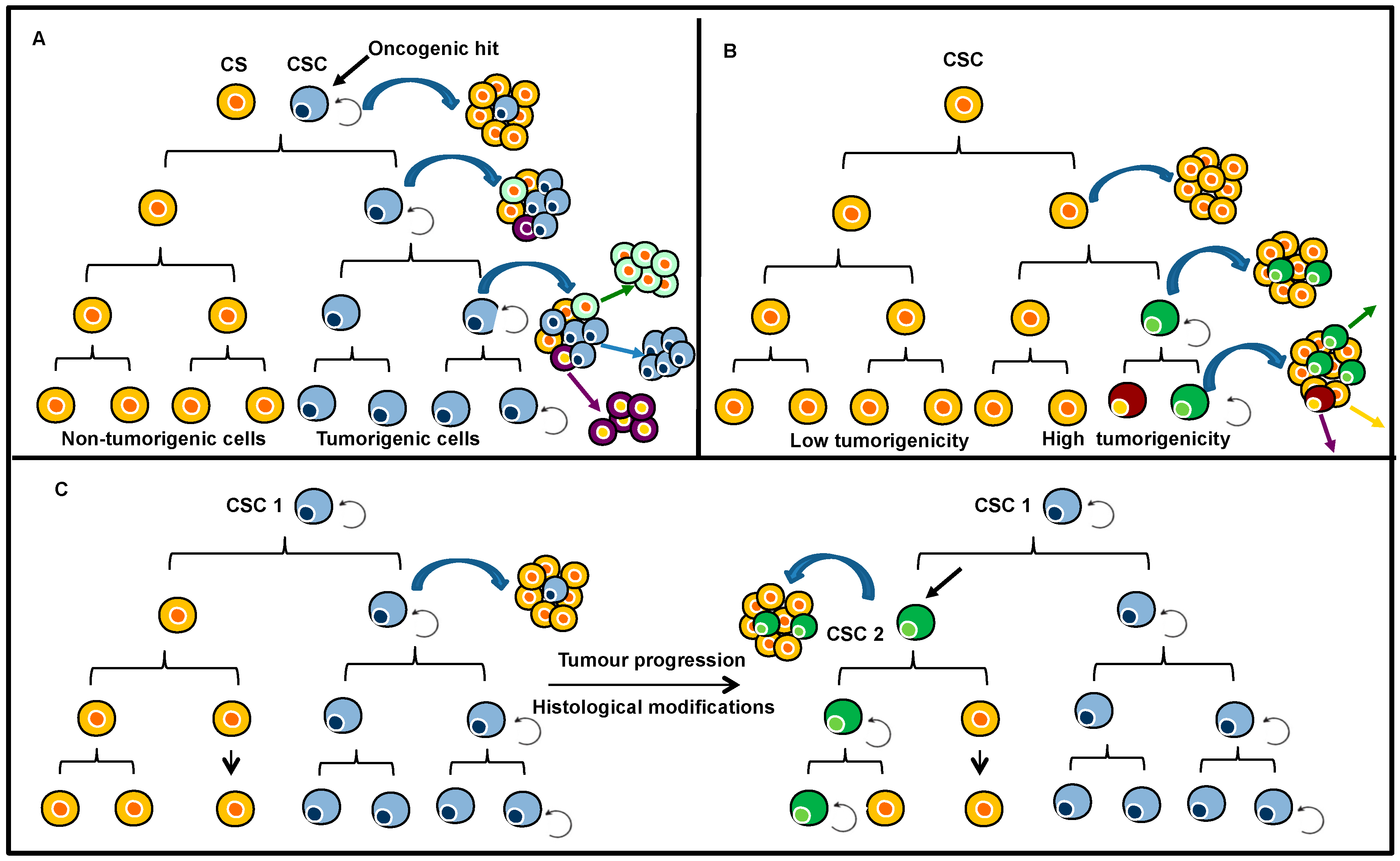
2.1. Intra-Tumour Heterogeneity
2.2. Inter-Tumour Heterogeneity
3. Sources of Heterogeneity
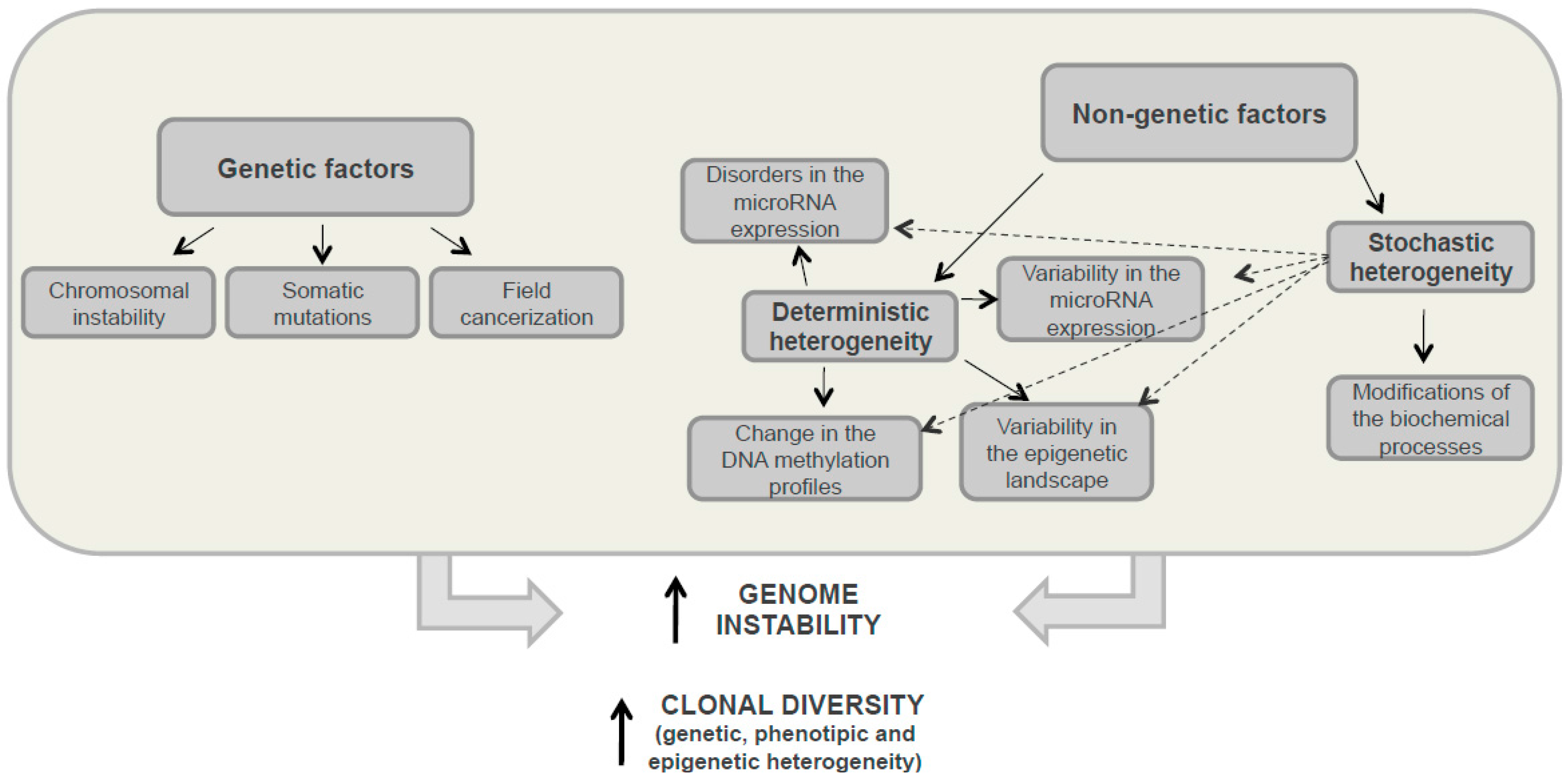
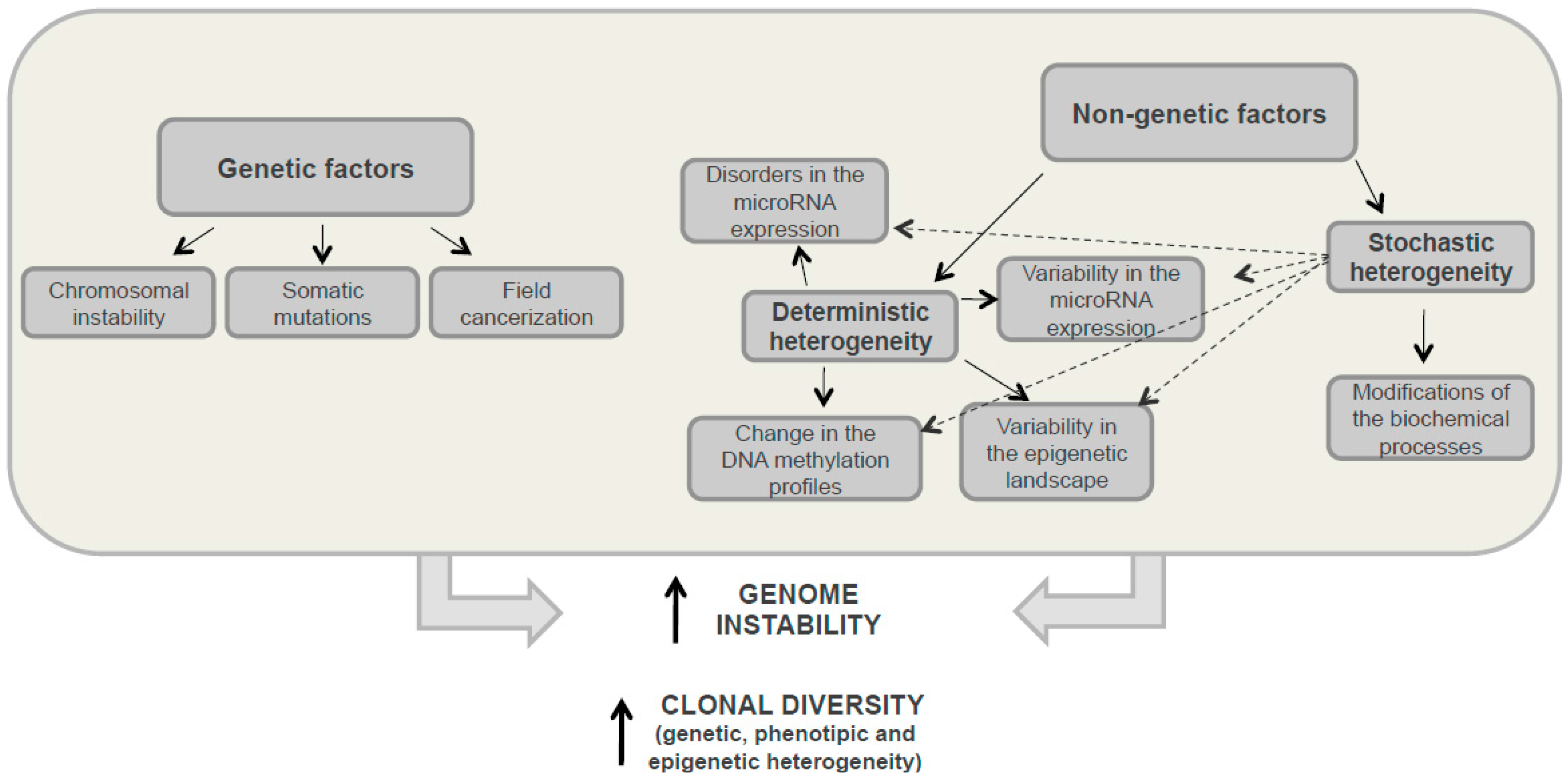
3.1. Genetic Heterogeneity
3.2. Nongenetic Heterogeneity
4. Heterogeneity of Distant Metastases: Circulating Tumour Cells
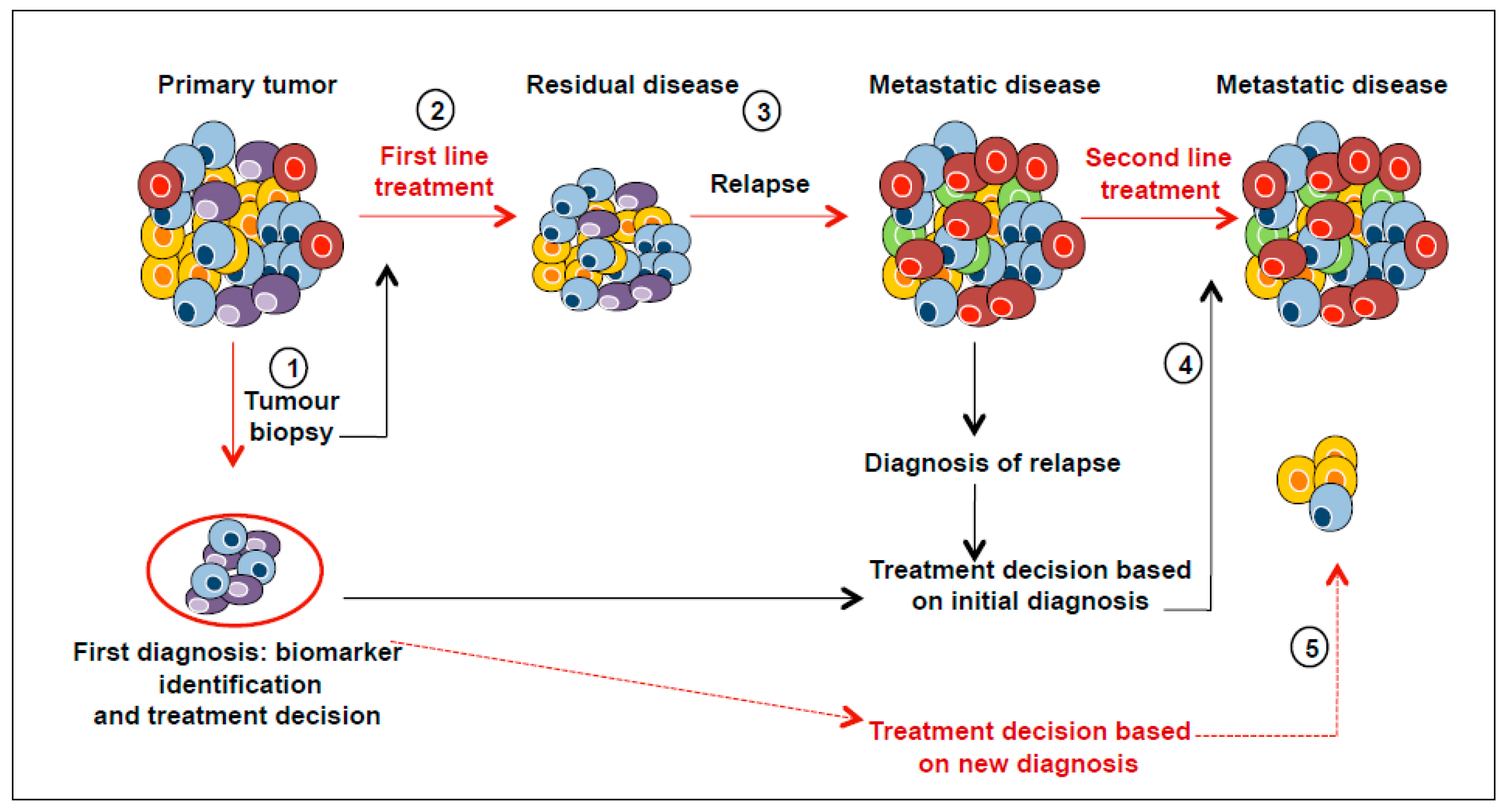
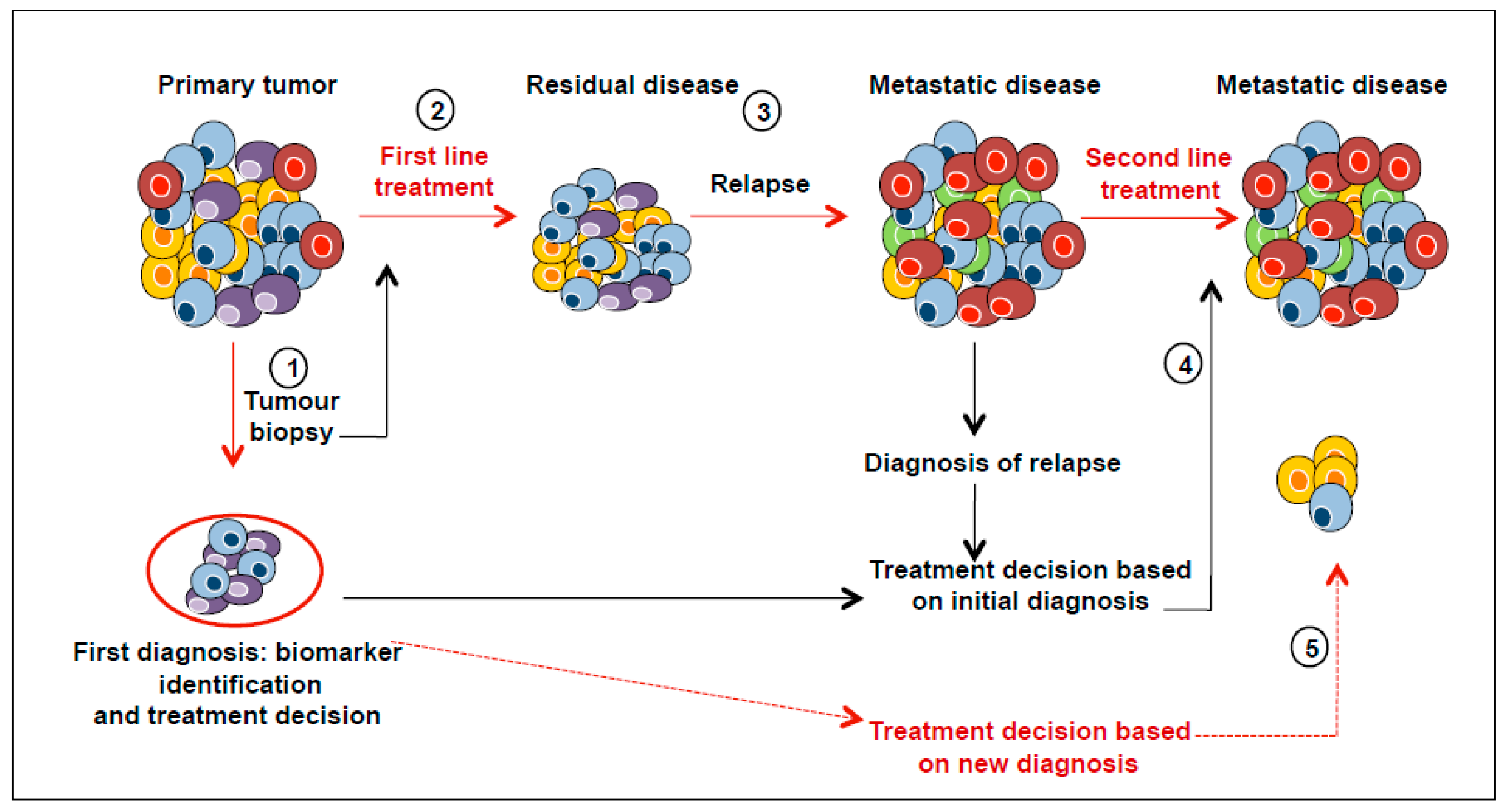
5. Clinical Implications of Tumour Heterogeneity
6. Methods for Studying Tumour Heterogeneity
6.1. Cell heterogeneity in Tissues
6.2. Cell Heterogeneity at the Single-Cell Level
6.2.1. Single-Cell Genomic and Transcriptomic Analyses
6.2.2. Single-Cell Proteomic Analysis
7. Conclusions and Future Direction
Acknowledgments
Conflicts of Interest
References
- Michor, F.; Polyak, K. The origins and implications of intratumor heterogeneity. Cancer Prev. Res. (Phila.) 2010, 3, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. The circulating tumor cells: Liquid biopsy of cancer. Klin. Lab. Diagn. 2014, 4, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M.T.; Calleja, L.R.; Chalopin, A.; Ory, B.; Heymann, D. Circulating Tumor Cells: A Review of Non-EpCAM-Based Approaches for Cell Enrichment and Isolation. Clin. Chem. 2016, 62, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.F.; Paoletti, C. Circulating tumour cells: Insights into tumour heterogeneity. J. Intern. Med. 2013, 274, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Andrechek, E.R.; Nevins, J.R. Mouse models of cancers: Opportunities to address heterogeneity of human cancer and evaluate therapeutic strategies. J. Mol. Med. (Berl.) 2010, 88, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Villanueva, J.; Herlyn, M. Intratumoral heterogeneity as a therapy resistance mechanism: Role of melanoma subpopulations. Adv. Pharmacol. 2012, 65, 335–359. [Google Scholar] [PubMed]
- Nassar, A.; Radhakrishnan, A.; Cabrero, I.A.; Cotsonis, G.A.; Cohen, C. Intratumoral heterogeneity of immunohistochemical marker expression in breast carcinoma: A tissue microarray-based study. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Chekhun, V.F.; Demash, D.V.; Nalieskina, L.A. Evaluation of biological effects and possible mechanisms of action of static magnetic field. Fiziol. Zh. 2012, 58, 85–94. [Google Scholar]
- Cassidy, J.W.; Caldas, C.; Bruna, A. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer Res. 2015, 75, 2963–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Reis-Filho, J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012, 13, e178–e185. [Google Scholar] [CrossRef]
- Wu, J.M.; Fackler, M.J.; Halushka, M.K.; Molavi, D.W.; Taylor, M.E.; Teo, W.W.; Griffin, C.; Fetting, J.; Davidson, N.E.; de Marzo, A.M.; et al. Heterogeneity of breast cancer metastases: Comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin. Cancer Res. 2008, 14, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S. Cancer stem cell heterogeneity in hereditary breast cancer. Breast Cancer Res. 2008, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Guidi, A.J.; Berry, D.A.; Broadwater, G.; Perloff, M.; Norton, L.; Barcos, M.P.; Hayes, D.F. Association of angiogenesis in lymph node metastases with outcome of breast cancer. J. Natl. Cancer Inst. 2000, 92, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Gundem, G.; van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.K.; Macintyre, G.; Wedge, D.C.; van Loo, P.; Patel, K.; Lunke, S.; Alexandrov, L.B.; Sloggett, C.; Cmero, M.; Marass, F.; et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 2015, 6, 6605. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 14, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Dakubo, G.D.; Jakupciak, J.P.; Birch-Machin, M.A.; Parr, R.L. Clinical implications and utility of field cancerization. Cancer Cell Int. 2007, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.V. Clonal heterogeneity of tumor may be due to continuous influx of newly transformed cells. Cancer Biol. Ther. 2006, 5, 1263–1264. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Assie, G.; Letouze, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updates 2012, 15, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, A.H.; Andersen, R.F.; Nielsen, B.S.; Sorensen, F.B.; Appelt, A.L.; Jakobsen, A.; Hansen, T.F. Intratumoral Heterogeneity of MicroRNA Expression in Rectal Cancer. PLoS ONE 2016, 11, e0156919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Vermaat, J.S.; Nijman, I.J.; Koudijs, M.J.; Gerritse, F.L.; Scherer, S.J.; Mokry, M.; Roessingh, W.M.; Lansu, N.; de Bruijn, E.; van Hillegersberg, R.; et al. Primary colorectal cancers and their subsequent hepatic metastases are genetically different: Implications for selection of patients for targeted treatment. Clin. Cancer Res. 2012, 18, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Vakiani, E.; Janakiraman, M.; Shen, R.; Sinha, R.; Zeng, Z.; Shia, J.; Cercek, A.; Kemeny, N.; D’Angelica, M.; Viale, A.; et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J. Clin. Oncol. 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Siyar Ekinci, A.; Demirci, U.; Cakmak Oksuzoglu, B.; Ozturk, A.; Esbah, O.; Ozatli, T.; Celik, B.; Budakoglu, B.; Turker, I.; Bal, O.; et al. KRAS discordance between primary and metastatic tumor in patients with metastatic colorectal carcinoma. J. BUON 2015, 20, 128–135. [Google Scholar] [PubMed]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; de Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wu, X.Y.; Yang, Z.Y.; Threapleton, D.E.; Yuan, J.Q.; Yu, Y.Y.; Tang, J.L. Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci. Rep. 2015, 5, 8065. [Google Scholar] [CrossRef] [PubMed]
- Cejas, P.; Lopez-Gomez, M.; Aguayo, C.; Madero, R.; Moreno-Rubio, J.; de Castro Carpeno, J.; Belda-Iniesta, C.; Barriuso, J.; Moreno Garcia, V.; Diaz, E.; et al. Analysis of the concordance in the EGFR pathway status between primary tumors and related metastases of colorectal cancer patients: Implications for cancer therapy. Curr. Cancer Drug Targets 2012, 12, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Yang, X.N.; Chen, Z.H.; Yang, J.J.; Zhou, Q.; Yan, H.H.; An, S.J.; et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist 2012, 17, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef] [PubMed]
- Mostert, B.; Jiang, Y.; Sieuwerts, A.M.; Wang, H.; Bolt-de Vries, J.; Biermann, K.; Kraan, J.; Lalmahomed, Z.; van Galen, A.; de Weerd, V.; et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int. J. Cancer 2013, 133, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Mohamed Suhaimi, N.A.; Foong, Y.M.; Lee, D.Y.; Phyo, W.M.; Cima, I.; Lee, E.X.; Goh, W.L.; Lim, W.Y.; Chia, K.S.; Kong, S.L.; et al. Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol. Oncol. 2015, 9, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Solakoglu, O.; Maierhofer, C.; Lahr, G.; Breit, E.; Scheunemann, P.; Heumos, I.; Pichlmeier, U.; Schlimok, G.; Oberneder, R.; Kollermann, M.W.; et al. Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Kraus, J.; Pantel, K.; Pinkel, D.; Albertson, D.G.; Speicher, M.R. High-resolution genomic profiling of occult micrometastatic tumor cells. Genes Chromosomes Cancer 2003, 36, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Pailler, E.; Auger, N.; Lindsay, C.R.; Vielh, P.; Islas-Morris-Hernandez, A.; Borget, I.; Ngo-Camus, M.; Planchard, D.; Soria, J.C.; Besse, B.; et al. High level of chromosomal instability in circulating tumor cells of ROS1-rearranged non-small-cell lung cancer. Ann. Oncol. 2015, 26, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Middleton, G.; Crack, L.R.; Popat, S.; Swanton, C.; Hollingsworth, S.J.; Buller, R.; Walker, I.; Carr, T.H.; Wherton, D.; Billingham, L.J.; et al. The National Lung Matrix Trial: Translating the biology of stratification in advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 2464–2469. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Hackshaw, A.; Ngai, Y.; Shaw, J.; Dive, C.; Quezada, S.; Middleton, G.; de Bruin, E.; Le Quesne, J.; Shafi, S.; et al. Tracking genomic cancer evolution for precision medicine: The lung TRACERx study. PLoS Biol. 2014, 12, e1001906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosse, S.A.; Gorges, T.M.; Pantel, K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Storey, B.D.; Moore, J.C.; Tang, Q.; Chen, Y.L.; Javaid, S.; Sarioglu, A.F.; Sullivan, R.; Madden, M.W.; O’Keefe, R.; et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. USA 2016, 113, 4947–4952. [Google Scholar] [CrossRef] [PubMed]
- Place, A.E.; Jin Huh, S.; Polyak, K. The microenvironment in breast cancer progression: Biology and implications for treatment. Breast Cancer Res. 2011, 13, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabieres, C.; Pantel, K. Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Pantel, K.; Riethdorf, S. Whole Genome Amplification in Genomic Analysis of Single Circulating Tumor Cells. Methods Mol. Biol. 2015, 1347, 221–232. [Google Scholar] [PubMed]
- Toss, A.; Mu, Z.; Fernandez, S.; Cristofanilli, M. CTC enumeration and characterization: Moving toward personalized medicine. Ann. Transl. Med. 2014, 2, 108. [Google Scholar] [PubMed]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, S.V.; Bingham, C.; Fittipaldi, P.; Austin, L.; Palazzo, J.; Palmer, G.; Alpaugh, K.; Cristofanilli, M. TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res. 2014, 16, 445. [Google Scholar] [CrossRef] [PubMed]
- Pestrin, M.; Salvianti, F.; Galardi, F.; de Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Sakaizawa, K.; Goto, Y.; Kiniwa, Y.; Uchiyama, A.; Harada, K.; Shimada, S.; Saida, T.; Ferrone, S.; Takata, M.; Uhara, H.; et al. Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br. J. Cancer 2012, 106, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Lu, C.; Chen, Y.; Paller, C.J.; Carducci, M.A.; Eisenberger, M.A.; Luo, J.; Antonarakis, E.S. Serial blood-based analysis of AR-V7 in men with advanced prostate cancer. Ann. Oncol. 2015, 26, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Canepa, H.M.; Bailey, A.S.; Nakayama, S.; Yamaguchi, N.; Goldstein, M.A.; Huberman, M.S.; Costa, D.B. Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J. Thorac. Oncol. 2013, 8, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Markou, A.; Farkona, S.; Schiza, C.; Efstathiou, T.; Kounelis, S.; Malamos, N.; Georgoulias, V.; Lianidou, E. PIK3CA mutational status in circulating tumor cells can change during disease recurrence or progression in patients with breast cancer. Clin. Cancer Res. 2014, 20, 5823–5834. [Google Scholar] [CrossRef] [PubMed]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular heterogeneity and molecular evolution in cancer. Annu. Rev. Pathol. 2013, 8, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Itzkovitz, S.; van Oudenaarden, A. Validating transcripts with probes and imaging technology. Nat. Methods 2011, 8 (Suppl. 4), S12–S19. [Google Scholar] [CrossRef] [PubMed]
- Summersgill, B.; Clark, J.; Shipley, J. Fluorescence and chromogenic in situ hybridization to detect genetic aberrations in formalin-fixed paraffin embedded material, including tissue microarrays. Nat. Protoc. 2008, 3, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Gonen, M.; Kim, H.J.; Michor, F.; Polyak, K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J. Clin. Investig. 2010, 120, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Fiegler, H.; Geigl, J.B.; Langer, S.; Rigler, D.; Porter, K.; Unger, K.; Carter, N.P.; Speicher, M.R. High resolution array-CGH analysis of single cells. Nucleic Acids Res. 2007, 35, e15. [Google Scholar] [CrossRef] [PubMed]
- Oostlander, A.E.; Meijer, G.A.; Ylstra, B. Microarray-based comparative genomic hybridization and its applications in human genetics. Clin. Genet. 2004, 66, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Ferrante, T.C.; Terry, R.; Turczyk, B.M.; Yang, J.L.; Lee, H.S.; Aach, J.; et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 2015, 10, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Liu, L.; Almendro, V.; Kuang, Y.; Paweletz, C.; Sakr, R.A.; Weigelt, B.; Hanker, A.B.; Chandarlapaty, S.; King, T.A.; et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat. Genet. 2015, 47, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Setou, M.; Shrivas, K.; Sroyraya, M.; Yang, H.; Sugiura, Y.; Moribe, J.; Kondo, A.; Tsutsumi, K.; Kimura, Y.; Kurabe, N.; et al. Developments and applications of mass microscopy. Med. Mol. Morphol. 2010, 43, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Hicks, J. Future medical applications of single-cell sequencing in cancer. Genome Med. 2011, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Nelson, P.; Fang, M. Genomic profiling defines subtypes of prostate cancer with the potential for therapeutic stratification. Clin. Cancer Res. 2013, 19, 4058–4066. [Google Scholar] [CrossRef] [PubMed]
- Thege, F.I.; Lannin, T.B.; Saha, T.N.; Tsai, S.; Kochman, M.L.; Hollingsworth, M.A.; Rhim, A.D.; Kirby, B.J. Microfluidic immunocapture of circulating pancreatic cells using parallel EpCAM and MUC1 capture: Characterization, optimization and downstream analysis. Lab Chip 2014, 14, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.R.; Pantel, K.; Riethdorf, S. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser capture microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tang, F. Single-cell sequencing in stem cell biology. Genome Biol. 2016, 17, 71. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Farlik, M.; Sheffield, N.C.; Nuzzo, A.; Datlinger, P.; Schonegger, A.; Klughammer, J.; Bock, C. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015, 10, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Doherty, R.; Couldrey, C. Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: A technical assessment. Front. Genet. 2014, 5, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, T.J.; Beck, S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods 2015, 72, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Wlodkowic, D.; Darzynkiewicz, Z. Rise of the micromachines: Microfluidics and the future of cytometry. Methods Cell Biol. 2011, 102, 105–125. [Google Scholar] [PubMed]
- Werner, M.; Merenda, F.; Piguet, J.; Salathe, R.P.; Vogel, H. Microfluidic array cytometer based on refractive optical tweezers for parallel trapping, imaging and sorting of individual cells. Lab Chip 2011, 11, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Miller, A.M.; Brogi, E.; Sui, Y.; Armenia, J.; McDonough, E.; Santamaria-Pang, A.; Carlin, S.; Stamper, A.; Campos, C.; et al. Multiplexed immunofluorescence delineates proteomic cancer cell states associated with metabolism. JCI Insight 2016, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hou, Y.; Yin, X.; Bao, L.; Tang, A.; Song, L.; Li, F.; Tsang, S.; Wu, K.; Wu, H.; et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 2012, 148, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Technologies for detection of circulating tumor cells: Facts and vision. Lab Chip 2014, 14, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc. Natl. Acad. Sci. USA 2014, 111, 17947–17952. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.Y.; Fraley, C.; Murua, A.; Raftery, A.E.; Ruzzo, W.L. Model-based clustering and data transformations for gene expression data. Bioinformatics 2001, 17, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. Cancer genomics: One cell at a time. Genome Biol. 2014, 15, 452. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [PubMed]
- Jiang, R.; Lu, Y.T.; Ho, H.; Li, B.; Chen, J.F.; Lin, M.; Li, F.; Wu, K.; Wu, H.; Lichterman, J.; et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget 2015, 6, 44781–44793. [Google Scholar] [PubMed]
- Powell, A.A.; Talasaz, A.H.; Zhang, H.; Coram, M.A.; Reddy, A.; Deng, G.; Telli, M.L.; Advani, R.H.; Carlson, R.W.; Mollick, J.A.; et al. Single cell profiling of circulating tumor cells: Transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE 2012, 7, e33788. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.E.; Scott, J.H.; Wolf, D.M.; Novak, P.; Punj, V.; Magbanua, M.J.; Zhu, W.; Mineyev, N.; Haqq, C.M.; Crothers, J.R.; et al. Expression profiling of circulating tumor cells in metastatic breast cancer. Breast Cancer Res. Treat. 2015, 149, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Masterman-Smith, M.D.; Graham, N.A.; Jiao, J.; Mottahedeh, J.; Laks, D.R.; Ohashi, M.; DeJesus, J.; Kamei, K.; Lee, K.B.; et al. A microfluidic platform for systems pathology: Multiparameter single-cell signaling measurements of clinical brain tumor specimens. Cancer Res. 2010, 70, 6128–6138. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, N.; Flores, N.J.; Irish, J.M.; Simonds, E.F.; Sakai, D.S.; Archambeault, S.; Diaz-Flores, E.; Coram, M.; Shannon, K.M.; Nolan, G.P.; et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell 2008, 14, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Irish, J.M.; Myklebust, J.H.; Alizadeh, A.A.; Houot, R.; Sharman, J.P.; Czerwinski, D.K.; Nolan, G.P.; Levy, R. B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 12747–12754. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.L.; Evensen, E.; Huang, Y.W.; Cesano, A.; Nolan, G.P.; Fantl, W.J. Association of reactive oxygen species-mediated signal transduction with in vitro apoptosis sensitivity in chronic lymphocytic leukemia B cells. PLoS ONE 2011, 6, e24592. [Google Scholar] [CrossRef] [PubMed]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Simonds, E.F.; Bendall, S.C.; Gibbs, K.D., Jr.; Bruggner, R.V.; Linderman, M.D.; Sachs, K.; Nolan, G.P.; Plevritis, S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011, 29, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Qiu, P.; Zeng, Z.; Jorgensen, J.L.; Mak, D.H.; Burks, J.K.; Schober, W.; McQueen, T.J.; Cortes, J.; Tanner, S.D.; et al. Single-cell mass cytometry reveals intracellular survival/proliferative signaling in FLT3-ITD-mutated AML stem/progenitor cells. Cytom. A 2015, 87, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.L.; Rader, J.; Ruden, J.; Rappaport, E.F.; Hunter, K.N.; Hallberg, P.L.; Krytska, K.; O’Dwyer, P.J.; Mosse, Y.P. Dielectrophoretic capture and genetic analysis of single neuroblastoma tumor cells. Front. Oncol. 2014, 4, 201. [Google Scholar] [CrossRef] [PubMed]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Murlidhar, V.; Ramnath, N.; Nagrath, S.; Reddy, R.M. Optimizing the Detection of Circulating Markers to Aid in Early Lung Cancer Detection. Cancers (Basel) 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Martelotto, L.G.; Ng, C.K.; Piscuoglio, S.; Weigelt, B.; Reis-Filho, J.S. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014, 16, 210. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015, 33, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, M.J.; Sood, A.; Sevinsky, C.; Pris, A.D.; Zavodszky, M.I.; Ginty, F. Emerging understanding of multiscale tumor heterogeneity. Front. Oncol. 2014, 4, 366. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Applications | Main Advantages and Drawbacks | References |
|---|---|---|---|
| Immunohistochemistry (IHC) and Immunofluorescence (IF) | Protein | - Preserved tissue context - Difficult to quantify and to compare between samples - Limitation in the number of analysed markers | [69,70] |
| Fluorescence in situ Hybridization (FISH) | DNA or RNA | ||
| Immuno-FISH | Genome imbalances and DNA translocations + antigenic markers | - High sensitivity and specificity - Reproducibility - Can be easily automated - Dependent on available probes - No available for high throughput - Only for the analysis of low cell density tumours | [71] |
| Comparative Genomic Hybridization Array (a-CGH) | DNA copy number variations | - High resolution - Analysis of whole genome - High specificity and sensitivity - Fast technique - Detects only, the copy number changes - No detection mosaicism | [72,73] |
| RNAscope | RNA | - Compatible with clinical routine practice - Multiple RNA probes can be used at the same time - High sensitivity and specificity - High time-consuming - Complex procedures | [74] |
| Fluorescent in situ Sequencing (FISSEQ) | mRNA | - Allow the detection of RNA splicing and post-transcriptional modifications (with preservation of their spatial context) - Discrimination of RNA with a small number of reads - Expensive equipment for analysing the results | [75] |
| Specific-To-Allele PCR–FISH (STARFISH) | Single nucleotide and DNA copy number alterations | - Relative moderated cost - Easy interpretation - Recommended for suspected mutations - Limited number of fluorochromes - Tissue handling affects mRNA expression | [76] |
| Matrix assisted laser desorption/ionization-imaging mass spectrometry (MALDI-IMS) | Proteins, lipids, metabolites | - Low amount of material can be analysed - Keep the spatial localization information - High sensitivity and molecular specificity - Require accurate sample - Difficulty to control the methods of preparation | [77] |
| Whole Genome Sequencing (WGS) | DNA: single nucleotide variants, copy number variants, non-coding and structural variants | - Single-base resolution - Deliver large volumes of data in a short amount of time - Suitable for discovering of new markers - Require high skills for data handling and interpretation - Relatively high cost | [78,79,80,81,82,83,84] |
| Whole Transcriptome Sequencing (WTS) | mRNA | - High throughput analyses - Single-base resolution - Low amount of sample required - Sample handling must be accurate - Complex procedures | [85,86] |
| Multiplexed error-robust FISH (MERFISH) | RNA | - Conservation of the cell spatial information - High throughput analyses - Predesigned probes (limited discovery capacity) | [87] |
| Chromatin ImmunoPrecipitation Sequencing (ChIP-Seq) | DNA/protein binding, histone marks | - Single nucleotide resolution - Repetitive regions in the genome can be analysed - Require large amounts of starting material - Low sensitivity and high technical read variance - Relatively high cost | [88] |
| Whole Genome Bisulfite Sequencing (WGBS) | Methylation of whole genome | - Low quantity of starting material - Relatively high cost | [89] |
| Reduced Representation Bisulfite Sequencing (RRBS) | Methylation of whole genome | - High throughput analyses - Low amounts of starting material - Multiple step procedure (Risk: accumulation of errors) | [90] |
| Flow Cytometry into lab-on-a-chip | Protein | - Quantitative technic - High throughput analyses - Multiparameter measurements - Some pathways can be disrupted when cells are taken out of context | [91,92,93] |
| Mass Cytometry (CyTOF) | Protein | - No spectral overlap of detectors - Up to 32 proteins can be detected simultaneously - Slow acquisition - Limited commercially labelled antibodies - Complex data to analyse | [91,92,93] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tellez-Gabriel, M.; Ory, B.; Lamoureux, F.; Heymann, M.-F.; Heymann, D. Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis. Int. J. Mol. Sci. 2016, 17, 2142. https://doi.org/10.3390/ijms17122142
Tellez-Gabriel M, Ory B, Lamoureux F, Heymann M-F, Heymann D. Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis. International Journal of Molecular Sciences. 2016; 17(12):2142. https://doi.org/10.3390/ijms17122142
Chicago/Turabian StyleTellez-Gabriel, Marta, Benjamin Ory, Francois Lamoureux, Marie-Francoise Heymann, and Dominique Heymann. 2016. "Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis" International Journal of Molecular Sciences 17, no. 12: 2142. https://doi.org/10.3390/ijms17122142