
Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment

Abstract
:1. Introduction
2. Ovarian Cancer Screening
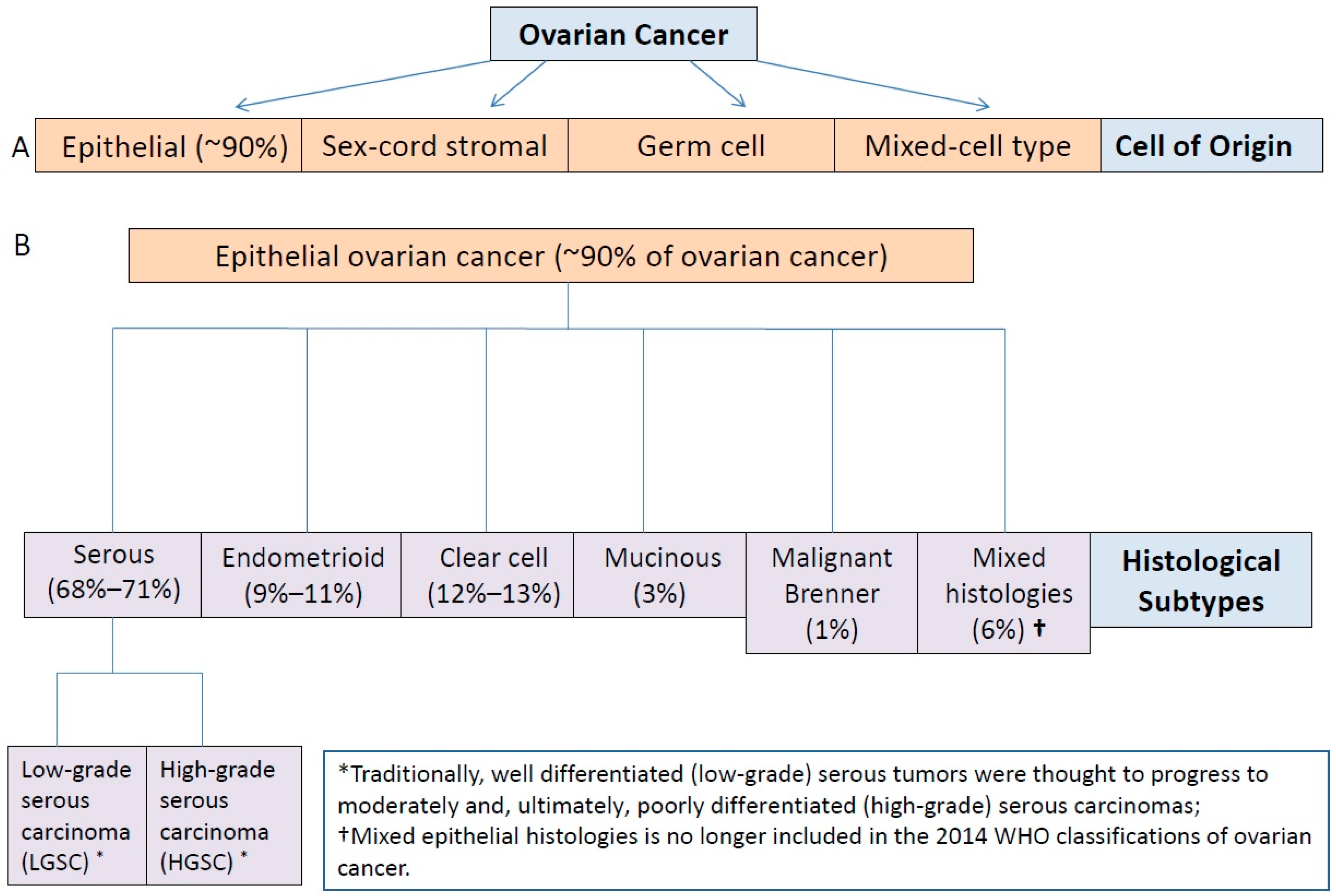
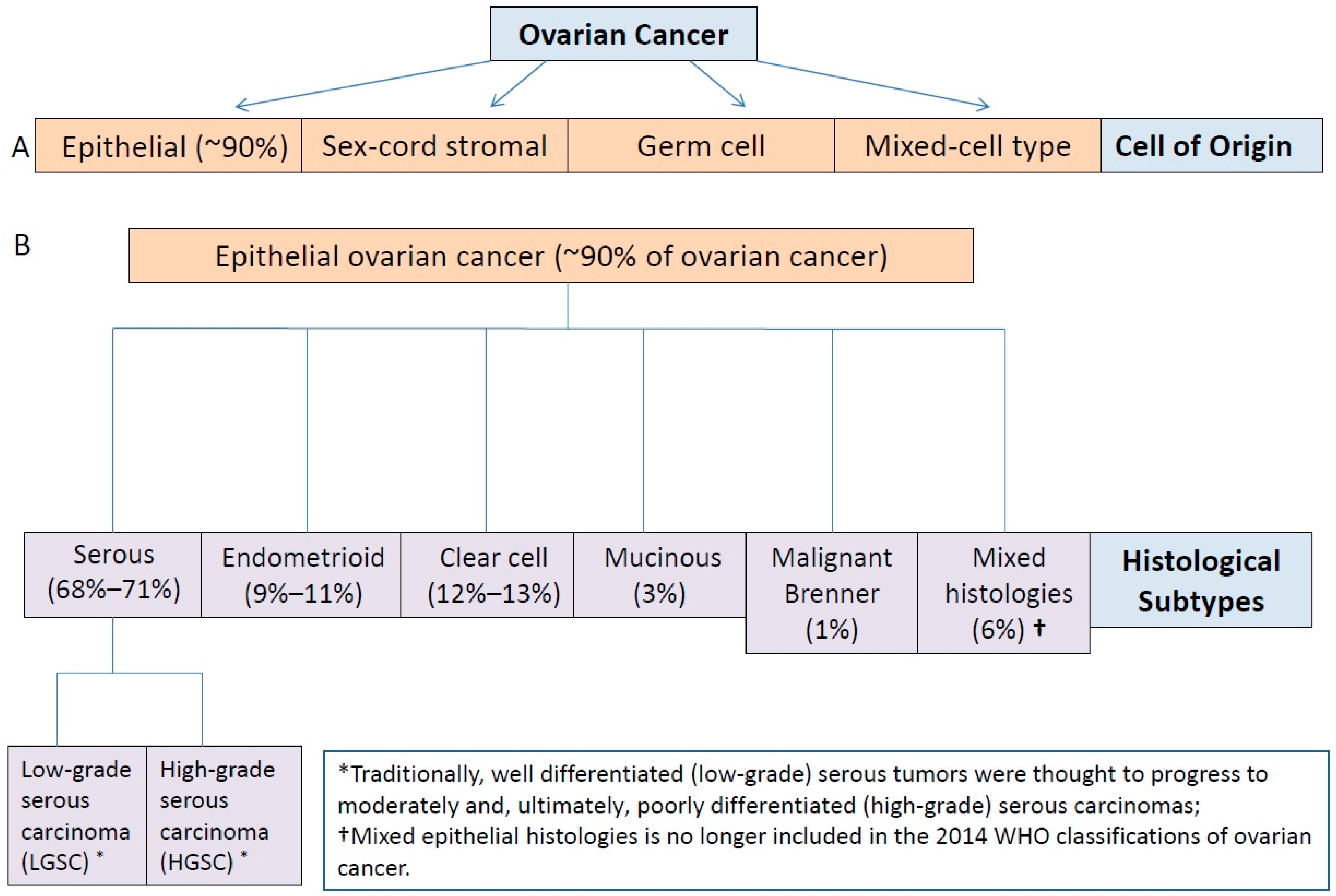
3. Ovarian Cancer as a Heterogeneous Disease
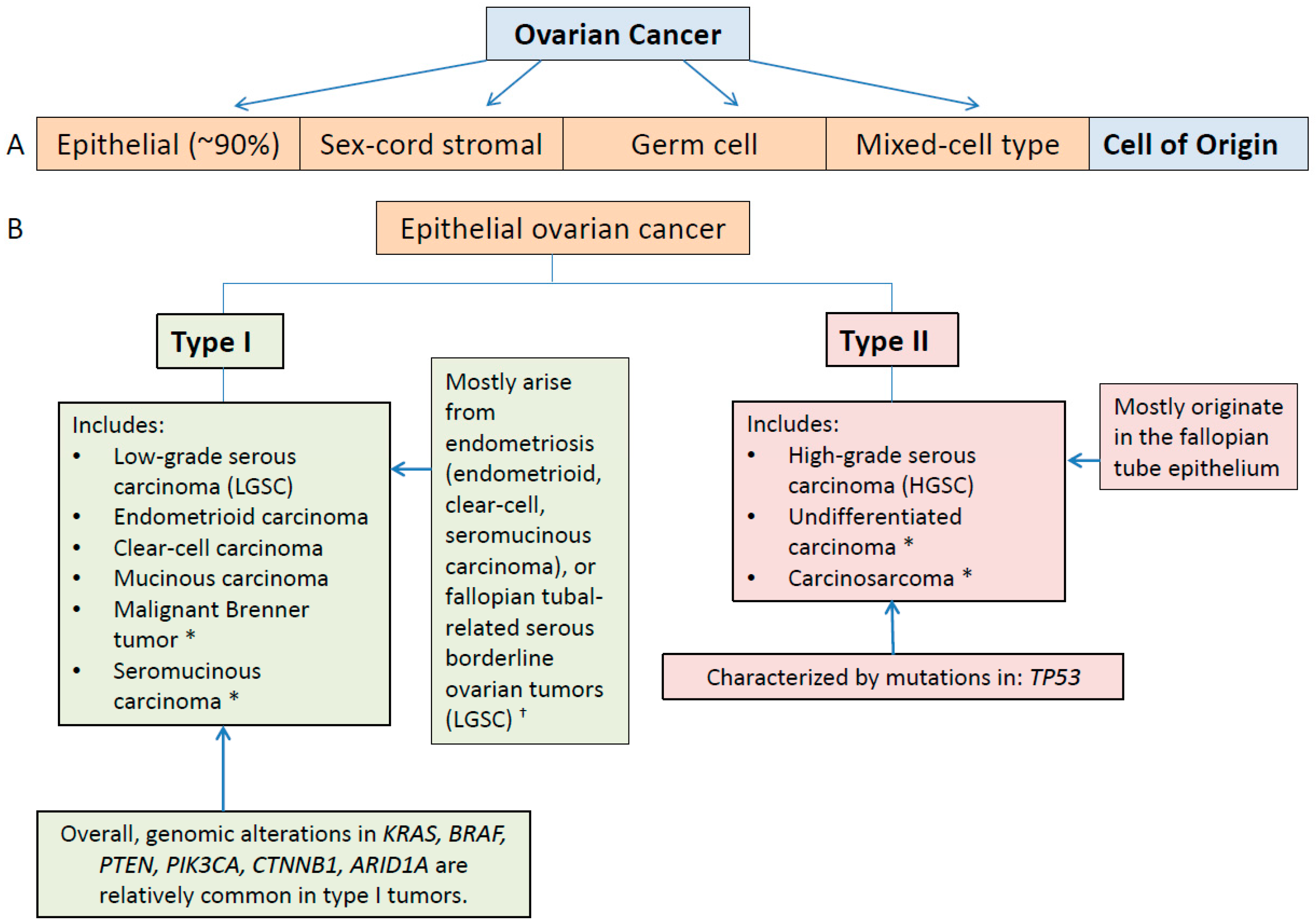
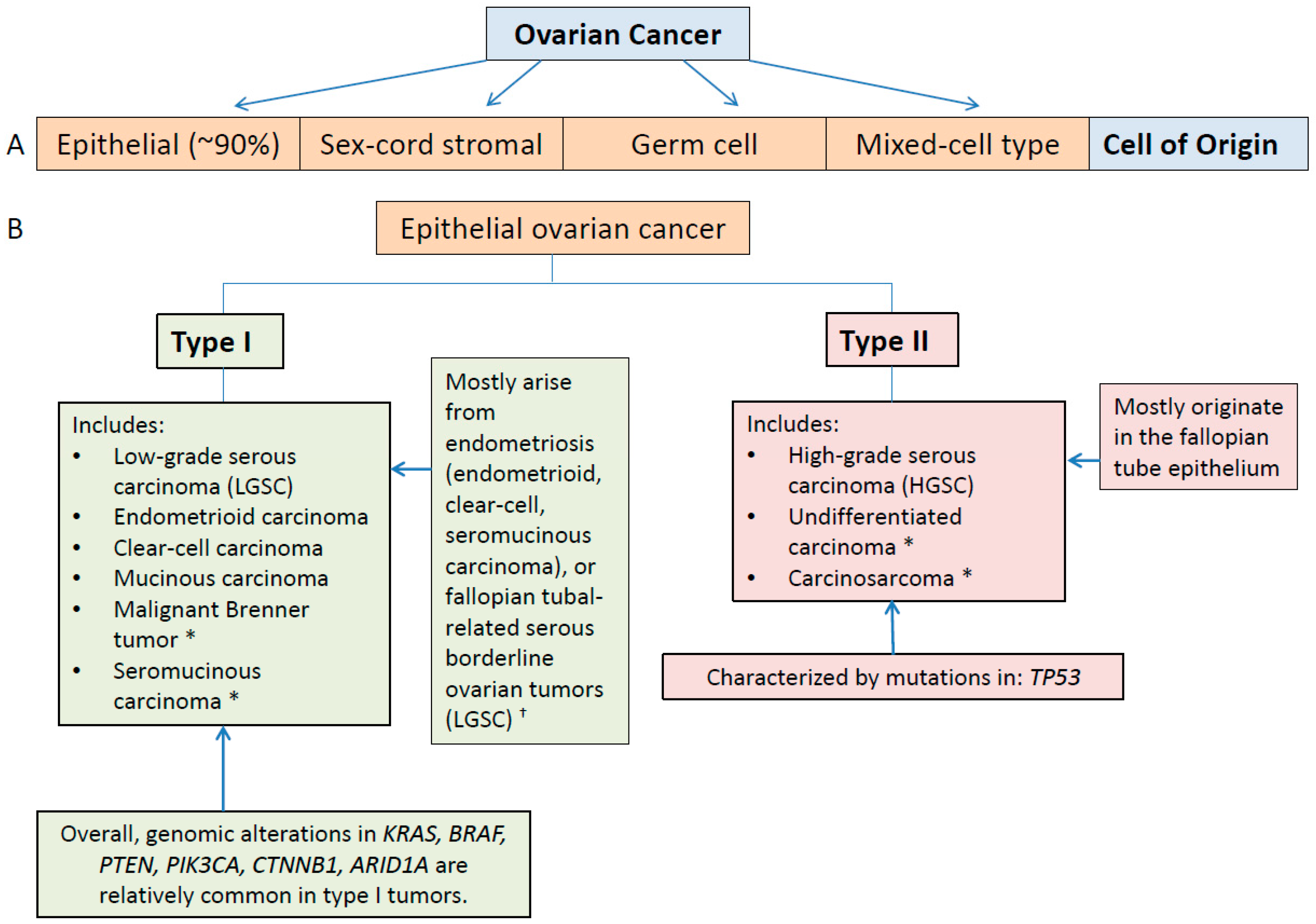
3.1. Type I and II Epithelial Ovarian Cancers
3.2. Molecular Classification and Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis
3.3. Inter- and Intratumoral Heterogeneity in Ovarian Cancer
3.4. Molecular Classification of Chemoresistant Epithelial Ovarian Cancer
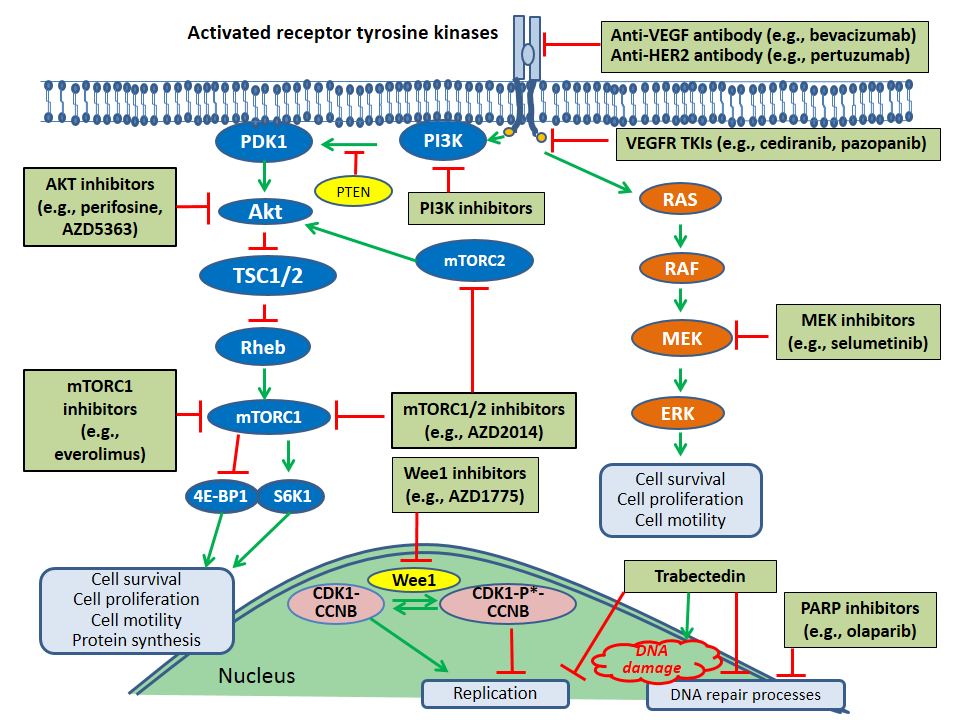
4. Implications of the Molecular Characterization of Epithelial Ovarian Cancer for Selection of Targeted Therapy
4.1. Molecular Targets for Therapy in Ovarian Cancer
4.1.1. BRCA1/2 Mutations
4.1.2. VEGF Pathway
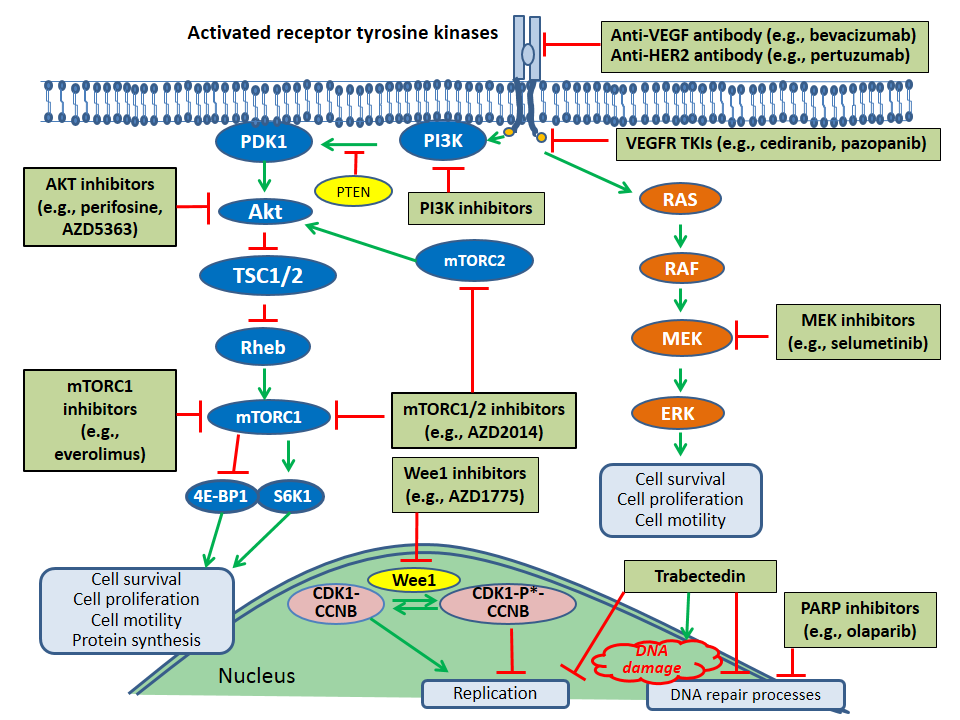
4.1.3. PI3K/Akt/mTOR Pathway
4.1.4. TP53 Mutations
4.1.5. RAS/RAF/MEK/ERK Pathway
4.1.6. Other Receptor Tyrosine Kinase Pathways: HER2
4.1.7. MicroRNAs
4.1.8. Immunologic Pathways
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Global Cancer Facts & Figures, 3rd Edition. Available online: http://www.cancer.org/acs/groups/content/@research/documents/document/acspc-044738.pdf (accessed on 12 December 2016).
- National Comprehensive Cancer Network (NCCN). Ovarian Cancer Guidelines V1 2016. Available online: https://www.nccn.org/ (accessed on 1 July 2016).
- Bell, R.; Petticrew, M.; Luengo, S.; Sheldon, T.A. Screening for ovarian cancer: A systematic review. Health Technol. Assess. 1998, 2, 1–84. [Google Scholar]
- Buys, S.S.; Partridge, E.; Black, A.; Johnson, C.C.; Lamerato, L.; Isaacs, C.; Reding, D.J.; Greenlee, R.T.; Yokochi, L.A.; Kessel, B.; et al. Effect on screening on ovarian cancer mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) cancer screening randomized controlled trial. JAMA 2011, 305, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, P.F.; Yu, K.; Kramer, B.S.; Black, A.; Buys, S.S.; Partridge, E.; Gohagan, J.; Berg, C.D.; Prorok, P.C. Extended mortality results for ovarian cancer screening in the PLCO trial with median 15 years follow up. Gynecol. Oncol. 2016, 143, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (ULCTOCS): A randomized controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef]
- Blagden, S.P. Harnessing Pandemonium: The clinical implications of tumor heterogeneity in ovarian cancer. Front. Oncol. 2015, 5, 149. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. Morphological subtypes of ovarian carcinoma: A review with emphasis on new developments and pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Meinhold-Heerlein, I.; Fotopoulou, C.; Harter, P.; Kurzeder, C.; Mustea, A.; Wimberger, P.; Hauptmann, S.; Sehouli, J. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch. Gynecol. Obstet. 2016, 293, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kamura, T.; Kigawa, J.; Terakawa, N.; Kikuchi, Y.; Kita, T.; Suzuki, M.; Sato, I.; Taguchi, K. Clinical characteristics of clear cell carcinoma of the ovary: A distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer 2000, 88, 2584–2589. [Google Scholar] [CrossRef]
- Itamochi, H.; Kigawa, J.; Terakawa, N. Mechanisms of chemoresistance and poor prognosis in ovarian clear cell carcinoma. Cancer Sci. 2008, 99, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Horkayne-Szakaly, I.; Haiba, M.; Boice, C.R.; Kurman, R.J.; Ronnett, B.M. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 2004, 23, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Kobel, M.; Kalloger, S.E.; Huntsman, D.G.; Santos, J.L.; Swenerton, K.D.; Seidman, J.D.; Gilks, C.B.; on behalf of Cheryl Brown Ovarian Cancer Outcomes Unit of the British Columbia Cancer Agency. Differences in tumor type in low-stage versus high-stage ovarian carcinomas. Int. J. Gynecol. Pathol. 2010, 29, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 2013, 24, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [Green Version]
- Ross, J.S.; Ali, S.M.; Wang, K.; Palmer, G.; Yelensky, R.; Lipson, D.; Miller, V.A.; Zajchowski, D.; Shawver, L.K.; Stephens, P.J. Comprehensive genomic profiling of epithelial ovarian cancer by next generation sequencing-based diagnostic assay reveals new routes to targeted therapies. Gynecol. Oncol. 2013, 130, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The dualistic model of ovarian carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Della Pepa, C.; Tonini, G.; Santini, D.; Losito, S.; Pisano, C.; di Napoli, M.; Cecere, S.C.; Gargiulo, P.; Pignata, S. Low grade serous ovarian carcinoma: From the molecular characterization to the best therapeutic strategy. Cancer Treat. Rev. 2015, 41, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Kurman, R.J.; Nakayama, K.; Velculescu, V.E.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; Shih Ie, M. Low-grade serous carcinomas of the ovary contain very few point mutations. J. Pathol. 2012, 226, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Vereczkey, I.; Serester, O.; Dobos, J.; Gallai, M.; Szakacs, O.; Szentirmay, Z.; Toth, E. Molecular characterization of 103 ovarian serous and mucinous tumors. Pathol. Oncol. Res. 2011, 17, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.; Iravani, M.; McCluggage, W.G.; Lambros, M.B.; Milanezi, F.; Mackay, A.; Gourley, C.; Geyer, F.C.; Vatcheva, R.; Millar, J.; et al. Genomic analysis reveals the molecular heterogeneity of ovarian clear cell carcinomas. Clin. Cancer Res. 2011, 17, 1521–1534. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N., Jr.; Birrer, M.J.; Sood, A.K. Early events in the pathogenesis of epithelial ovarian cancer. J. Clin. Oncol. 2008, 26, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.T.; Mao, T.L.; Jones, S.; Veras, E.; Ayhan, A.; Wang, T.L.; Glas, R.; Slamon, D.; Velculescu, V.E.; Kuman, R.J.; et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am. J. Pathol. 2009, 174, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Willner, J.; Wurz, K.; Allison, K.H.; Galic, V.; Garcia, R.L.; Goff, B.A.; Swisher, E.M. Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Hum. Pathol. 2007, 38, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Fadare, O.; Khadbele, D. Molecular profing of epithelial ovarian cancer, My Cancer Genome. Available online: http://www.mycancergenome.org/content/disease/ovarian-cancer (accessed on 27 October 2016).
- Singer, G.; Oldt, R., 3rd; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih Ie, M. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Gemignani, M.L.; Schlaerth, A.C.; Bogomolniy, F.; Barakat, R.R.; Lin, O.; Soslow, R.; Venkatraman, E.; Boyd, J. Role of KRAS and BRAF gene mutations in mucinous ovarian carcinoma. Gynecol. Oncol. 2003, 90, 378–381. [Google Scholar] [CrossRef]
- Cuatrecasas, M.; Villanueva, A.; Matias-Guiu, X.; Prat, J. K-ras mutations in mucinous ovarian tumors: A clinicopathologic and molecular study of 95 cases. Cancer 1997, 79, 1581–1586. [Google Scholar] [CrossRef]
- Hunter, S.M.; Anglesio, M.S.; Ryland, G.L.; Sharma, R.; Chiew, Y.E.; Rowley, S.M.; Doyle, M.A.; Li, J.; Gilks, C.B.; Moss, P.; et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget 2015, 6, 37663–37677. [Google Scholar] [PubMed]
- Bell, D.A. Origins and molecular pathology of ovarian cancer. Mod. Pathol. 2005, 18, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Catasus, L.; Bussaglia, E.; Rodriguez, I.; Gallardo, A.; Pons, C.; Irving, J.A.; Prat, J. Molecular genetic alterations in endometrioid carcinomas of the ovary: Similar frequency of beta-catenin abnormalities but lower rate of microsatellite instability and PTEN alterations than in uterine endometrioid carcinomas. Hum. Pathol. 2004, 35, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Caramia, F.; Li, J.; Rowley, S.M.; Christie, M.; Allan, P.E.; Stephens, A.N.; Bowtell, D.D.; et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; Garcia, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer 2015, 15, 415. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Kommoss, S.; Tolcher, M.C.; Clarke, B.; Galletta, L.; Porter, H.; Damaraju, S.; Fereday, S.; Winterhoff, B.J.; Kalloger, S.E.; et al. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. J. Pathol. 2013, 229, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian fallopian tube, and peritoneal carcinomas. Clin. Cancer. Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Beltrame, L.; di Marino, M.; Fruscio, R.; Calura, E.; Chapman, B.; Clivio, L.; Sina, F.; Mele, C.; Iatropoulos, P.; Grassi, T.; et al. Profiling cancer gene mutations in longitudinal epithelial ovarian cancer biopsies by targeted next-generation sequencing: A retrospective study. Ann. Oncol. 2015, 26, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Paracchini, L.; Mannarino, L.; Craparotta, I.; Romualdi, C.; Fruscio, R.; Grassi, T.; Fotia, V.; Caratti, G.; Perego, P.; Calura, E.; et al. Regional and temporal heterogeneity of epithelial ovarian cancer tumor biopsies: Implications for therapeutic strategies. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih Ie, M.; Kurman, R.J. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: A rereview of cases lacking TP53 mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih Ie, M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Folkins, A.K.; Jarboe, E.A.; Saleemuddin, A.; Lee, Y.; Callahan, M.J.; Drapkin, R.; Garber, J.E.; Muto, M.G.; Tworoger, S.; Crum, C.P. A candidate precursor to pelvic serous cancer (p53 signature) and its prevalence in ovaries and fallopian tubes from women with BRCA mutations. Gynecol. Oncol. 2008, 109, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N. Ovarian surface epithelium as a source of ovarian cancers: Unwarranted speculation or evidence-based hypothesis? Gynecol. Oncol. 2013, 130, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Chene, G.; Ouellet, V.; Rahimi, K.; Barres, V.; Caceres, K.; Meunier, L.; Cyr, L.; de Ladurantaye, M.; Provencher, D.; Mes Masson, A.M. DNA damage signaling and apoptosis in preinvasive tubal lesions of ovarian carcinoma. Int. J. Gynecol. Cancer 2015, 25, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Zeppernick, F.; Meinhold-Heerlein, I.; Shih Ie, M. Precursors of ovarian cancer in the fallopian tube: Serous tubal intraepithelial carcinoma--an update. J. Obstet. Gynaecol. Res. 2015, 41, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V. Next-generation clinical trials: Novel strategies to address the challenge of tumor molecular heterogeneity. Mol. Oncol. 2015, 9, 967–996. [Google Scholar] [CrossRef] [PubMed]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Milne, K.; Zeng, T.; Tse, K.; Mayo, M.; Zhao, Y.; Webb, J.R.; Watson, P.H.; Nelson, B.H.; Holt, R.A. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J. Pathol. 2013, 229, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Cowin, P.A.; George, J.; Fereday, S.; Loehrer, E.; van Loo, P.; Cullinane, C.; Etemadmoghadam, D.; Ftouni, S.; Galletta, L.; Anglesio, M.S.; et al. LRP1B deletion in high-grade serous ovarian cancers is associated with acquired chemotherapy resistance to liposomal doxorubicin. Cancer Res. 2012, 72, 4060–4073. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yoon, J.K.; Kim, B.; Kim, S.; Kim, M.A.; Lim, H.; Bang, D.; Song, Y.S. Tumor evolution and intratumor heterogeneity of an epithelial ovarian cancer investigated using next-generation sequencing. BMC Cancer 2015, 15, 85. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraat, M.; de Pagter, M.S.; Cirkel, G.A.; van Roosmalen, M.J.; Harkins, T.T.; Duran, K.; Kreeftmeijer, J.; Renkens, I.; Witteveen, P.O.; Lee, C.C.; et al. Genomic and transcriptomic plasticity in treatment-naive ovarian cancer. Genome Res. 2014, 24, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, W.; Leng, B.; Zheng, W.; He, Z.; Zuo, M.; Chen, A. Circulating cell free DNA as the diagnostic marker for ovarian cancer: A systematic review and meta-analysis. PLoS ONE 2016, 11, e0155495. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Huang, R.Y.; Wong, M.K.; Ye, J.; Lau, J.A.; Wu, M.C.; Bin Abdul Hadi, L.H.; Soong, R.; Choolani, M.; et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 2013, 5, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Galletta, L.; Etemadmoghadam, D.; George, J.; Australian Ovarian Cancer Study; Kobel, M.; Ramus, S.J.; Bowtell, D. Efficient molecular subtype classification of high-grade serous ovarian cancer. J. Pathol. 2015, 236, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.L.; DeBoever, C.; Jepsen, K.; Saenz, C.C.; Carson, D.A.; Frazer, K.A. Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proc. Natl. Acad. Sci. USA 2015, 112, 3050–3057. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, S.; Smeets, D.; Moisse, M.; Braicu, E.I.; Vanderstichele, A.; Zhao, H.; van Nieuwenhuysen, E.; Berns, E.; Sehouli, J.; Zeillinger, R.; et al. Genetic heterogeneity after first-line chemotherapy in high-grade serous ovarian cancer. Eur. J. Cancer 2016, 53, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Bhang, H.E.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Avastin (bevacizumab). [package insert]. Genentech, Inc.: South San Francisco, CA, 2014. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125085s0169lbl.pdf (accessed on 12 December 2016).
- Lynparza (olaparib). [package insert]. AztraZeneca Pharmaceuticals: Wilmington, DE, 2014. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206162lbl.pdf (accessed on 12 December 2016).
- European Medicines Agency. Available online: http://www.ema.europa.eu/ema/ (accessed on 21 November 2016).
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Arkun, Y. Dynamic modeling and analysis of the cross-talk between Insulin/AKT and MAPK/ERK signaling pathways. PLoS ONE 2016, 11, e0149684. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2014, 25, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Spacek, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2016, 140, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Sill, M.W.; Bell-McGuinn, K.; Aghajanian, C.; Gray, H.J.; Tewari, K.S.; Rubin, S.C.; Rutherford, T.J.; Chan, J.K.; Chen, A.; et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation-An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2015, 137, 386–391. [Google Scholar] [PubMed]
- Liu, J.F.; Matulonis, U.A. What is the place of PARP inhibitors in ovarian cancer treatment? Curr. Oncol. Rep. 2016, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, B.J.; Ghatage, P.; Parekh, T.; Henitz, E.; Knoblauch, R.; Matos-Pita, A.S.; Nieto, A.; Park, Y.C.; Cheng, P.S.; Li, W.; et al. Effect of BRCA1 and XPG mutations on treatment response to trabectedin and pegylated liposomal doxorubicin in patients with advanced ovarian cancer: Exploratory analysis of the phase 3 OVA-301 study. Ann. Oncol. 2015, 26, 914–920. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. NCT01846611. A Study Comparing the Combination of Trabectedin (YONDELIS) and DOXIL/CAELYX With DOXIL/CAELYX for the Treatment of Advanced-Relapsed Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01846611 (accessed on 3 August 2016).
- Siddiqui, G.K.; Maclean, A.B.; Elmasry, K.; Wong Te Fong, A.; Morris, R.W.; Rashid, M.; Begent, R.H.; Boxer, G.M. Immunohistochemical expression of VEGF predicts response to platinum based chemotherapy in patients with epithelial ovarian cancer. Angiogenesis 2011, 14, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Goff, B.; Nycum, L.R.; Wang, Y.V.; Husain, A.; Blank, S.V. Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol. 2015, 139, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.Y.; Urban, R.R.; Liao, J.B.; Goff, B.A. Bevacizumab toxicity in heavily pretreated recurrent epithelial ovarian, fallopian tube, and primary peritoneal cancers. J. Gynecol. Oncol. 2016, 27, 47. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Rhee, J.; Sovak, M.A.; Kong, G.; Nguyen, H.P.; Bookman, M.A. Independent radiologic review of the Gynecologic Oncology Group Study 0218, a phase III trial of bevacizumab in the primary treatment of advanced epithelial ovarian, primary peritoneal, or fallopian tube cancer. Gynecol. Oncol. 2013, 131, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Amadio, G.; Salutari, V.; Paris, I.; di Stefano, M.G.; Ferandina, G.; Scambia, G.; Fagotti, A. Impact of bevacizumab containing first line chemotherapy on recurrent disease in epithelial ovarian cancer: A case-control study. Gynecol. Oncol. 2016, 142, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ferriss, J.S.; Java, J.J.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Walker, J.L.; Homesley, H.D.; Fowler, J.; Greer, B.E.; Boente, M.P.; et al. Ascites predicts treatment benefit of bevacizumab in front-line therapy of advanced epithelial ovarian, fallopian tube and peritoneal cancers: An NRG Oncology/GOG study. Gynecol. Oncol. 2015, 139, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Secord, A.A.; Nixon, A.B.; Hurwitz, H.I. The search for biomarkers to direct antiangiogenic treatment in epithelial ovarian cancer. Gynecol. Oncol. 2014, 135, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Gourley, C.; McCavigan, A.; Perren, T.; Paul, J.; Michie, C.O.; Churchman, M.; Williams, A.; McCluggage, W.G.; Parmar, M.; Kaplan, R.S.; et al. Molecular subgroup of high-grade serous ovarian cancer (HGSOC) as a predictor of outcome following bevacizumab. J. Clin. Oncol. 2014, 32, 5502. [Google Scholar]
- Jackson, A.L.; Eisenhauer, E.L.; Herzog, T.J. Emerging therapies: Angiogenesis inhibitors for ovarian cancer. Expert Opin. Emerg. Drugs 2015, 20, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Embleton, A.C.; Raja, F.; Perren, T.J.; Jayson, G.C.; Rustin, G.J.; Kaye, S.B.; Hirte, H.; Eisenhauer, E.; Vaughan, M.; et al. Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 387, 1066–1074. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I.; et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Behbakht, K.; Sill, M.W.; Darcy, K.M.; Rubin, S.C.; Mannel, R.S.; Waggoner, S.; Schilder, R.J.; Cai, K.Q.; Godwin, A.K.; Alpaugh, R.K. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: A Gynecologic Oncology Group study. Gynecol. Oncol. 2011, 123, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Kurzeder, C.; Schmalfeldt, B.; Neuser, P.; de Gregorio, N.; Pfisterer, J.; Park-Simon, T.W.; Mahner, S.; Schroder, W.; Luck, H.J.; et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol. Oncol. 2016, 140, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Husseinzadeh, N.; Husseinzadeh, H.D. mTOR inhibitors and their clinical application in cervical, endometrial and ovarian cancers: A critical review. Gynecol. Oncol. 2014, 133, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.J.; Choueiri, T.K.; Fay, A.P.; Rini, B.I.; Thorner, A.R.; de Velasco, G.; Tyburczy, M.E.; Hamieh, L.; Albiges, L.; Agarwal, N.; et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2016, 22, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Park, H.S.; Kim, S.; Kim, S.; Ali, S.M.; Greenbowe, J.R.; Yang, I.S.; Kwon, N.J.; Lee, J.L.; Ryu, M.H.; et al. Next-generation sequencing reveals somatic mutations that confer exceptional response to everolimus. Oncotarget 2016, 7, 10547–10556. [Google Scholar] [PubMed]
- ClinicalTrials.gov. NCT01031381. Study of RAD001 and Bevacizumab in Recurrent Ovarian, Peritoneal, and Fallopian Tube Cancer (RADBEV). Available online: https://clinicaltrials.gov/ct2/show/NCT01031381 (accessed on 3 August 2016).
- ClinicalTrials.gov. NCT02188550. Single Arm Trial with Combination of Everolimus and Letrozole in Treatment of Platinum Resistant Relapse or Refractory or Persistant Ovarian Cancer. Endometrial Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02188550 (accessed on 27 October 2016).
- ClinicalTrials.gov. NCT02283658. Everolimus and Letrozole in Treating Patients with Recurrent Hormone Receptor Positive Ovarian, Fallopian Tube, or Primary Peritoneal Cavity Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02283658 (accessed on 27 October 2016).
- ClinicalTrials.gov. NCT01623349. Phase I study of the oral PI3kinase Inhibitor BKM120 or BYL719 and the oral PARP Inhibitor Olaparib in Patients with Recurrent Triple Negative Breast Cancer or High Grade Serous Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01623349 (accessed on 11 August 2016).
- Michalarea, V.; Lorente, D.; Lopez, J. Accelerated phase I trial of 2 schedules of the combination of the PARP inhibitor olaparib and AKT inhibitor AZD5363 using a novel intrapatient dose escalation design in advanced cancer patients. In Proceedings of the American Association for Cancer Research Annual Meeting, Philadelphia, PA, USA, 18–22 April 2015. Abstract Number 8529.
- Fu, S.; Hennessy, B.T.; Ng, C.S.; Ju, Z.; Coombes, K.R.; Wolf, J.K.; Sood, A.K.; Levenback, C.F.; Coleman, R.L.; Kavanagh, J.J.; et al. Perifosine plus docetaxel in patients with platinum and taxane resistant or refractory high-grade epithelial ovarian cancer. Gynecol. Oncol. 2012, 126, 47–53. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. NCT02208375. mTOR1/2 Inhibitor AZD2014 or the Oral AKT Inhibitor AZD5363 for Recurrent Endometrial and ovarian cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02208375 (accessed on 28 October 2016).
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. p53 as a target for the treatment of cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.D.; Rose, S.; Lee, M.A.; et al. Phase II study of Wee1 inhibitor AZD1775 plus carboplatin in patients with p53 mutated ovarian cancer refractory or resistant (<3 months) to standard first-line therapy. J. Clin. Oncol. 2015, 33, 2507. [Google Scholar]
- Oza, A.M.; Weberpals, J.I.; Provencher, D.M.; Grischke, E.-M.; Hall, M.; Uyar, D.; Estevez-Diz, M.D.; Marmé, F.; Kuzmin, A.; Rosenberg, P.; et al. An international, biomarker-directed, randomized, phase II trial of AZD1775 plus paclitaxel and carboplatin (P/C) for the treatment of women with platinum-sensitive TP53-mutant ovarian cancer. J. Clin. Oncol. 2015, 33, 5506. [Google Scholar]
- Mueller, S.; Haas-Kogan, D.A. WEE1 kinase as a target for cancer therapy. J. Clin. Oncol. 2015, 33, 3485–3487. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Beijnen, J.H.; Schellens, J.H. Abrogation of the G2 checkpoint by inhibition of Wee-1 kinase results in sensitization of p53-deficient tumor cells to DNA-damaging agents. Curr. Clin. Pharmacol. 2010, 5, 86–91. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. NCT0166400. A Safety, Pharmacokinetic and Pharmacodynamic Study of Kevetrin in Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT01664000 (accessed on 28 November 2016).
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. NCT02012192. GANNET53: Ganetespib in Metastatic, p.53 Mutant, Platinum-Resitant Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02012192 (accessed on 11 August 2016).
- Farley, J.; Brady, W.E.; Vathipadiekal, V.; Lankes, H.A.; Coleman, R.; Morgan, M.A.; Mannel, R.; Yamada, S.D.; Mutch, D.; Rodgers, W.H.; et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet Oncol. 2013, 14, 134–140. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. NCT01849874. A Study of MEK162 vs. Physician’s Choice Chemotherapy in Patients with Low-Grade Serous Ovarian, Fallopian Tube or Peritoneal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01849874 (accessed on 27 October 2016).
- MEK Inhibitor Misses Mark in Phase III Ovarian Cancer Study. Available online: http://www.onclive.com/web-exclusives/mek-inhibitor-misses-mark-in-phase-iii-ovarian-cancer-study (accessed on 29 July 2016).
- ClinicalTrials.gov. NCT02101788. Trametinib in Treating Patients with Recurrent or Progressive Low-Grade Ovarian Cancer or Peritoneal Cavity Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02101788 (accessed on 29 July 2016).
- Teplinsky, E.; Muggia, F. Targeting HER2 in ovarian and uterine cancers: challenges and future directions. Gynecol. Oncol. 2014, 135, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Bookman, M.A.; Darcy, K.M.; Clarke-Pearson, D.; Boothby, R.A.; Horowitz, I.R. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: A phase II trial of the Gynecologic Oncology Group. J. Clin. Oncol. 2003, 21, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.P.; Faratian, D.; Nagumo, Y.; Mullen, P.; Harrison, D.J. Pertuzumab for the treatment of ovarian cancer. Expert Opin. Biol. Ther. 2010, 10, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.A.; Sill, M.W.; Lankes, H.A.; Godwin, A.K.; Mannel, R.S.; Armstrong, D.K.; Carolla, R.L.; Liepman, M.K.; Spirtos, N.M.; Fischer, E.G.; et al. A phase II evaluation of lapatinib in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2012, 124, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Poole, C.J.; Danska-Bidzinska, A.; Gianni, L.; del Conte, G.; Gorbunova, V.; Novikova, E.; Strauss, A.; Moczko, M.; McNally, V.A.; et al. A randomized phase II study evaluating the combination of carboplatin-based chemotherapy with pertuzumab versus carboplatin-based therapy alone in patients with relapsed, platinum-sensitive ovarian cancer. Ann. Oncol. 2013, 24, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Makhija, S.; Amler, L.C.; Glenn, D.; Ueland, F.R.; Gold, M.A.; Dizon, D.S.; Paton, V.; Lin, C.Y.; Januario, T.; Ng, K.; et al. Clinical activity of gemcitabine plus pertuzumab in platinum-resistant ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. J. Clin. Oncol. 2010, 28, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Mahdian-Shakib, A.; Dorostkar, R.; Tat, M.; Hashemzadeh, M.S.; Saidi, N. Differential role of microRNAs in prognosis, diagnosis, and therapy of ovarian cancer. Biomed. Pharmacother. 2016, 84, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Miles, G.D.; Seiler, M.; Rodriguez, L.; Rajagopal, G.; Bhanot, G. Identifying microRNA/mRNA dysregulations in ovarian cancer. BMC Res. Notes 2012, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, M.; Canevan, S.; Califano, D.; Losito, S.; Maio, M.D.; Raspagliesi, F.; Carcangiu, M.L.; Toffoli, G.; Cecchin, E.; Sorio, R.; et al. Development and validation of a microRNA-based signature (MiROvaR) to predict early relapse or progression of epithelial ovarian cancer: A cohort study. Lancet 2016, 17, 1137–1146. [Google Scholar] [CrossRef]
- Calura, E.L.; Paracchini, L.R.; Fruscio, R.A.; DiFeo, A.; Ravaggi, A.; Peronne, J.; Martini, P.; Sales, G.; Beltrame, L.; Bignotti, E.; et al. A prognostic regulatory pathway in stage I epithelial ovarian cancer: New hints for the poor prognosis assessment. Ann. Oncol. 2016, 27, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, Y.; Hu, L.; Zheng, H.; Ji, P.; Pecot, C.V.; Zhao, Y.; Reynolds, S.; Cheng, H.; Rupaimoole, R.; et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell 2013, 23, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sawada, K.; Yoshimura, A.; Kinose, Y.; Nakatsuka, E.; Kimura, T. Clinical relevance of circulating cell-free microRNAs in ovarian cancer. Mol. Cancer 2016, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Traver, S.; Assou, S.; Scalici, E.; Haouzi, D.; Al-Edani, T.; Belloc, S.; Hamamah, S. Cell-free nucleic acids as non-invasive biomarkers of gynecologic cancers, ovarian, endometrial and obstetric disorders and fetal aneuploidy. Hum. Reprod. Update 2014, 20, 905–923. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Konishi, I. Immune checkpoint inhibition in ovarian cancer. Int. Immunol. 2016, 28, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Mittica, G.; Genta, S.; Aglietta, M.; Valabrega, G. Immune checkpoint inhibitors: a new opportunity in the treatment of ovarian cancer? Int. J. Mol. Sci. 2016, 17, 1169. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.R.; Wang, J.; Silk, A.W.; Ganesan, S.; Kaufman, H.L.; Mehnert, J.M. Biomarkers for immunotherapy: Current developments and challenges. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 493–503. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. NCT02718417. A Randomized, Open-LABEL., Multicenter, Phase 3 Study to Evaluate the Efficacy and Safety of Avelumab (msb0010718c) in Combination with and/or Following Chemotherapy in Patients with Previously Untreated Epithelial Ovarian Cancer, J Javelin Ovarian 100. Available online: https://clinicaltrials.gov/ct2/show/NCT02718417 (accessed on 9 September 2016).
- Coukos, G.; Tanyi, J.; Kandalaft, L.E. Opportunities in immunotherapy of ovarian cancer. Ann. Oncol. 2016, 27, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Minko, T.; Rodriguez-Rodriguez, L.; Pozharov, V. Nanotechnology approaches for personalized treatment of multidrug resistant cancers. Adv. Drug Deliv. Rev. 2013, 65, 1880–1895. [Google Scholar] [CrossRef] [PubMed]
- Molecular Analysis for Therapy Choice (NCI- MATCH Trial). Available online: http://www.cancer.gov/about-cancer/treatment/clinical-trials/nci-supported/nci-match (accessed on 14 August 2016).
- Lung-MAP Trial. Available online: http://www.lung-map.org/ (accessed on 14 August 2016).

{kind=link}
{kind=link}
{kind=link}
| Gene Alterations | Low-Grade Serous Cancer | Ovarian Clear Cell Carcinoma | Endometrioid | Mucinous |
|---|---|---|---|---|
| Mutations | ||||
| BRAF | 33% a; 38% b; 16% c | 0% e; 1% f | 24% a | 0% k; 23% l; 5% m; |
| KRAS | 19% b; 35% a; 21% c | <1% a; 7% f | <1% a | 50% k; 68% n; 65% m |
| PIK3CA | 11% b | 25% e; 33% f | 12% e | 14% m |
| PTEN | 20% d | 0% e; 5% f | 14% j; 31% e | 3% m |
| ARID1A | -- | 46% g; 57% h | 30% g | 9% l |
| CTNNB1 | -- | 0% e; 3% f | 23% e; 24% j | 5% m |
| CDKN2A | -- | -- | -- | 19% m |
| TP53 | -- | -- | -- | 57% m; 52% l |
| Copy number alterations | ||||
| ERBB2 (HER2; gain) | -- | 14% i | -- | 12% m; 19% o |
| Gene | Frequency of Mutations | Frequency of Copy Number Alterations b |
|---|---|---|
| TP53 | 96% | 0.9% |
| BRCA1 c | 12% | 0.6% |
| BRCA2 | 11% | 2% |
| MYC | 0% | 31% |
| MECOM | 0.6% | 22% |
| CCNE1 | 0% | 20% |
| PRKCI | 0.6% | 19% |
| EIF5A2 | 0% | 18% |
| PIK3CA | 0.6% | 17% |
| NOTCH3 | 0.9% | 11% |
| KRAS | 0.6% | 11% |
| RAB25 | 0% | 7% |
| AKT2 | 0% | 6% |
| AURKA | 0% | 3% |
| PIK3R1 | 0.3% | 2% d |
| AKT1 | 0% | 3% |
| ERBB2 | 0.9% | 2% |
| KIT | 2% | 1% |
| FGF1 | 0% | 1% |
| EGFR | 2% | 0.4% |
| BRAF | 0.6% | 5% |
| PTEN | 0.6% | 6% d |
| RB1 | 2% | 7% d |
| NF1 | 4% | 6% d |
| ETV4 | 0% | 0.5% |
| FOXM1 | 0% | 5% |
| LSR | 0% | 8% |
| CD9 | 0.3% | 6% |
| RAB11FIP4 | 0% | 3% d |
| FGFRL1 | 0% | 3% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. https://doi.org/10.3390/ijms17122113
Rojas V, Hirshfield KM, Ganesan S, Rodriguez-Rodriguez L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. International Journal of Molecular Sciences. 2016; 17(12):2113. https://doi.org/10.3390/ijms17122113
Chicago/Turabian StyleRojas, Veronica, Kim M. Hirshfield, Shridar Ganesan, and Lorna Rodriguez-Rodriguez. 2016. "Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment" International Journal of Molecular Sciences 17, no. 12: 2113. https://doi.org/10.3390/ijms17122113