
Checkpoints to the Brain: Directing Myeloid Cell Migration to the Central Nervous System


Abstract
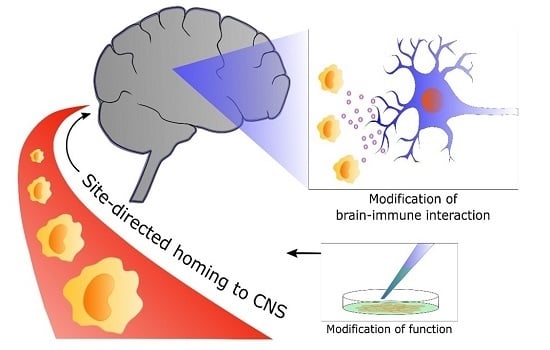
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Contribution of Myeloid Cells to Central Nervous System (CNS) Homeostasis
2.1. The Resident CNS Myeloid Cell, the Microglia
2.2. Non-Microglial Myeloid Cells: Location and Transport in and out of the CNS
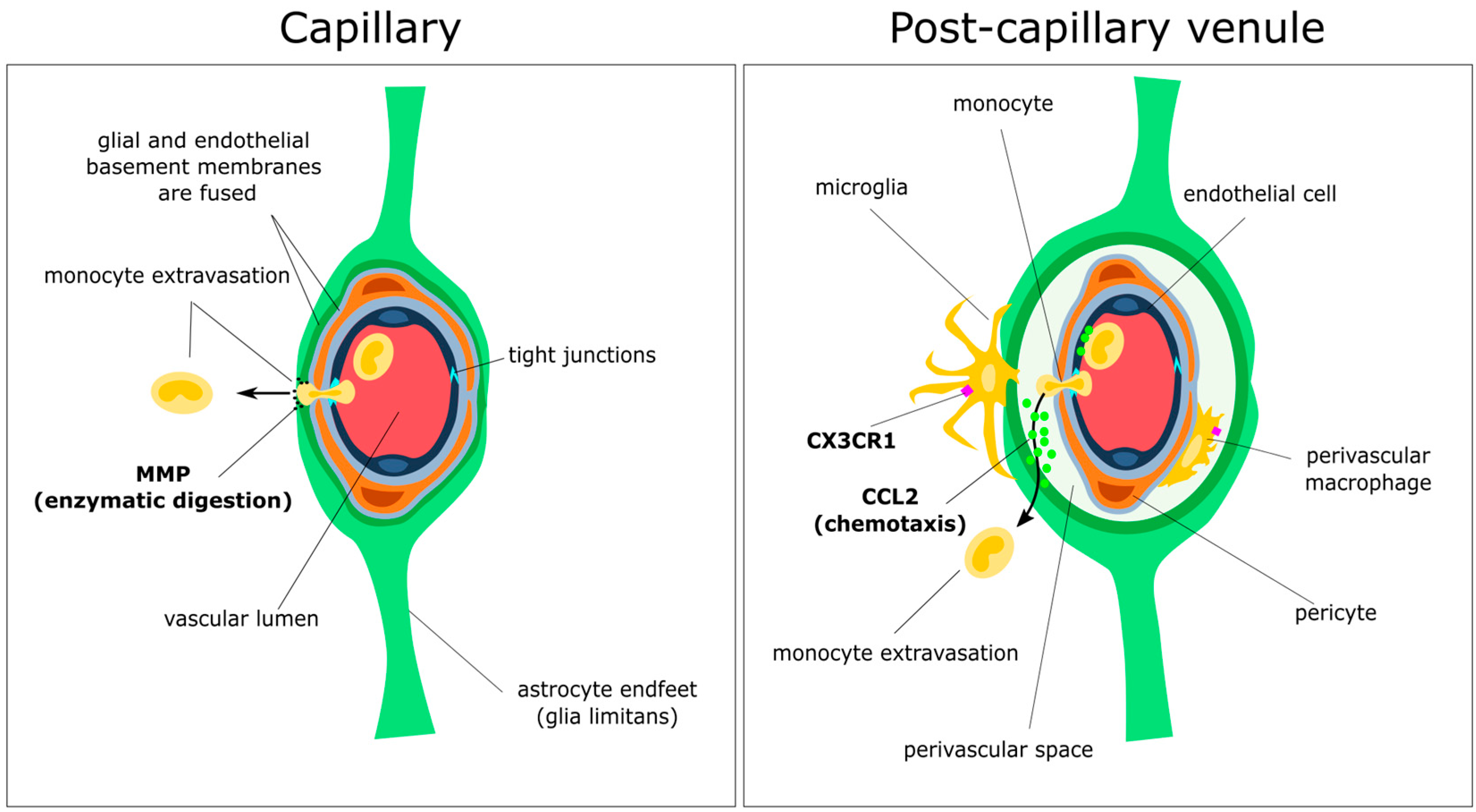
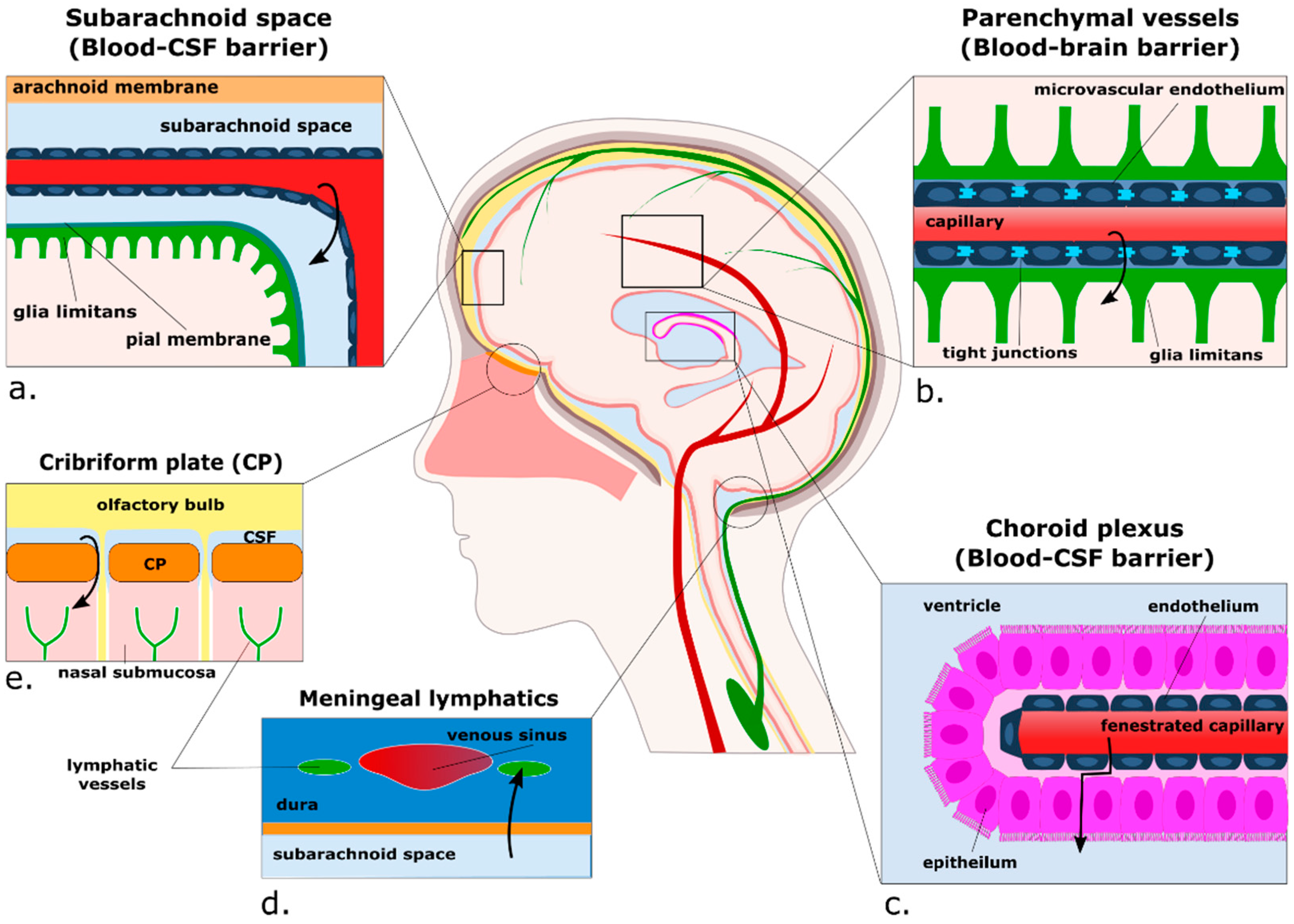
3. Passing the Checkpoints to the Parenchyma: A Focus on the Neurovascular Unit
3.1. Anatomy of the Perivascular Niche
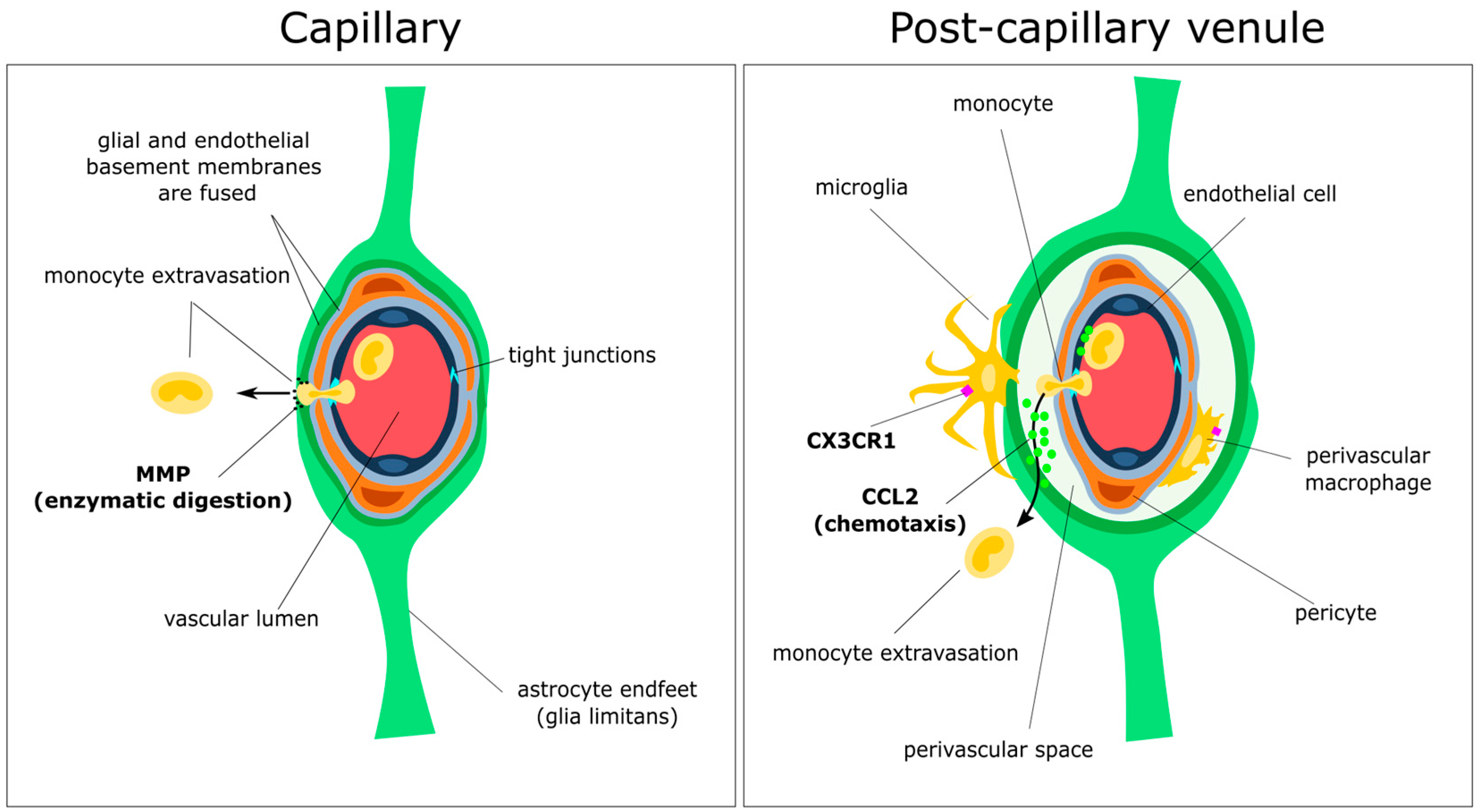
3.2. Initial Attraction: Monocyte–Endothelium Interactions
3.3. Gatekeepers: Astrocytes and the Glia Limitans
3.4. Spotlight on Coordinated Chemotaxis: The CCL2/CCR2 Pathway
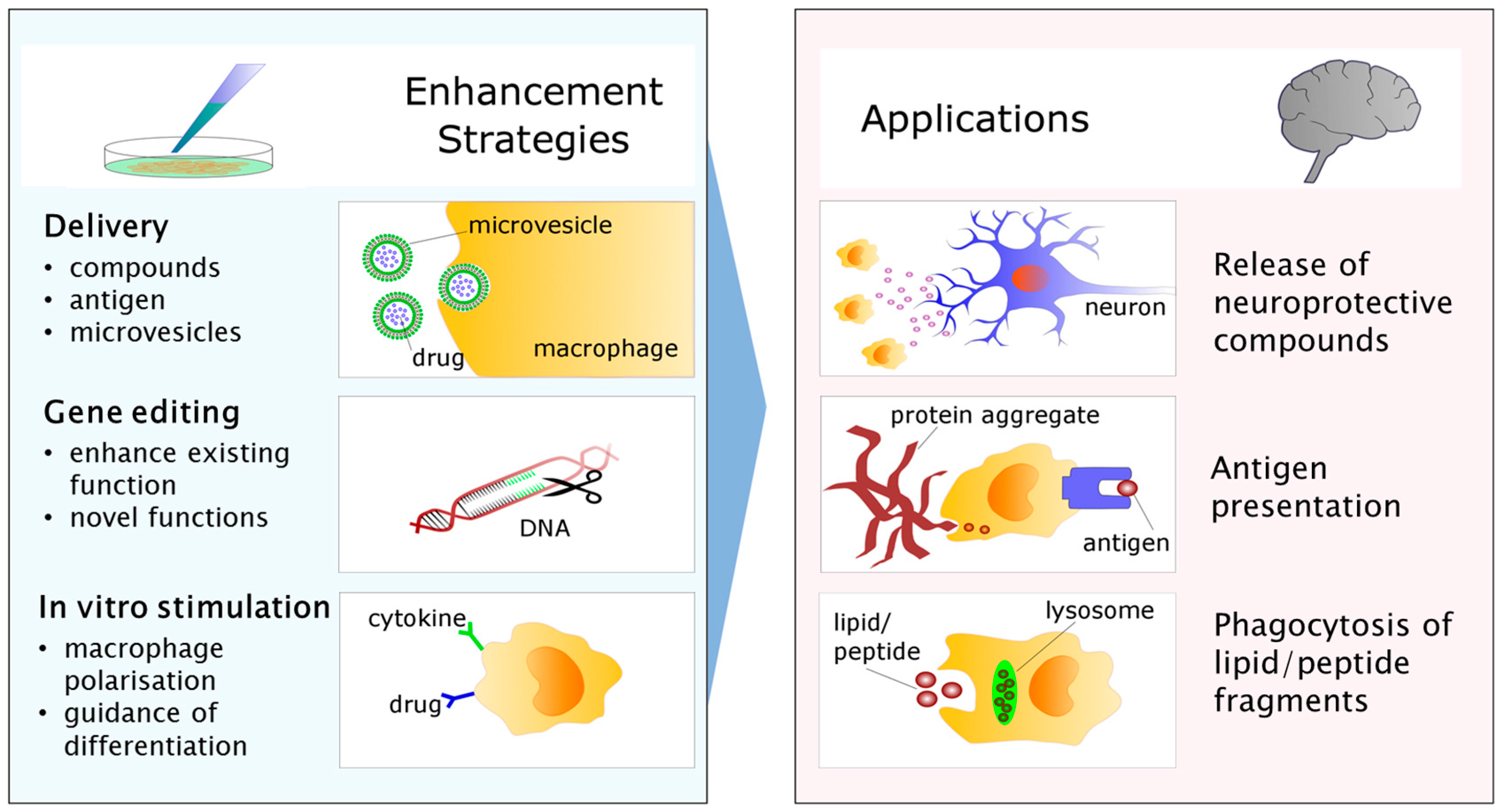
4. Myeloid Cells: The White Knights of CNS Therapy?
4.1. Genetic Deficiencies: Lysosomal/Peroxisomal Storage Diseases
4.2. Neurodegenerative Diseases
4.2.1. Alzheimer’s Disease
4.2.2. Amyotrophic Lateral Sclerosis (ALS)
4.3. Autoimmune Diseases: Multiple Sclerosis
4.4. Brain Tumours: Glioma
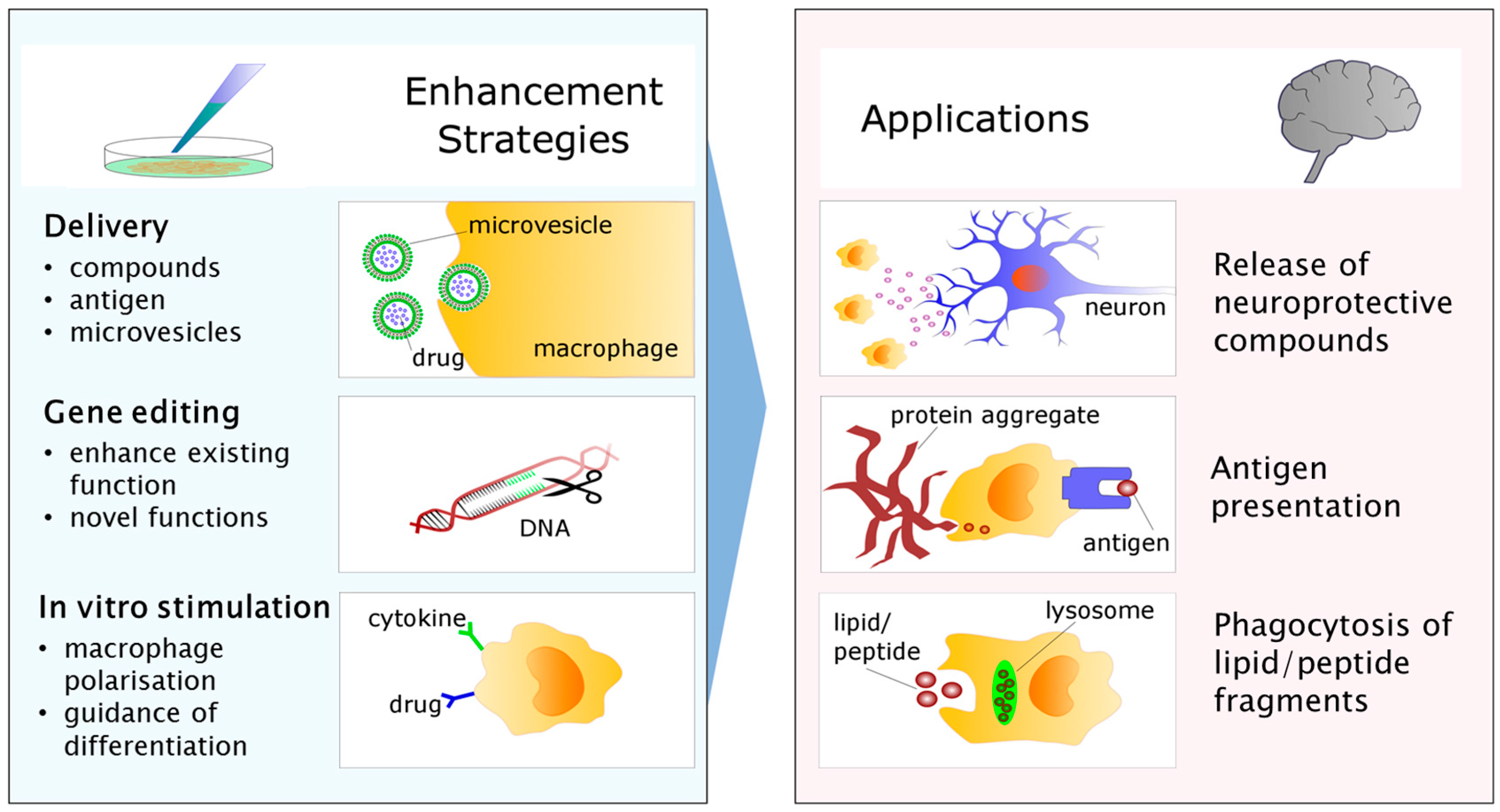
5. From Bench to Bedside: Targets for Future Research
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| BBB | Blood–brain barrier |
| CSF | Cerebrospinal fluid |
| ISF | Interstitial fluid |
| EAE | Experimental autoimmune encephalitis |
| CAA | Cerebral amyloid angiopathy |
| AD | Alzheimer’s disease |
| MS | Multiple sclerosis |
| LSD | Lysosomal storage disease |
| TSPO | Translocator protein (18kDa) |
| HSC | Hematopoietic stem cell |
References
- Collins, P.Y.; Patel, V.; Joestl, S.S.; March, D.; Insel, T.R.; Daar, A.S. Grand challenges in global mental health: A consortium of researchers, advocates and clinicians announces here research priorities for improving the lives of people with mental illness around the world, and calls for urgent action and investment. Nature 2011, 475, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Fineberg, N.A.; Haddad, P.M.; Carpenter, L.; Gannon, B.; Sharpe, R.; Young, A.H.; Joyce, E.; Rowe, J.; Wellsted, D.; Nutt, D.J.; et al. The size, burden and cost of disorders of the brain in the UK. J. Psychopharmacol. 2013, 27, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittchen, H.U.; Jacobi, F.; Rehm, J.; Gustavsson, A.; Svensson, M.; Jönsson, B.; Olesen, J.; Allgulander, C.; Alonso, J.; Faravelli, C.; et al. The size and burden of mental disorders and other disorders of the brain in europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 655–679. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hickey, W.F. Basic principles of immunological surveillance of the normal central nervous system. Glia 2001, 36, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flügel, A.; Laman, J.D.; Weller, R.O. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016, 132, 317–338. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Menasria, R.; Canivet, C.; Piret, J.; Boivin, G. Infiltration pattern of blood monocytes into the central nervous system during experimental herpes simplex virus encephalitis. PLoS ONE 2015, 10, e0145773. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Lelios, I.; Croxford, A.L. Microglia versus myeloid cell nomenclature during brain inflammation. Front. Immunol. 2015, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Lessard, M.; Rivest, S. Effects of myeloablation, peripheral chimerism, and whole-body irradiation on the entry of bone marrow-derived cells into the brain. Cell Transplant. 2012, 21, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Jung, S. Monocytes: Subsets, origins, fates and functions. Curr. Opin. Hematol. 2010, 17, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Brown, B.D.; Merad, M. Studying the mononuclear phagocyte system in the molecular age. Nat. Rev. Immunol. 2011, 11, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Merad, M. Microglia arise from extra-embryonic yolk sac primitive progenitors. Med. Sci. 2011, 27, 719. [Google Scholar]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.-K.; Mack, M.; Heikenwalder, M.; Bruck, W.; Priller, J.; Prinz, M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Katzmarski, N.; Haas, C.A.; Prinz, M. Bone marrow cell recruitment to the brain in the absence of irradiation or parabiosis bias. PLoS ONE 2013, 8, e58544. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Beutner, C.; Linnartz-Gerlach, B.; Schmidt, S.V.; Beyer, M.; Mallmann, M.R.; Staratschek-Jox, A.; Schultze, J.L.; Neumann, H. Unique transcriptome signature of mouse microglia. Glia 2013, 61, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.S.; Ase, A.R.; Kinsara, A.; Rao, V.T.S.; Michell-Robinson, M.; Leong, S.Y.; Butovsky, O.; Ludwin, S.K.; Séguéla, P.; Bar-Or, A.; et al. P2Y12 expression and function in alternatively activated human microglia. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e80. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Xavier, A.L.; Kress, B.T.; Goldman, S.A.; Lacerda de Menezes, J.R.; Nedergaard, M. A distinct population of microglia supports adult neurogenesis in the subventricular zone. J. Neurosci. 2015, 35, 11848–11861. [Google Scholar] [CrossRef] [PubMed]
- Kesler, C.T.; Liao, S.; Munn, L.L.; Padera, T.P. Lymphatic vessels in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Sorokin, L. The blood–brain and the blood–cerebrospinal fluid barriers: Function and dysfunction. Semin. Immunopathol. 2009, 31, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.R.; Perry, V.H.; Weller, R.O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates csf flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Zeppenfeld, D.M.; Venkataraman, A.; Plog, B.A.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral arterial pulsation drives paravascular CSF–interstitial fluid exchange in the murine brain. J. Neurosci. 2013, 33, 18190–18199. [Google Scholar] [CrossRef] [PubMed]
- Shechter, R.; London, A.; Schwartz, M. Orchestrated leukocyte recruitment to immune-privileged sites: Absolute barriers versus educational gates. Nat. Rev. Immunol. 2013, 13, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, H.R.; Ruitenberg, M.J.; McMenamin, P.G. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J. Neuropathol. Exp. Neurol. 2010, 69, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Jordao, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Prodinger, C.; Bunse, J.; Krüger, M.; Schiefenhövel, F.; Brandt, C.; Laman, J.D.; Greter, M.; Immig, K.; Heppner, F.; Becher, B.; et al. CD11c-expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol. 2011, 121, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, D.S.; Evans, T.A.; Myers, J.; Petrosiute, A.; Silver, J.; Huang, A.Y. Extravascular CX3CR1+ cells extend intravascular dendritic processes into intact central nervous system vessel lumen. Microsc. Microanal. 2013, 19, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Zimprich, F.; Vass, K.; Hickey, W.F. Microglial cells are a component of the perivascular glia limitans. J. Neurosci. Res. 1991, 28, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Kida, S.; Pantazis, A.; Weller, R.O. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol. Appl. Neurobiol. 1993, 19, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Hohsfield, L.A.; Humpel, C. Migration of blood cells to β-amyloid plaques in alzheimer’s disease. Exp. Gerontol. 2015, 65, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B. Molecular mechanisms involved in T cell migration across the blood–brain barrier. J. Neural Transm. 2006, 113, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.L.; Holman, D.W.; Klein, R.S. Chemokines in the balance: Maintenance of homeostasis and protection at cns barriers. Front. Cell. Neurosci. 2014, 8, 336. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Ge, S.; Lemire, Y.; Jellison, E.R.; Serwanski, D.R.; Ruddle, N.H.; Pachter, J.S. Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation. J. Neuroinflamm. 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Anderson, P.; Durbeej, M.; van Rooijen, N.; Ivars, F.; Opdenakker, G.; Sorokin, L.M. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Fut. Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood–brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF β receptor in mouse models with normal and deficient PDGF β receptor signaling. Mol. Neurodegener. 2010, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Nisancioglu, M.H.; Mahoney, W.M.; Kimmel, D.D.; Schwartz, S.M.; Betsholtz, C.; Genové, G. Generation and characterization of rgs5 mutant mice. Mol. Cell. Biol. 2008, 28, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.; Krause, D.; Kremer, M.; Dermietzel, R. The 140-kDa protein of blood-brain barrier-associated pericytes is identical to aminopeptidase N. J. Neurochem. 1994, 62, 2375–2386. [Google Scholar] [CrossRef] [PubMed]
- Trost, A.; Lange, S.; Schroedl, F.; Bruckner, D.; Motloch, K.A.; Bogner, B.; Kaser-Eichberger, A.; Strohmaier, C.; Runge, C.; Aigner, L.; et al. Brain and retinal pericytes: Origin, function and role. Front. Cell. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.; Bechmann, I. Cns pericytes: Concepts, misconceptions, and a way out. Glia 2010, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, R.; Kawahara, M.; Nakano-Doi, A.; Takahashi, A.; Tanaka, Y.; Narita, A.; Kuwahara-Otani, S.; Hayakawa, T.; Yagi, H.; Matsuyama, T.; et al. Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J. Neuroinflamm. 2016, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain microvascular pericytes are immunoactive in culture: Cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflamm. 2011, 8, 139. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Nakano-Doi, A.; Kawamura, M.; Matsuyama, T. Do vascular pericytes contribute to neurovasculogenesis in the central nervous system as multipotent vascular stem cells? Stem Cells Dev. 2015, 24, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Özen, I.; Deierborg, T.; Miharada, K.; Padel, T.; Englund, E.; Genové, G.; Paul, G. Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol. 2014, 128, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Cucullo, L. Pericytes and astrocytes crosstalk: Understanding perivascular synergism at the bbb. FASEB J. 2013, 27, lb720. [Google Scholar]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Vallières, L.; Sawchenko, P.E. Bone marrow-derived cells that populate the adult mouse brain preserve their hematopoietic identity. J. Neurosci. 2003, 23, 5197–5207. [Google Scholar] [PubMed]
- Bechmann, I.; Priller, J.; Kovac, A.; Bontert, M.; Wehner, T.; Klett, F.F.; Bohsung, J.; Stuschke, M.; Dirnagl, U.; Nitsch, R. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur. J. Neurosci. 2001, 14, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, I.; Kwidzinski, E.; Kovac, A.D.; Simbürger, E.; Horvath, T.; Gimsa, U.; Dirnagl, U.; Priller, J.; Nitsch, R. Turnover of rat brain perivascular cells. Exp. Neurol. 2001, 168, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jorstad, N.L.; Shiao, C.; Cherne, M.K.; Khademi, S.B.; Montine, K.S.; Montine, T.J.; Keene, C.D. Perivascular, but not parenchymal, cerebral engraftment of donor cells after non-myeloablative bone marrow transplantation. Exp. Mol. Pathol. 2013, 95, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hickey, W.F.; Kimura, H. Perivascular microglial cells of the cns are bone marrow-derived and present antigen in vivo. Science 1988, 239, 290. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Man, S.; Ubogu, E.E.; Ransohoff, R.M. Inflammatory cell migration into the central nervous system: A few new twists on an old tale. Brain Pathol. 2007, 17, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Winger, R.C.; Koblinski, J.E.; Kanda, T.; Ransohoff, R.M.; Muller, W.A. Rapid remodeling of tight junctions during paracellular diapedesis in a human model of the blood–brain barrier. J. Immunol. 2014, 193, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.R.; Walker, D.C.; Brown, E.S.; Thurmon, L.T.; Bowden, R.A.; Keese, C.R.; Simon, S.I.; Entman, M.L.; Smith, C.W. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J. Immunol. 1997, 159, 2893–2903. [Google Scholar] [PubMed]
- Von Wedel-Parlow, M.; Schrot, S.; Lemmen, J.; Treeratanapiboon, L.; Wegener, J.; Galla, H.J. Neutrophils cross the BBB primarily on transcellular pathways: An in vitro study. Brain Res. 2011, 1367, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Wewer, C.; Seibt, A.; Wolburg, H.; Greune, L.; Schmidt, M.A.; Berger, J.; Galla, H.J.; Quitsch, U.; Schwerk, C.; Schroten, H.; et al. Transcellular migration of neutrophil granulocytes through the blood-cerebrospinal fluid barrier after infection with streptococcus suis. J. Neuroinflamm. 2011, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Buhler, L.A.; Samara, R.; Guzman, E.; Wilson, C.L.; Krizanac-Bengez, L.; Janigro, D.; Ethell, D.W. Matrix metalloproteinase-7 facilitates immune access to the CNS in experimental autoimmune encephalomyelitis. BMC Neurosci. 2009, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Babcock, A.A.; Kuziel, W.A.; Rivest, S.; Owens, T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J. Neurosci. 2003, 23, 7922. [Google Scholar] [PubMed]
- Hsieh, C.L.; Niemi, E.C.; Wang, S.H.; Lee, C.C.; Bingham, D.; Zhang, J.; Cozen, M.L.; Charo, I.; Huang, E.J.; Liu, J.; et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J. Neurotrauma 2014, 31, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wang, J.; Kivisakk, P.; Rollins, B.J.; Ransohoff, R.M. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific t helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 2001, 193, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Dogan, R.-N.E.; Elhofy, A.; Karpus, W.J. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of tnf- and inos-expressing macrophages and myeloid dendritic cells. J. Immunol. 2008, 180, 7376–7384. [Google Scholar] [CrossRef] [PubMed]
- Stowe, A.M.; Wacker, B.K.; Cravens, P.D.; Perfater, J.L.; Li, M.K.; Hu, R.; Freie, A.B.; Stüve, O.; Gidday, J.M. CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J. Neuroinflamm. 2012, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Toft-Hansen, H.; Buist, R.; Sun, X.-J.; Schellenberg, A.; Peeling, J.; Owens, T. Metalloproteinases control brain inflammation induced by pertussis toxin in mice overexpressing the chemokine CCL2 in the central nervous system. J. Immunol. 2006, 177, 7242–7249. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.C.; Krivacic, K.; Kirby, B.; Vaccariello, S.A.; Wei, T.; Ransohoff, R.M.; Zigmond, R.E. Monocyte chemoattractant protein (MCP)-1 is rapidly expressed by sympathetic ganglion neurons following axonal injury. NeuroReport 2001, 12, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Flügel, A.; Hager, G.; Horvat, A.; Spitzer, C.; Singer, G.M.A.; Graeber, M.B.; Kreutzberg, G.W.; Schwaiger, F.W. Neuronal MCP-1 expression in response to remote nerve injury. J. Cereb. Blood Flow Metab. 2001, 21, 69–76. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factorα signaling during peripheral organ inflammation. J. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Krivit, W.; Sung, J.H.; Shapiro, E.G.; Lockman, L.A. Microglia: The effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transpl. 1995, 4, 385–392. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723. [Google Scholar] [CrossRef] [PubMed]
- Archer, L.D.; Langford-Smith, K.J.; Bigger, B.W.; Fildes, J.E. Mucopolysaccharide diseases: A complex interplay between neuroinflammation, microglial activation and adaptive immunity. J. Inherit. Metab. Dis. 2014, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Aubourg, P. Hematopoietic stem cell gene therapy in hurler syndrome, globoid cell leukodystrophy, metachromatic leukodystrophy and X-adrenoleukodystrophy. Curr. Opin. Mol. Ther. 2008, 10, 471–478. [Google Scholar] [PubMed]
- Krall, W.J.; Challita, P.M.; Perlmutter, L.S.; Skelton, D.C.; Kohn, D.B. Cells expressing human glucocerebrosidase from a retroviral vector repopulate macrophages and central nervous system microglia after murine bone marrow transplantation. Blood 1994, 83, 2737–2748. [Google Scholar] [PubMed]
- Biffi, A.; de Palma, M.; Quattrini, A.; del Carro, U.; Amadio, S.; Visigalli, I.; Sessa, M.; Fasano, S.; Brambilla, R.; Marchesini, S.; et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Investig. 2004, 113, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Miyake, K.; Karlsson, S.; Shimada, T. Successful treatment of metachromatic leukodystrophy using bone marrow transplantation of HoxB4 overexpressing cells. Mol. Ther. 2010, 18, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Rozengurt, N.; Ryazantsev, S.; Kohn, D.B.; Satake, N.; Neufeld, E.F. Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol. Genet. Metab. 2003, 79, 233–244. [Google Scholar] [CrossRef]
- Sergijenko, A.; Langford-Smith, A.; Liao, A.Y.; Pickford, C.E.; McDermott, J.; Nowinski, G.; Langford-Smith, K.J.; Merry, C.L.R.; Jones, S.A.; Wraith, J.E.; et al. Myeloid/microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol. Ther. 2013, 21, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Cyr, D.M.; Lindquist, S. Amyloid deposits: Protection against toxic protein species? Cell Cycle 2009, 8, 1668–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agadjanyan, M.G.; Petrovsky, N.; Ghochikyan, A. A fresh perspective from immunologists and vaccine researchers: Active vaccination strategies to prevent and reverse alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R. Alzheimer’s disease: Epidemiology. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 89, pp. 195–205. [Google Scholar]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.-S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.-S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Audoy-Rémus, J.; Bozoyan, L.; Dumas, A.; Filali, M.; Lecours, C.; Lacroix, S.; Rivest, S.; Tremblay, M.-E.; Vallières, L. Gpr84 deficiency reduces microgliosis, but accelerates dendritic degeneration and cognitive decline in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 2015, 46, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.-P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Laouar, Y.; Pittenger, C.; Mori, T.; Szekely, C.A.; Tan, J.; Duman, R.S.; Flavell, R.A. Blocking tgf-β–smad2/3 innate immune signaling mitigates alzheimer-like pathology. Nat. Med. 2008, 14, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Naert, G.; Rivest, S. Cc chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6208–6220. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Chung, H.; Dolios, G.; Wang, R.; Asamoah, N.; Lobel, P.; Maxfield, F.R. Degradation of fibrillar forms of Alzheimer’s amyloid β-peptide by macrophages. Neurobiol. Aging 2008, 29, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. Trem2 variants in Alzheimer’s disease. N. Engl. J. Med. 2012, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Murcia, J.D.; Schmutz, C.; Munger, C.; Perkes, A.; Gustin, A.; Peterson, M.; Ebbert, M.T.W.; Norton, M.C.; Tschanz, J.T.; Munger, R.G.; et al. Assessment of TREM2 rs75932628 association with Alzheimer’s disease in a population-based sample: The cache county study. Neurobiol. Aging 2013, 34, 2889.e2811–2889.e2813. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tan, L.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Tan, M.-S.; Gu, L.-Z.; Wang, H.-F.; Ding, Z.-Z.; Zhang, Y.-D.; et al. Upregulation of trem2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Wang, Y. Trem2 variants: New keys to decipher alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243–286. [Google Scholar] [CrossRef] [PubMed]
- Keage, H.A.D.; Matthews, F.E.; Yip, A.; Gao, L.; McCracken, C.; McKeith, I.G.; Rubinsztein, D.C.; Brayne, C.; Function, M.R.C.C.; Ageing, S. APOE and ACE polymorphisms and dementia risk in the older population over prolonged follow-up: 10 years of incidence in the MRC CFA study. Age Ageing 2010, 39, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lin, S.; Bales, K.R.; Gelfanova, V.; Koger, D.; DeLong, C.; Hale, J.; Liu, F.; Hunter, J.M.; Paul, S.M. Macrophage-mediated degradation of β-amyloid via an apolipoprotein E isoform-dependent mechanism. J. Neurosci. 2009, 29, 3603–3612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cudaback, E.; Jorstad, N.L.; Hemingway, J.F.; Hagan, C.E.; Melief, E.J.; Li, X.; Yoo, T.; Khademi, S.B.; Montine, K.S.; et al. APOE3, but not APOE4, bone marrow transplantation mitigates behavioral and pathological changes in a mouse model of Alzheimer disease. Am. J. Pathol. 2013, 183, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Ibach, B.; Haen, E. Acetylcholinesterase inhibition in Alzheimer’s disease. Curr. Pharm. Des. 2004, 10, 231–251. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kitamura, Y.; Saeki, M.; Terada, M.; Kagitani, S.; Kitamura, R.; Fujikawa, Y.; Maelicke, A.; Tomimoto, H.; Taniguchi, T.; et al. Galantamine-induced amyloid-β clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J. Biol. Chem. 2010, 285, 40180–40191. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Kim, S.Y.; Lee, H.G.; Kim, S.U.; Lee, Y.B. Activation of nicotinic acetylcholine receptor prevents the production of reactive oxygen species in fibrillar β amyloid peptide (1-42)-stimulated microglia. Exp. Mol. Med. 2008, 40, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.-M.; Gilman, S.; Dartigues, J.-F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with ad after aβ42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.J.; Bullock, R.; Jones, R.W.; Wilkinson, D.; Paterson, K.R.; Jenkins, L.; Millais, S.B.; Donoghue, S. Evaluation of the safety and immunogenicity of synthetic aβ42 (an1792) in patients with ad. Neurology 2005, 64, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; Barton, E.; Boche, D.; Neal, J.W.; Ferrer, I.; Thompson, P.; Vlachouli, C.; Wilkinson, D.; Bayer, A.; Games, D.; et al. Aβ species removal after aβ42 immunization. J. Neuropathol. Exp. Neurol. 2006, 65, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Patton, R.L.; Kalback, W.M.; Esh, C.L.; Kokjohn, T.A.; Van Vickle, G.D.; Luehrs, D.C.; Kuo, Y.-M.; Lopez, J.; Brune, D.; Ferrer, I.; et al. Amyloid-β peptide remnants in an-1792-immunized alzheimer’s disease patients: A biochemical analysis. Am. J. Pathol. 2006, 169, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; Andreasen, N.; Riviere, M.-E.; Vostiar, I.; Vitaliti, A.; Sovago, J.; Caputo, A.; Winblad, B.; Graf, A. Long-term treatment with active aβ immunotherapy with cad106 in mild Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, M.; Boncristiano, S.; Bondolfi, L.; Stalder, A.; Deller, T.; Staufenbiel, M.; Mathews, P.M.; Jucker, M. Cerebral hemorrhage after passive anti-aβ immunotherapy. Science 2002, 298, 1379. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Boyd, T.D.; Bennett, S.P.; Mori, T.; Governatori, N.; Runfeldt, M.; Norden, M.; Padmanabhan, J.; Neame, P.; Wefes, I.; Sanchez-Ramos, J.; et al. GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J. Alzheimer’s Dis.: JAD 2010, 21, 507–518. [Google Scholar] [PubMed]
- Vogel, D.Y.S.; Kooij, G.; Heijnen, P.D.A.M.; Breur, M.; Peferoen, L.A.N.; van der Valk, P.; de Vries, H.E.; Amor, S.; Dijkstra, C.D. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur. J. Immunol. 2015, 45, 1808–1819. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- McGoldrick, P.; Joyce, P.I.; Fisher, E.M.C.; Greensmith, L. Rodent models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2013, 1832, 1421–1436. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Zondler, L.; Müller, K.; Khalaji, S.; Bliederhäuser, C.; Ruf, W.P.; Grozdanov, V.; Thiemann, M.; Fundel-Clemes, K.; Freischmidt, A.; Holzmann, K.; et al. Peripheral monocytes are functionally altered and invade the cns in als patients. Acta Neuropathol. 2016, 132, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Doble, A. The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, S233–S241. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.S.; Ferreira, R.; Lively, S.; Schlichter, L.C. Microglial SK3 and SK4 currents and activation state are modulated by the neuroprotective drug, riluzole. J. Neuroimmune Pharmacol. 2013, 8, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Nagai, M.; Miyazaki, K.; Tanaka, N.; Kawai, H.; Mimoto, T.; Morimoto, N.; Kurata, T.; Ikeda, Y.; Matsuura, T.; et al. Neuroprotective and angiogenic effects of bone marrow transplantation combined with granulocyte colony-stimulating factor in a mouse model of amyotrophic lateral sclerosis. Cell Med. 2011, 2, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Zhang, R.; Block, G.; Katz, J.; Barohn, R.; Kasarskis, E.; Forshew, D.; Gopalakrishnan, V.; McGrath, M.S. NP001 regulation of macrophage activation markers in ALS: A phase I clinical and biomarker study. Amyotroph. Lateral Scler. Frontotemporal Degener. 2014, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Van der Valk, P.; de Groot, C.J.A. Staging of multiple sclerosis (MS) lesions: Pathology of the time frame of MS. Neuropathol. Appl. Neurobiol. 2000, 26, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Brendecke, S.M.; Prinz, M. Do not judge a cell by its cover—Diversity of CNS resident, adjoining and infiltrating myeloid cells in inflammation. Semin. Immunopathol. 2015, 37, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating monocytes trigger eae progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Mack, M.; Schmidt, H.; Bruck, W.; Djukic, M.; Zabel, M.D.; Hille, A.; Priller, J.; Prinz, M. Ccr2+ly-6chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 2009, 132, 2487–2500. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia/macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Ruckh, J.M.; Zhao, J.-W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J.M. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Kivisäkk, P.; Imitola, J.; Rasmussen, S.; Elyaman, W.; Zhu, B.; Ransohoff, R.M.; Khoury, S.J. Localizing CNS immune surveillance: Meningeal APCs activate T cells during eae. Ann. Neurol. 2009, 65, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Shemer, A.; Jung, S. Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin. Immunopathol. 2015, 37, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Franklin, R.J.M. Macrophages and cns remyelination. J. Neurochem. 2014, 130, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflamm. 2013, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4βl integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Brakebusch, C.; Coisne, C.; Sixt, M.; Wekerle, H.; Engelhardt, B.; Fässler, R. Β1 integrins differentially control extravasation of inflammatory cell subsets into the cns during autoimmunity. Proc. Natl. Acad. Sci. USA 2009, 106, 1920–1925. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Alon, R.; Moser, B.; Springer, T. Sequential regulation of α4β1 and α5β1 integrin avidity by cc chemokines in monocytes: Implications for transendothelial chemotaxis. J. Cell Biol. 1996, 134, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R.; Koralnik, I.J. Progressive multifocal leukoencephalopathy and natalizumab—Unforeseen consequences. N. Engl. J. Med. 2005, 353, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Nikfar, S.; Rahimi, R.; Abdollahi, M. A meta-analysis of the efficacy and tolerability of interferon-β in multiple sclerosis, overall and by drug and disease type. Clin. Ther. 2010, 32, 1871–1888. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Reder, A.T. Immunomodulatory activity of interferon-beta. Ann. Clin. Transl. Neurol. 2014, 1, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Floris, S.; Ruuls, S.R.; Wierinckx, A.; van der Pol, S.M.; Dopp, E.; van der Meide, P.H.; Dijkstra, C.D.; de Vries, H.E. Interferon-β directly influences monocyte infiltration into the central nervous system. J. Neuroimmunol. 2002, 127, 69–79. [Google Scholar] [CrossRef]
- Jiang, H.; Milo, R.; Swoveland, P.; Johnson, K.P.; Panitch, H.; Dhib-Jalbut, S. Interferon β-1b reduces interferon γ-induced antigen-presenting capacity of human glial and b cells. J. Neuroimmunol. 1995, 61, 17–25. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Kocur, M.; Schneider, R.; Pulm, A.-K.; Bauer, J.; Kropp, S.; Gliem, M.; Ingwersen, J.; Goebels, N.; Alferink, J.; Prozorovski, T.; et al. IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol. Commun. 2015, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Hosmane, S.; Tegenge, M.A.; Rajbhandari, L.; Uapinyoying, P.; Kumar, N.G.; Thakor, N.; Venkatesan, A. Toll/interleukin-1 receptor domain-containing adapter inducing interferon-β mediates microglial phagocytosis of degenerating axons. J. Neurosci. 2012, 32, 7745–7757. [Google Scholar] [CrossRef] [PubMed]
- Ekta Franscina, P.; Chittaranjan, A. Interferon-related depression: A primer on mechanisms, treatment, and prevention of a common clinical problem. Curr. Neuropharmacol. 2016, 14, 743–748. [Google Scholar]
- Burt, R.K.; Balabanov, R.; Han, X.; Sharrack, B.; Morgan, A.; Quigley, K.; Yaung, K.; Helenowski, I.B.; Jovanovic, B.; Spahovic, D. Association of nonmyeloablative hematopoietic stem cell transplantation with neurological disability in patients with relapsing-remitting multiple sclerosis. JAMA 2015, 313, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.C.; Jonuleit, H.; Wiendl, H. Fulfilling the dream: Tolerogenic dendritic cells to treat multiple sclerosis. Eur. J. Immunol. 2012, 42, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Raϊch-Regué, D.; Grau-López, L.; Naranjo-Gómez, M.; Ramo-Tello, C.; Pujol-Borrell, R.; Martínez-Cáceres, E.; Borràs, F.E. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur. J. Immunol. 2012, 42, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, M.J.; Sellès-Moreno, C.; Fàbregas-Puig, S.; Amoedo, J.; Navarro-Barriuso, J.; Teniente-Serra, A.; Grau-López, L.; Ramo-Tello, C.; Martínez-Cáceres, E.M. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci. Ther. 2015, 21, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, M.J.; Contreras-Cardone, R.; Navarro-Barriuso, J.; Cools, N.; Berneman, Z.; Ramo-Tello, C.; Martínez-Cáceres, E.M. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. J. Neuroinflamm. 2016, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma. Cancer Res. 2004, 64, 6892. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Wang, Y.; Peng, X.; You, G.; Zhang, W.; Yan, W.; Bao, Z.; Wang, Y.; Qiu, X.; Jiang, T. Management and survival rates in patients with glioma in China (2004–2010): A retrospective study from a single-institution. J. Neuro-Oncol. 2013, 113, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro-Oncology 2010, 12, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Rumiato, E.; Bertorelle, R.; Saggioro, D.; Farina, P.; Della Puppa, A.; Zustovich, F.; Berti, F.; Sacchetto, V.; Marcato, R.; et al. Clinical and genetic factors associated with severe hematological toxicity in glioblastoma patients during radiation plus temozolomide treatment: A prospective study. Am. J. Clin. Oncol. 2015, 38, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Alizadeh, D.; Van Handel, M.; Kortylewski, M.; Yu, H.; Badie, B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia 2009, 57, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, H.; Villeneuve, J.; Gowing, G.; Julien, J.-P.; Vallières, L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007, 67, 8874. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuño, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004, 64, 4973. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M.; et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: Results of a clinical phase I/II trial. Clin. Cancer Res. 2005, 11, 4160. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Wang, X.; Soto, H.; Young, E.; Lisiero, D.N.; Fong, B.; Everson, R.; Yong, W.H.; Lai, A.; Li, G.; et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J. Immunother. 2013, 36, 152–157. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Rapp, M.; Sorg, R.V.; Steiger, H.-J.; Stummer, W.; van Gool, S.; Sabel, M. Dendritic cell vaccination in patients with malignant gliomas: Current status and future directions. Neurosurgery 2006, 59, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Holsinger, R.M.D.; Kruse, C.A.; Flügel, A.; Graeber, M.B. The potential for genetically altered microglia to influence glioma treatment. CNS Neurol. Disord. Drug Targets 2013, 12, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.E.; Beard, B.C.; Trobridge, G.D.; Neff, T.; Rockhill, J.K.; Silbergeld, D.L.; Mrugala, M.; Kiem, H.-P. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci. Transl. Med. 2012, 4, 133–157. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.E.; Johnston, S.K.; Mrugala, M.M.; Beard, B.C.; Guyman, L.A.; Baldock, A.L.; Bridge, C.A.; Hawkins-Daarud, A.; Gori, J.L.; Born, D.E.; et al. Gene therapy enhances chemotherapy tolerance and efficacy in glioblastoma patients. J. Clin. Investig. 2014, 124, 4082–4092. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Ginhoux, F.; Greter, M.; Bruttger, J. Homeostasis of microglia in the adult brain: Review of novel microglia depletion systems. Trends Immunol. 2015, 36, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Betlazar, C.; Middleton, R.J.; Banati, R.B.; Liu, G.-J. The impact of high and low dose ionising radiation on the central nervous system. Redox Biol. 2016, 9, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Shechter, R.; Schwartz, M. Harnessing monocyte-derived macrophages to control central nervous system pathologies: No longer ‘if’ but ‘how’. J. Pathol. 2013, 229, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Aarntzen, E.H.J.G.; Bulte, J.W.M.; Oyen, W.J.; Heerschap, A.; de Vries, I.J.M.; Figdor, C.G. Imaging of cellular therapies. Adv. Drug Deliv. Rev. 2010, 62, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-J.; Middleton, R.J.; Hatty, C.R.; Kam, W.W.-Y.; Chan, R.; Pham, T.; Harrison-Brown, M.; Dodson, E.; Veale, K.; Banati, R.B. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. 2014, 24, 631–653. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In Vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Banati, R.B.; Newcombe, J.; Gunn, R.N.; Cagnin, A.; Turkheimer, F.; Heppner, F.; Price, G.; Wegner, F.; Giovannoni, G.; Miller, D.H.; et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis. Brain 2000, 123, 2321. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, P.; Politis, M.; Su, P.; Turkheimer, F.; Malik, O.; Keihaninejad, S.; Wu, K.; Reynolds, R.; Nicholas, R.; Piccini, P. Microglia activation in multiple sclerosis black holes predicts outcome in progressive patients: An in vivo [(11)C](R)-PK11195-PET pilot study. Neurobiol. Dis. 2014, 65, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef] [PubMed]
- Roncaroli, F.; Su, Z.; Herholz, K.; Gerhard, A.; Turkheimer, F.E. TSPO expression in brain tumours: Is TSPO a target for brain tumour imaging? Clin. Transl. Imaging 2016, 4, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Fais, S.; Logozzi, M.; Lugini, L.; Federici, C.; Azzarito, T.; Zarovni, N.; Chiesi, A. Exosomes: The ideal nanovectors for biodelivery. Biol. Chem. 2013, 394, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yim, N.; Ryu, S.-W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.-H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.-L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar]
- Klyachko, N.L.; Haney, M.J.; Zhao, Y.; Manickam, D.S.; Mahajan, V.; Suresh, P.; Hingtgen, S.D.; Mosley, R.L.; Gendelman, H.E.; Kabanov, A.V.; et al. Macrophages offer a paradigm switch for CNS delivery of therapeutic proteins. Nanomedicine 2014, 9, 1403–1422. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison-Brown, M.; Liu, G.-J.; Banati, R. Checkpoints to the Brain: Directing Myeloid Cell Migration to the Central Nervous System. Int. J. Mol. Sci. 2016, 17, 2030. https://doi.org/10.3390/ijms17122030
Harrison-Brown M, Liu G-J, Banati R. Checkpoints to the Brain: Directing Myeloid Cell Migration to the Central Nervous System. International Journal of Molecular Sciences. 2016; 17(12):2030. https://doi.org/10.3390/ijms17122030
Chicago/Turabian StyleHarrison-Brown, Meredith, Guo-Jun Liu, and Richard Banati. 2016. "Checkpoints to the Brain: Directing Myeloid Cell Migration to the Central Nervous System" International Journal of Molecular Sciences 17, no. 12: 2030. https://doi.org/10.3390/ijms17122030