
Immunopathogenic Mechanisms of Autoimmune Hepatitis: How Much Do We Know from Animal Models?


{kind=link}
Abstract
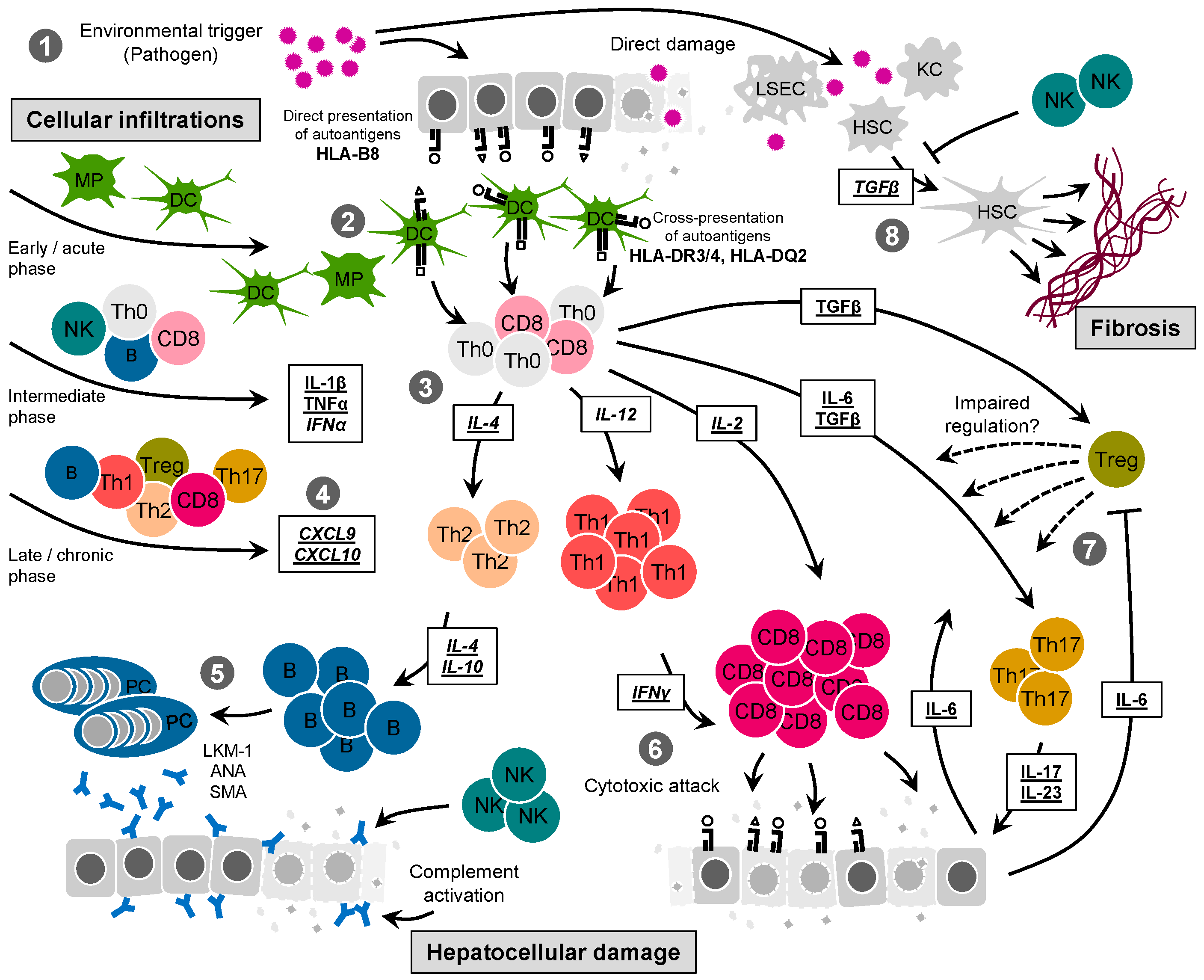
:1. Clinical Features of Autoimmune Hepatitis
2. Current Treatment
3. Mechanistic Insight from Clinical Observations
4. Mechanistic Insight from Animal Models
5. Insight from the CYP2D6 Mouse Model
6. Conclusions and Future Perspectives
Conflicts of Interest
Abbreviations
| AIH | autoimmune hepatitis |
| PBC | primary biliary cholangitis |
| PSC | primary sclerosing cholangitis |
| ANA | anti-nuclear antibody |
| SMA | anti-smooth muscle antibody |
| LKM-1 | type 1 liver/kidney microsomal autoantibodies |
| CYP2D6 | cytochrome P450 2D6 |
| LSP | liver-specific membrane lipoprotein |
| ASGPR | asialoglycoprotein receptor |
| MAPK | mitogen-activated protein kinase |
| NASH | non-alcoholic steatohepatitis |
| NAFLD | non-alcoholic fatty liver disease |
| PDH-E2 | E2-subunit of the pyruvate dehydrogenase complex |
| 2-OA | 2-octynoic acid |
| CMV | cytomegalovirus |
| ICP4 | infected cell protein 4 |
| HSV-1 | herpes simplex virus 1 |
| HHV-8 | Kaposi’s sarcoma-associated herpes virus |
| HHV-5 | human cytomegalovirus |
| EAH | experimental autoimmune hepatitis |
| S-100 | 100,000 g supernatant of syngeneic liver homogenate |
| NOD | non-obese diabetic |
| PD-1 | programmed cell death |
| NT | neonatal thymectomized |
| TLR | toll-like receptor |
| ConA | concanavalin A |
| NKT | natural killer T cells |
| HBsAg | hepatitis B virus surface antigen |
| CTL | cytotoxic T-lymphocytes |
| GP | glycoprotein |
| LCMV | lymphocytic choriomeningitis virus |
| Alb | albumin promoter |
| RIP | rat insulin promoter |
| LSEC | liver sinusoidal endothelial cells |
| HSC | hepatic stellate cell |
| CRP | carbon-reactive protein |
| TCR | T cell receptor |
| MBP | myelin basic protein |
| HA | hemagglutinin |
| mTEC | medullary thymic epithelial cells |
| AIRE | autoimmune regulator |
| APS-1 | autoimmune polyendocrine syndrome type 1 |
| TTR | transthyretin |
| NP | nucleoprotein |
| Ad-2D6 | adenovirus expressing CYP2D6 |
| αSMA | α-smooth muscle actin |
| FTCD | formiminotransferase cyclodeaminase |
References
- Czaja, A.J. Diagnosis and management of autoimmune hepatitis. Clin. Liver Dis. 2015, 19, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Grant, C.R.; Mieli-Vergani, G.; Vergani, D. Autoimmune hepatitis: A comprehensive review. J. Autoimmun. 2013, 41, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Vierling, J.M. Diagnosis and treatment of autoimmune hepatitis. Curr. Gastroenterol. Rep. 2012, 14, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Lohse, A.W.; Wiegard, C. Diagnostic criteria for autoimmune hepatitis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Lohse, A.W.; Vergani, D. Autoimmune hepatitis—Update 2015. J. Hepatol. 2015, 62, S100–S111. [Google Scholar] [CrossRef] [PubMed]
- Boberg, K.M. Prevalence and epidemiology of autoimmune hepatitis. Clin. Liver Dis. 2002, 6, 635–647. [Google Scholar] [CrossRef]
- Czaja, A.J. Autoimmune hepatitis—Approach to diagnosis. MedGenMed 2006, 8, 55. [Google Scholar] [PubMed]
- World Health Organization. Dept. of Protection of the Human Environment; Inter-Organization Programme for the Sound Management of Chemicals. Principles and Methods for Assessing Autoimmunity Associated with Exposure to Chemicals. World Health Organization: Geneva, Switzerland, 2006. Available online: http://www.who.int/iris/handle/10665/43603 (accessed on 23 November 2016).
- Manns, M.P.; Czaja, A.J.; Gorham, J.D.; Krawitt, E.L.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M. Diagnosis and management of autoimmune hepatitis. Hepatology 2010, 51, 2193–2213. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T.; Doherty, D.G.; Hayllar, K.M.; McFarlane, I.G.; Johnson, P.J.; Williams, R. Susceptibility to autoimmune chronic active hepatitis: Human leukocyte antigens DR4 and A1-B8-DR3 are independent risk factors. Hepatology 1991, 13, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D.G.; Donaldson, P.T.; Underhill, J.A.; Farrant, J.M.; Duthie, A.; Mieli-Vergani, G.; McFarlane, I.G.; Johnson, P.J.; Eddleston, A.L.; Mowat, A.P.; et al. Allelic sequence variation in the HLA class II genes and proteins in patients with autoimmune hepatitis. Hepatology 1994, 19, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Carpenter, H.A.; Santrach, P.J.; Moore, S.B. Significance of HLA DR4 in type 1 autoimmune hepatitis. Gastroenterology 1993, 105, 1502–1507. [Google Scholar] [CrossRef]
- Donaldson, P.T. Genetics of liver disease: Immunogenetics and disease pathogenesis. Gut 2004, 53, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Bogdanos, D.P.; Hussain, M.J.; Underhill, J.; Bansal, S.; Longhi, M.S.; Cheeseman, P.; Mieli-Vergani, G.; Vergani, D. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology 2006, 130, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Djilali-Saiah, I.; Renous, R.; Caillat-Zucman, S.; Debray, D.; Alvarez, F. Linkage disequilibrium between HLA class II region and autoimmune hepatitis in pediatric patients. J. Hepatol. 2004, 40, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Krawitt, E.L.; Vierling, J.M.; Manns, M.P.; Mieli-Vergani, G.; Vergani, D. Cutting edge issues in autoimmune hepatitis. J. Autoimmun. 2016, 75, 6–19. [Google Scholar] [CrossRef] [PubMed]
- De Boer, Y.S.; van Gerven, N.M.; Zwiers, A.; Verwer, B.J.; van Hoek, B.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; Drenth, J.P.; den Ouden, J.W.; et al. Genome-wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroenterology 2014, 147, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Strassburg, C.P. Autoimmune hepatitis: Clinical challenges. Gastroenterology 2001, 120, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Van Gerven, N.M.; de Boer, Y.S.; Zwiers, A.; Verwer, B.J.; Drenth, J.P.; van Hoek, B.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; den Ouden, J.W.; et al. HLA-DRB1*03:01 and HLA-DRB1*04:01 modify the presentation and outcome in autoimmune hepatitis type-1. Genes Immun. 2015, 16, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Metzler, F.; Geiger, E.; Heinrich, E.; Hallensleben, M.; Manns, M.P.; Vogel, A. Prediction of short- and long-term outcome in patients with autoimmune hepatitis. Hepatology 2015, 62, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Challenges in the diagnosis and management of autoimmune hepatitis. Can. J. Gastroenterol. 2013, 27, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Strassburg, C.P.; Manns, M.P. Therapy of autoimmune hepatitis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.M.; Zeniya, M.; Czaja, A.J.; Pares, A.; Dalekos, G.N.; Krawitt, E.L.; Bittencourt, P.L.; Porta, G.; Boberg, K.M.; Hofer, H.; et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008, 48, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Mieli-Vergani, G.; Vergani, D. Clinical significance of autoantibodies in autoimmune hepatitis. J. Autoimmun. 2013, 46, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Christen, U. The role of autoantibodies in autoimmune hepatitis type 2. Immunotherapy 2013, 5, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Krawitt, E.L. Autoimmune hepatitis. N. Engl. J. Med. 2006, 354, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Muratori, P.; Lalanne, C.; Fabbri, A.; Cassani, F.; Lenzi, M.; Muratori, L. Type 1 and type 2 autoimmune hepatitis in adults share the same clinical phenotype. Aliment. Pharmacol. Ther. 2015, 41, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Johnson, E.F.; Griffin, K.J.; Tan, E.M.; Sullivan, K.F. Major antigen of liver kidney microsomal autoantibodies in idiopathic autoimmune hepatitis is cytochrome P450db1. J. Clin. Investig. 1989, 83, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Hauri, H.P.; Loeper, J.; Homberg, J.C.; Meyer, U.A. Antibodies against human cytochrome P-450db1 in autoimmune hepatitis type II. Proc. Natl. Acad. Sci. USA 1988, 85, 8256–8260. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.M.; Cresteil, D.; Boniface, O.; Clerc, F.F.; Alvarez, F. Identification and analysis of cytochrome P450IID6 antigenic sites recognized by anti-liver-kidney microsome type-1 antibodies (LKM1). Eur. J. Immunol. 1993, 23, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Griffin, K.J.; Sullivan, K.F.; Johnson, E.F. LKM-1 autoantibodies recognize a short linear sequence in P450IID6, a cytochrome P-450 monooxygenase. J. Clin. Investig. 1991, 88, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Lohr, H.; Manns, M.; Kyriatsoulis, A.; Lohse, A.W.; Trautwein, C.; Meyer zum Buschenfelde, K.H.; Fleischer, B. Clonal analysis of liver-infiltrating T cells in patients with LKM-1 antibody-positive autoimmune chronic active hepatitis. Clin. Exp. Immunol. 1991, 84, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Lohr, H.F.; Schlaak, J.F.; Lohse, A.W.; Bocher, W.O.; Arenz, M.; Gerken, G.; Meyer Zum Buschenfelde, K.H. Autoreactive CD4+ LKM-specific and anticlonotypic T-cell responses in LKM-1 antibody-positive autoimmune hepatitis. Hepatology 1996, 24, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Advances in the current treatment of autoimmune hepatitis. Dig. Dis. Sci. 2012, 57, 1996–2010. [Google Scholar] [CrossRef] [PubMed]
- Zachou, K.; Gatselis, N.; Papadamou, G.; Rigopoulou, E.I.; Dalekos, G.N. Mycophenolate for the treatment of autoimmune hepatitis: Prospective assessment of its efficacy and safety for induction and maintenance of remission in a large cohort of treatment-naive patients. J. Hepatol. 2011, 55, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Weiler-Normann, C.; Schramm, C.; Quaas, A.; Wiegard, C.; Glaubke, C.; Pannicke, N.; Moller, S.; Lohse, A.W. Infliximab as a rescue treatment in difficult-to-treat autoimmune hepatitis. J. Hepatol. 2013, 58, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Burak, K.W.; Swain, M.G.; Santodomingo-Garzon, T.; Lee, S.S.; Urbanski, S.J.; Aspinall, A.I.; Coffin, C.S.; Myers, R.P. Rituximab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can. J. Gastroenterol. 2013, 27, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Boberg, K.M.; Chapman, R.W.; Hirschfield, G.M.; Lohse, A.W.; Manns, M.P.; Schrumpf, E. Overlap syndromes: The International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J. Hepatol. 2011, 54, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Okamoto, M.; Thomas, M.G.; Bogdanos, D.P.; Lopes, A.R.; Portmann, B.; Underhill, J.; Durr, R.; Mieli-Vergani, G.; Vergani, D. Antibodies to conformational epitopes of soluble liver antigen define a severe form of autoimmune liver disease. Hepatology 2002, 35, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Vergani, D.; Mieli-Vergani, G.; Mondelli, M.; Portmann, B.; Eddleston, A.L. Immunoglobulin on the surface of isolated hepatocytes is associated with antibody-dependent cell-mediated cytotoxicity and liver damage. Liver 1987, 7, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Muratori, L.; Parola, M.; Ripalti, A.; Robino, G.; Muratori, P.; Bellomo, G.; Carini, R.; Lenzi, M.; Landini, M.P.; Albano, E.; et al. Liver/kidney microsomal antibody type 1 targets CYP2D6 on hepatocyte plasma membrane. Gut 2000, 46, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Schuh, E.; Berer, K.; Mulazzani, M.; Feil, K.; Meinl, I.; Lahm, H.; Krane, M.; Lange, R.; Pfannes, K.; Subklewe, M.; et al. Features of Human CD3+CD20+ T Cells. J. Immunol. 2016, 197, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Senaldi, G.; Portmann, B.; Mowat, A.P.; Mieli-Vergani, G.; Vergani, D. Immunohistochemical features of the portal tract mononuclear cell infiltrate in chronic aggressive hepatitis. Arch. Dis. Child. 1992, 67, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Vergani, G.M.; Vergani, D.; Jenkins, P.J.; Portmann, B.; Mowat, A.P.; Eddleston, A.L.; Williams, R. Lymphocyte cytotoxicity to autologous hepatocytes in HBsAg-negative chronic active hepatitis. Clin. Exp. Immunol. 1979, 38, 16–21. [Google Scholar] [PubMed]
- Wen, L.; Peakman, M.; Lobo-Yeo, A.; McFarlane, B.M.; Mowat, A.P.; Mieli-Vergani, G.; Vergani, D. T-cell-directed hepatocyte damage in autoimmune chronic active hepatitis. Lancet 1990, 336, 1527–1530. [Google Scholar] [CrossRef]
- Hintermann, E.; Holdener, M.; Bayer, M.; Loges, S.; Pfeilschifter, J.M.; Granier, C.; Manns, M.P.; Christen, U. Epitope spreading of the anti-CYP2D6 antibody response in patients with autoimmune hepatitis and in the CYP2D6 mouse model. J. Autoimmun. 2011, 37, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Ehser, J.; Holdener, M.; Christen, S.; Bayer, M.; Pfeilschifter, J.M.; Hintermann, E.; Bogdanos, D.; Christen, U. Molecular mimicry rather than identity breaks T-cell tolerance in the CYP2D6 mouse model for human autoimmune hepatitis. J. Autoimmun. 2013, 42, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tang, Y.; You, Z.; Wang, Q.; Liang, S.; Han, X.; Qiu, D.; Wei, J.; Liu, Y.; Shen, L.; et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS ONE 2011, 6, e18909. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Hatton, R.D. Interplay between the TH17 and TReg cell lineages: A (co-)evolutionary perspective. Nat. Rev. Immunol. 2009, 9, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Ma, Y.; Bogdanos, D.P.; Cheeseman, P.; Mieli-Vergani, G.; Vergani, D. Impairment of CD4+CD25+ regulatory T-cells in autoimmune liver disease. J. Hepatol. 2004, 41, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Mitry, R.R.; Samyn, M.; Scalori, A.; Hussain, M.J.; Quaglia, A.; Mieli-Vergani, G.; Ma, Y.; Vergani, D. Vigorous activation of monocytes in juvenile autoimmune liver disease escapes the control of regulatory T-cells. Hepatology 2009, 50, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Sebode, M.; Franke, B.; Wortmann, F.; Schwinge, D.; Quaas, A.; Baron, U.; Olek, S.; Wiegard, C.; Lohse, A.W.; et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J. Hepatol. 2012, 57, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Tsuneyama, K.; Baba, H.; Kikuchi, K.; Nishida, T.; Nomoto, K.; Hayashi, S.; Miwa, S.; Nakajima, T.; Nakanishi, Y.; Masuda, S.; et al. Autoimmune features in metabolic liver disease: A single-center experience and review of the literature. Clin. Rev. Allergy Immunol. 2013, 45, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lindor, K.D.; Angulo, P. The prevalence of autoantibodies and autoimmune hepatitis in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2004, 99, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Yatsuji, S.; Hashimoto, E.; Kaneda, H.; Taniai, M.; Tokushige, K.; Shiratori, K. Diagnosing autoimmune hepatitis in nonalcoholic fatty liver disease: Is the International Autoimmune Hepatitis Group scoring system useful? J. Gastroenterol. 2005, 40, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Albano, E. Role of adaptive immunity in alcoholic liver disease. Int. J. Hepatol. 2012, 2012, 893026. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Messmer, M.; Bayer, M.; Pfeilschifter, J.M.; Hintermann, E.; Christen, U. Non-alcoholic fatty liver disease (NAFLD) potentiates autoimmune hepatitis in the CYP2D6 mouse model. J. Autoimmun. 2016, 69, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Gut, J.; Christen, U.; Frey, N.; Koch, V.; Stoffler, D. Molecular mimicry in halothane hepatitis: Biochemical and structural characterization of lipoylated autoantigens. Toxicology 1995, 97, 199–224. [Google Scholar] [CrossRef]
- Czaja, A.J. Drug-induced autoimmune-like hepatitis. Dig. Dis. Sci. 2011, 56, 958–976. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Leung, P.S.; Rieger, R.; Quan, C.; Wang, X.; Marik, J.; Suen, Y.F.; Kurth, M.J.; Nantz, M.H.; Ansari, A.A.; et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: Identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J. Immunol. 2005, 174, 5874–5883. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Lian, Z.X.; Leung, P.S.; Moritoki, Y.; Tsuneyama, K.; Kurth, M.J.; Lam, K.S.; Yoshida, K.; Yang, G.X.; Hibi, T.; et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology 2008, 48, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.A. Molecular mimicry and autoimmune disease. Cell 1987, 50, 819–820. [Google Scholar] [CrossRef]
- Christen, U. Molecular mimiry. In Autoantibodies, 3rd ed.; Shoenfeld, Y., Meroni, P.L., Gershwin, M.E., Eds.; Elsevier: Waltham, MA, USA, 2014; pp. 35–42. [Google Scholar]
- Christen, U.; Hintermann, E. Pathogen infection as a possible cause for autoimmune hepatitis. Int. Rev. Immunol. 2014, 33, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Marceau, G.; Lapierre, P.; Beland, K.; Soudeyns, H.; Alvarez, F. LKM1 autoantibodies in chronic hepatitis C infection: A case of molecular mimicry? Hepatology 2005, 42, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, N.; Choudhuri, K.; Ma, Y.; Mahmoud, A.; Bogdanos, D.P.; Muratori, L.; Bianchi, F.; Williams, R.; Mieli-Vergani, G.; Vergani, D. Cytochrome P4502D6193–212: A new immunodominant epitope and target of virus/self cross-reactivity in liver kidney microsomal autoantibody type 1-positive liver disease. J. Immunol. 2003, 170, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Christen, U. Pathogen infection and autoimmunity. Int. Rev. Immunol. 2014, 33, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.; Weismuller, T.J.; Lankisch, T.O.; Santer, D.M.; Tyrrell, D.L.; Manns, M.P.; Houghton, M. Differential serum levels of eosinophilic eotaxins in primary sclerosing cholangitis, primary biliary cirrhosis, and autoimmune hepatitis. J. Interferon Cytokine Res. 2014, 34, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Giuggioli, D.; Ferrannini, E.; Ferri, C.; Fallahi, P. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun. Rev. 2014, 13, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Current and prospective pharmacotherapy for autoimmune hepatitis. Expert Opin. Pharm. 2014, 15, 1715–1736. [Google Scholar] [CrossRef] [PubMed]
- Nishioji, K.; Okanoue, T.; Itoh, Y.; Narumi, S.; Sakamoto, M.; Nakamura, H.; Morita, A.; Kashima, K. Increase of chemokine interferon-inducible protein-10 (IP-10) in the serum of patients with autoimmune liver diseases and increase of its mRNA expression in hepatocytes. Clin. Exp. Immunol. 2001, 123, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Liu, N.; Zhao, D.T.; Li, Z.M.; Zhang, H.P.; Liu, Y.M.; Yan, H.P.; Zhao, Y. Investigate circulating levels of chemokines and evaluate the correlation between these chemokines and liver function indicators in autoimmune hepatitis. Zhonghua Gan Zang Bing Za Zhi 2013, 21, 299–303. [Google Scholar] [PubMed]
- Ikeda, A.; Aoki, N.; Kido, M.; Iwamoto, S.; Nishiura, H.; Maruoka, R.; Chiba, T.; Watanabe, N. Progression of autoimmune hepatitis is mediated by IL-18-producing dendritic cells and hepatic CXCL9 expression in mice. Hepatology 2014, 60, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christen, U. CXCR3 and Its Ligands. In Encyclopedia of Inflammatory Diseases; Parnham, M., Ed.; Springer: Basel, Switzerland, 2015; pp. 1–14. [Google Scholar]
- Yuksel, M.; Laukens, D.; Heindryckx, F.; van Vlierberghe, H.; Geerts, A.; Wong, F.S.; Wen, L.; Colle, I. Hepatitis mouse models: From acute-to-chronic autoimmune hepatitis. Int. J. Exp. Pathol. 2014, 95, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Hintermann, E. An update on animal models of autoimmune hepatitis: Are we there yet? Curr. Pharm. Des. 2015, 21, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Meyer zum Buschenfelde, K.H.; Kossling, F.K.; Miescher, P.A. Experimental chronic active hepatitis in rabbits following immunization with human liver proteins. Clin. Exp. Immunol. 1972, 11, 99–108. [Google Scholar]
- Kuriki, J.; Murakami, H.; Kakumu, S.; Sakamoto, N.; Yokochi, T.; Nakashima, I.; Kato, N. Experimental autoimmune hepatitis in mice after immunization with syngeneic liver proteins together with the polysaccharide of Klebsiella pneumoniae. Gastroenterology 1983, 84, 596–603. [Google Scholar] [PubMed]
- Lohse, A.W.; Manns, M.; Dienes, H.P.; Meyer zum Buschenfelde, K.H.; Cohen, I.R. Experimental autoimmune hepatitis: disease induction, time course and T-cell reactivity. Hepatology 1990, 11, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kawakami, H.; Kawamoto, H.; Ikemoto, Y.; Masuda, K.; Takezaki, E.; Nakanishi, T.; Kajiyama, G.; Takeno, H. Effect of neonatal thymectomy on experimental autoimmune hepatitis in mice. Clin. Exp. Immunol. 1987, 67, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Asano, M.; Toda, M.; Sakaguchi, N.; Sakaguchi, S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 1996, 184, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Lohse, A.W.; Dienes, H.P.; Meyer zum Buschenfelde, K.H. Suppression of murine experimental autoimmune hepatitis by T-cell vaccination or immunosuppression. Hepatology 1998, 27, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 1980, 29, 1–13. [Google Scholar] [PubMed]
- Mauad, T.H.; van Nieuwkerk, C.M.; Dingemans, K.P.; Smit, J.J.; Schinkel, A.H.; Notenboom, R.G.; van den Bergh Weerman, M.A.; Verkruisen, R.P.; Groen, A.K.; Oude Elferink, R.P.; et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 1994, 145, 1237–1245. [Google Scholar] [PubMed]
- Kido, M.; Watanabe, N.; Okazaki, T.; Akamatsu, T.; Tanaka, J.; Saga, K.; Nishio, A.; Honjo, T.; Chiba, T. Fatal autoimmune hepatitis induced by concurrent loss of naturally arising regulatory T cells and PD-1-mediated signaling. Gastroenterology 2008, 135, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, R.; Aoki, N.; Kido, M.; Iwamoto, S.; Nishiura, H.; Ikeda, A.; Chiba, T.; Watanabe, N. Splenectomy prolongs the effects of corticosteroids in mouse models of autoimmune hepatitis. Gastroenterology 2013, 145, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, N.; Liu, P.; Zhang, Y.; Wu, H.; Chen, Y.; Han, D. Development of a spontaneous liver disease resembling autoimmune hepatitis in mice lacking Tyro3, Axl and Mer receptor tyrosine kinases. PLoS ONE 2013, 8, e66604. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Kusters, S.; Gantner, F.; Kunstle, G.; Tiegs, G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996, 111, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Gantner, F.; Leist, M.; Lohse, A.W.; Germann, P.G.; Tiegs, G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: The role of tumor necrosis factor. Hepatology 1995, 21, 190–198. [Google Scholar] [PubMed]
- Gantner, F.; Leist, M.; Kusters, S.; Vogt, K.; Volk, H.D.; Tiegs, G. T cell stimulus-induced crosstalk between lymphocytes and liver macrophages results in augmented cytokine release. Exp. Cell Res. 1996, 229, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Hayakawa, Y.; van Kaer, L.; Matsuda, H.; Yagita, H.; Okumura, K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc. Natl. Acad. Sci. USA 2000, 97, 5498–5503. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Guidotti, L.G.; Wirth, S.; Ishikawa, T.; Missale, G.; Moriyama, T.; Schreiber, R.D.; Schlicht, H.J.; Huang, S.N.; Chisari, F.V. Class I-restricted cytotoxic T lymphocytes are directly cytopathic for their target cells in vivo. J. Immunol. 1994, 152, 3245–3253. [Google Scholar] [PubMed]
- Moriyama, T.; Guilhot, S.; Klopchin, K.; Moss, B.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L.; Kanagawa, O.; Chisari, F.V. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science 1990, 248, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Ferber, I.; Schoenrich, G.; Schenkel, J.; Mellor, A.L.; Haemmerling, G.J.; Arnold, B. Levels of peripheral T cell tolerance induced by different doses of tolerogen. Science 1994, 263, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Limmer, A.; Sacher, T.; Alferink, J.; Kretschmar, M.; Schonrich, G.; Nichterlein, T.; Arnold, B.; Hammerling, G.J. Failure to induce organ-specific autoimmunity by breaking of tolerance: Importance of the microenvironment. Eur. J. Immunol. 1998, 28, 2395–2406. [Google Scholar] [CrossRef]
- Voehringer, D.; Blaser, C.; Grawitz, A.B.; Chisari, F.V.; Buerki, K.; Pircher, H. Break of T cell ignorance to a viral antigen in the liver induces hepatitis. J. Immunol. 2000, 165, 2415–2422. [Google Scholar] [CrossRef] [PubMed]
- Sacher, T.; Knolle, P.; Nichterlein, T.; Arnold, B.; Hammerling, G.J.; Limmer, A. CpG-ODN-induced inflammation is sufficient to cause T-cell-mediated autoaggression against hepatocytes. Eur. J. Immunol. 2002, 32, 3628–3637. [Google Scholar] [CrossRef]
- Djilali-Saiah, I.; Lapierre, P.; Vittozi, S.; Alvarez, F. DNA vaccination breaks tolerance for a neo-self antigen in liver: A transgenic murine model of autoimmune hepatitis. J. Immunol. 2002, 169, 4889–4896. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, P.; Djilali-Saiah, I.; Vitozzi, S.; Alvarez, F. A murine model of type 2 autoimmune hepatitis: Xenoimmunization with human antigens. Hepatology 2004, 39, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, S.; Homma, S.; Enomoto, Y.; Komita, H.; Zeniya, M.; Ohno, T.; Toda, G. Autoimmune hepatic inflammation by vaccination of mice with dendritic cells loaded with well-differentiated hepatocellular carcinoma cells and administration of interleukin-12. Clin. Immunol. 2005, 117, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Derkow, K.; Loddenkemper, C.; Mintern, J.; Kruse, N.; Klugewitz, K.; Berg, T.; Wiedenmann, B.; Ploegh, H.L.; Schott, E. Differential priming of CD8 and CD4 T-cells in animal models of autoimmune hepatitis and cholangitis. Hepatology 2007, 46, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Zierden, M.; Kuhnen, E.; Odenthal, M.; Dienes, H.P. Effects and regulation of autoreactive CD8+ T cells in a transgenic mouse model of autoimmune hepatitis. Gastroenterology 2010, 139, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Von Herrath, M.G.; Fujinami, R.S.; Whitton, J.L. Microorganisms and autoimmunity: Making the barren field fertile. Nat. Rev. Microbiol. 2003, 1, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.A.; Nerenberg, M.; Southern, P.; Price, J.; Lewicki, H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: Role of anti-self (virus) immune response. Cell 1991, 65, 319–331. [Google Scholar] [CrossRef]
- Ohashi, P.; Oehen, S.; Buerki, K.; Pircher, H.; Ohashi, C.; Odermatt, B.; Malissen, B.; Zinkernagel, R.; Hengartner, H. Ablation of tolerance and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 1991, 65, 305–317. [Google Scholar] [CrossRef]
- Von Herrath, M.G.; Dockter, J.; Oldstone, M.B.A. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity 1994, 1, 231–242. [Google Scholar] [CrossRef]
- Christen, U.; Bender, C.; von Herrath, M.G. Infection as a cause of type 1 diabetes? Curr. Opin. Rheumatol. 2012, 24, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.G.; Zen, M.; Holz, L.; Davis, T.; McCaughan, G.W.; Bertolino, P. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J. Clin. Investig. 2004, 114, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Hepatic T cells and liver tolerance. Nat. Rev. Immunol. 2003, 3, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Luth, S.; Huber, S.; Schramm, C.; Buch, T.; Zander, S.; Stadelmann, C.; Bruck, W.; Wraith, D.C.; Herkel, J.; Lohse, A.W. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J. Clin. Investig. 2008, 118, 3403–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Kuo, L.M.; Chang, Y.; Wu, W.; Goldbach, C.; Ross, M.A.; Stolz, D.B.; Chen, L.; Fung, J.J.; Lu, L.; et al. In vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology 2006, 44, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Guo, Z.; Xu, X.; Yi, H.; Wang, Q.; Cao, X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood 2008, 112, 3175–3185. [Google Scholar] [CrossRef] [PubMed]
- Alexandropoulos, K.; Bonito, A.J.; Weinstein, E.G.; Herbin, O. Medullary thymic epithelial cells and central tolerance in autoimmune hepatitis development: Novel perspective from a new mouse model. Int. J. Mol. Sci. 2015, 16, 1980–2000. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Taubert, R.; Noyan, F.; Sievers, M.; Dywicki, J.; Schlue, J.; Falk, C.S.; Ardesjo Lundgren, B.; Scott, H.S.; Pich, A.; et al. Autoimmune hepatitis in a murine autoimmune polyendocrine syndrome type 1 model is directed against multiple autoantigens. Hepatology 2015, 61, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, E.; Ehser, J.; Christen, U. The CYP2D6 Animal Model: How to Induce Autoimmune Hepatitis in Mice. J. Vis. Exp. 2012, 60, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Holdener, M.; Hintermann, E.; Bayer, M.; Rhode, A.; Rodrigo, E.; Hintereder, G.; Johnson, E.F.; Gonzalez, F.J.; Pfeilschifter, J.; Manns, M.P.; et al. Breaking tolerance to the natural human liver autoantigen cytochrome P450 2D6 by virus infection. J. Exp. Med. 2008, 205, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.P.; Lenzi, M.; Okamoto, M.; Rigopoulou, E.I.; Muratori, P.; Ma, Y.; Muratori, L.; Tsantoulas, D.; Mieli-Vergani, G.; Bianchi, F.B.; et al. Multiple viral/self immunological cross-reactivity in liver kidney microsomal antibody positive hepatitis C virus infected patients is associated with the possession of HLA B51. Int. J. Immunopathol. Pharmacol. 2004, 17, 83–92. [Google Scholar] [PubMed]
- Gueguen, M.; Boniface, O.; Bernard, O.; Clerc, F.; Cartwright, T.; Alvarez, F. Identification of the main epitope on human cytochrome P450 IID6 recognized by anti-liver kidney microsome antibody. J. Autoimmun. 1991, 4, 607–615. [Google Scholar] [CrossRef]
- Kitazawa, E.; Igarashi, T.; Kawaguchi, N.; Matsushima, H.; Kawashima, Y.; Hankins, R.W.; Miyakawa, H. Differences in Anti-LKM-1 Autoantibody Immunoreactivity to CYP2D6 Antigenic Sites Between Hepatitis C Virus-negative and -positive Patients. J. Autoimmun. 2001, 17, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Ehser, J.; Müller, P.; Bayer, M.; Pfeilschifter, J.M.; Hintermann, E.; Christen, U. Absence of CXCL10 or CCL5 has opposite effects on the severity of autoimmune hepatitis in the CYP2D6 mouse model. Unpublished work. 2016. [Google Scholar]
- Milks, M.W.; Cripps, J.G.; Lin, H.; Wang, J.; Robinson, R.T.; Sargent, J.L.; Whitfield, M.L.; Gorham, J.D. The role of Ifng in alterations in liver gene expression in a mouse model of fulminant autoimmune hepatitis. Liver Int. 2009, 29, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, A.; Wegscheid, C.; Claass, B.; Carambia, A.; Herkel, J.; Mittrucker, H.W.; Panzer, U.; Tiegs, G. CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J. Immunol. 2011, 186, 5284–5293. [Google Scholar] [CrossRef] [PubMed]
- Colvin, R.A.; Campanella, G.S.; Manice, L.A.; Luster, A.D. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol. Cell. Biol. 2006, 26, 5838–5849. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, E.; Ehser, J.; Bayer, M.; Pfeilschifter, J.M.; Christen, U. Mechanism of autoimmune hepatic fibrogenesis induced by an adenovirus encoding the human liver autoantigen cytochrome P450 2D6. J. Autoimmun. 2013, 44, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Fischer, K.; Noyan, F.; Schlue, J.; Falk, C.S.; Stahlhut, M.; Woller, N.; Kuehnel, F.; Taubert, R.; Manns, M.P.; et al. Genetic predisposition and environmental danger signals initiate chronic autoimmune hepatitis driven by CD4+ T cells. Hepatology 2013, 58, 718–728. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christen, U.; Hintermann, E. Immunopathogenic Mechanisms of Autoimmune Hepatitis: How Much Do We Know from Animal Models? Int. J. Mol. Sci. 2016, 17, 2007. https://doi.org/10.3390/ijms17122007
Christen U, Hintermann E. Immunopathogenic Mechanisms of Autoimmune Hepatitis: How Much Do We Know from Animal Models? International Journal of Molecular Sciences. 2016; 17(12):2007. https://doi.org/10.3390/ijms17122007
Chicago/Turabian StyleChristen, Urs, and Edith Hintermann. 2016. "Immunopathogenic Mechanisms of Autoimmune Hepatitis: How Much Do We Know from Animal Models?" International Journal of Molecular Sciences 17, no. 12: 2007. https://doi.org/10.3390/ijms17122007