
Prediction of Protein–Protein Interactions by Evidence Combining Methods


 ,
,
Abstract
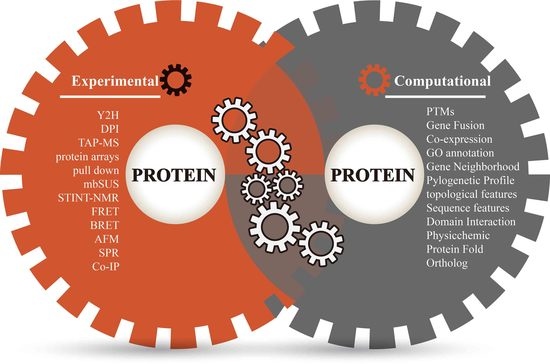
:
1. Introduction
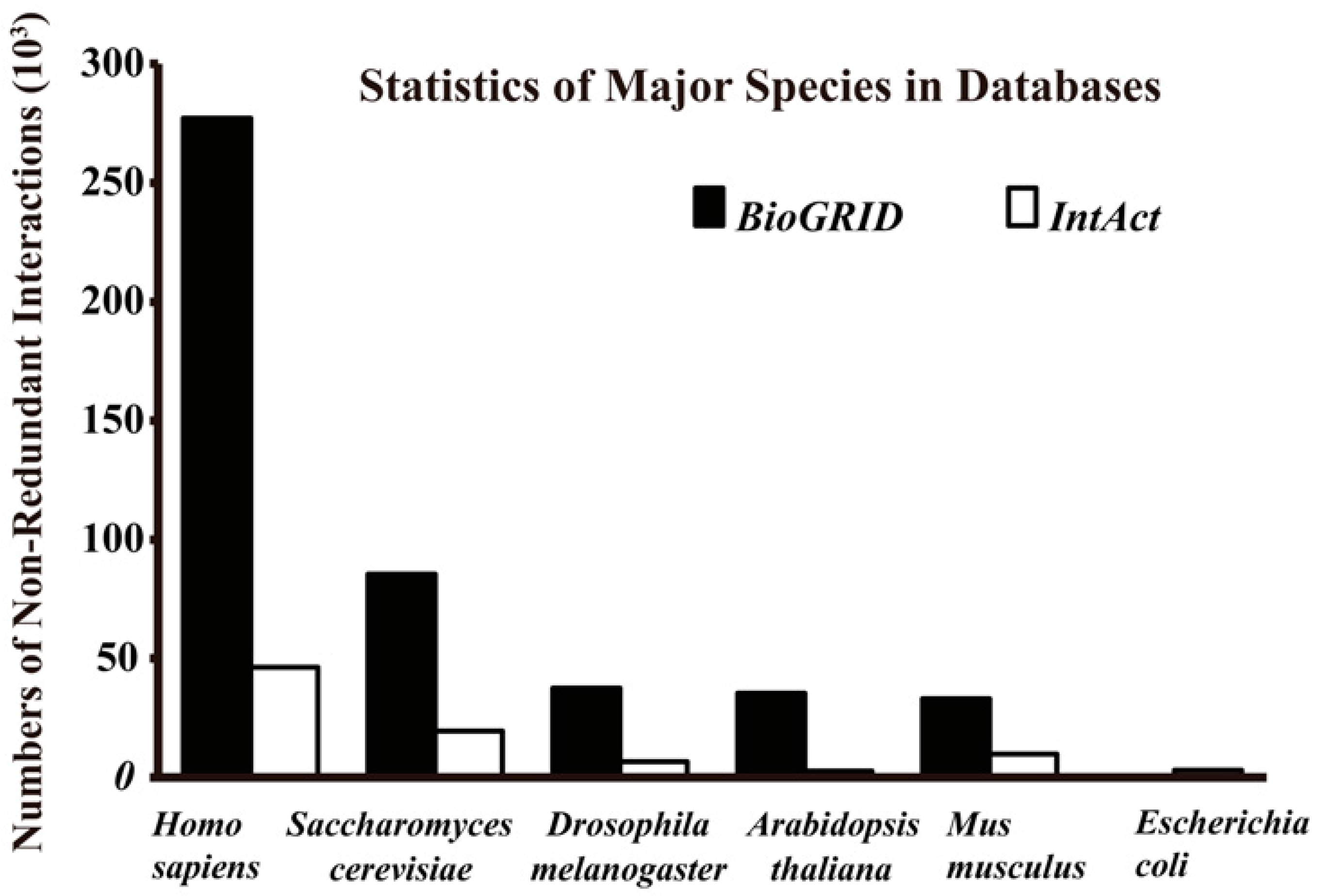
2. Defining Gold Standard Datasets
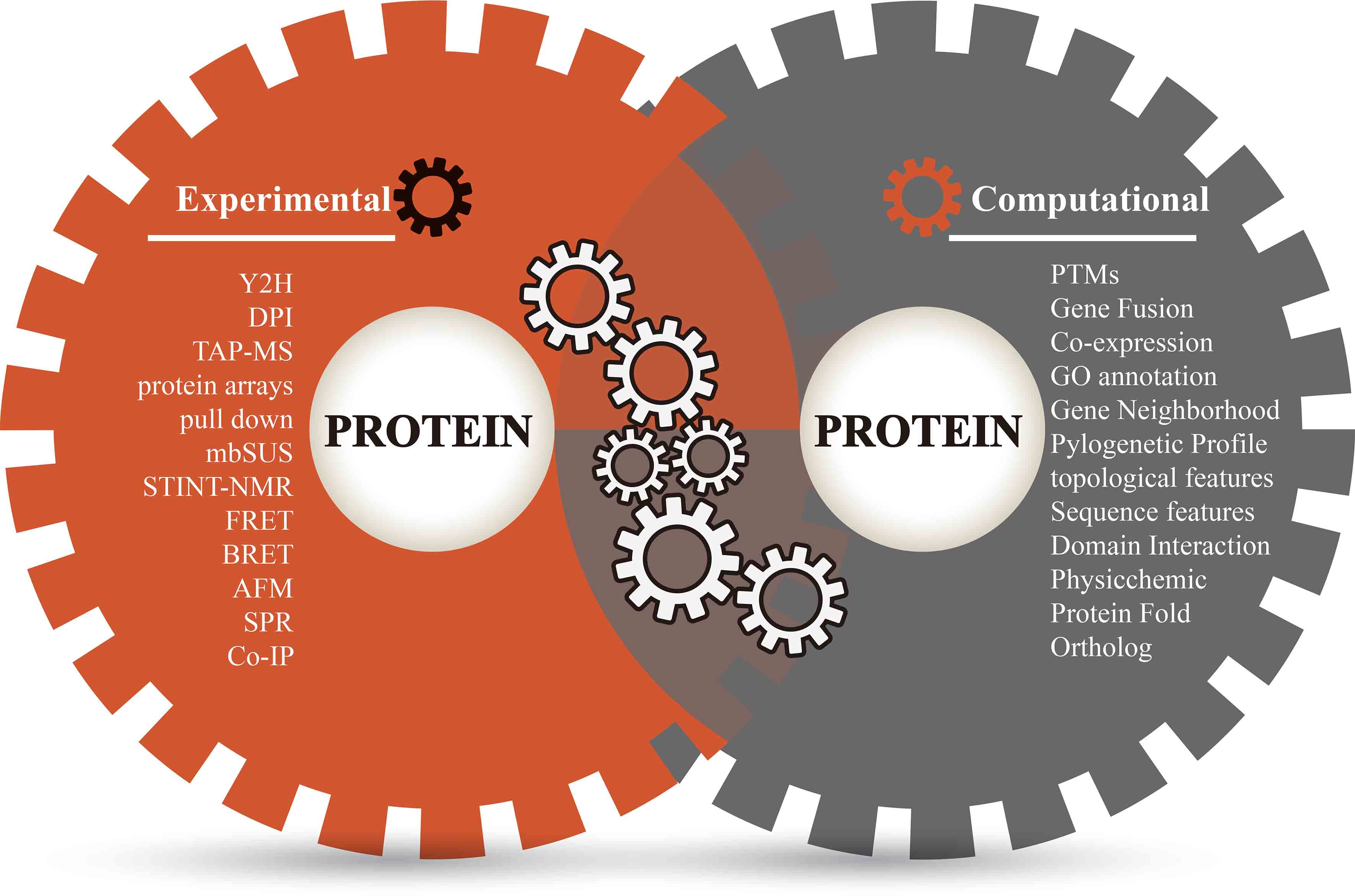
3. Annotate Protein Pairs with Diverse Evidence
4. Strategy for Integrative Analysis
4.1. Exploratory PPI Predictions Using Combinated Vector Descriptors
4.2. Exploratory PPI Predictions Using Probabilistic Classification Scoring
4.3. Prediction of Protein–Protein Interaction Sites
5. Performance Evaluation of PPI Prediction
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kotlyar, M.; Pastrello, C.; Pivetta, F.; lo Sardo, A.; Cumbaa, C.; Li, H.; Naranian, T.; Niu, Y.; Ding, Z.; Vafaee, F.; et al. In silico prediction of physical protein interactions and characterization of interactome orphans. Nat. Methods 2015, 12, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, N.L.; Pavlopoulos, G.A.; Theodosiou, T.; Iliopoulos, I. Protein–protein interaction predictions using text mining methods. Methods 2015, 74, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, S.; Ehrhardt, D.W.; Loque, D.; Chen, J.; Rhee, S.Y.; Frommer, W.B. Molecular and cellular approaches for the detection of protein–protein interactions: Latest techniques and current limitations. Plant J. 2008, 53, 610–635. [Google Scholar] [CrossRef] [PubMed]
- Ngounou Wetie, A.G.; Sokolowska, I.; Woods, A.G.; Roy, U.; Deinhardt, K.; Darie, C.C. Protein–protein interactions: Switch from classical methods to proteomics and bioinformatics-based approaches. Cell. Mol. Life Sci. 2014, 71, 205–228. [Google Scholar] [CrossRef] [PubMed]
- Piehler, J. New methodologies for measuring protein interactions in vivo and in vitro. Curr. Opin. Struct. Biol. 2005, 15, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Pastrello, C.; Pasini, E.; Kotlyar, M.; Otasek, D.; Wong, S.; Sangrar, W.; Rahmati, S.; Jurisica, I. Integration, visualization and analysis of human interactome. Biochem. Biophys. Res. Commun. 2014, 445, 757–773. [Google Scholar] [PubMed]
- Giot, L.; Bader, J.S.; Brouwer, C.; Chaudhuri, A.; Kuang, B.; Li, Y.; Hao, Y.L.; Ooi, C.E.; Godwin, B.; Vitols, E.; et al. A protein interaction map of Drosophila melanogaster. Science 2003, 302, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [PubMed]
- Huang, X.T.; Zhu, Y.; Chan, L.L.H.; Zhao, Z.Y.; Yan, H. An integrative C. elegans protein–protein interaction network with reliability assessment based on a probabilistic graphical model. Mol. Biosyst. 2016, 12, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Byron, O.; Vestergaard, B. Protein–protein interactions: A supra-structural phenomenon demanding trans-disciplinary biophysical approaches. Curr. Opin. Struct. Biol. 2015, 35, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Marcotte, E.M.; Thompson, M.J.; Eisenberg, D.; Yeates, T.O. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 1999, 96, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.T.; Guo, M.Z.; Needham, C.J.; Huang, Y.C.; Cai, L.; Westhead, D.R. Simple sequence-based kernels do not predict protein–protein interactions. Bioinformatics 2010, 26, 2610–2614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Jiao, X.; Song, J.; Chang, S. Prediction of human protein–protein interaction by a domain-based approach. J. Theor. Biol. 2016, 396, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Greenbaum, D.; Gerstein, M. Relating whole-genome expression data with protein–protein interactions. Genome Res. 2002, 12, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Z.; Shen, X.L.; Chen, X. PAIR: The predicted Arabidopsis interactome resource. Nucleic Acids Res. 2011, 39, D1134–D1140. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kim, I.; Zhao, H. Protein interaction predictions from diverse sources. Drug Discov. Today 2008, 13, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Tomlins, S.A.; Varambally, S.; Mahavisno, V.; Barrette, T.; Kalyana-Sundaram, S.; Ghosh, D.; Pandey, A.; Chinnaiyan, A.M. Probabilistic model of the human protein–protein interaction network. Nat. Biotechnol. 2005, 23, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, H.; Chang, J.W.; Lei, Y.; Li, S.; Chen, L.L. Prediction and characterization of protein–protein interaction network in Xanthomonas oryzae pv. oryzae PXO99 A. Res. Microbiol. 2013, 164, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Zubek, J.; Tatjewski, M.; Boniecki, A.; Mnich, M.; Basu, S.; Plewczynski, D. Multi-level machine learning prediction of protein–protein interactions in Saccharomyces cerevisiae. PeerJ 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Sun, Y.; Ding, G.H.; Liu, Q.; Wang, C.; He, Y.Y.; Shi, T.L.; Li, Y.X.; Zhao, Z.M. InPrePPI: An integrated evaluation method based on genomic context for predicting protein–protein interactions in prokaryotic genomes. BMC Bioinform. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, G.A.; Zhao, C.; Li, Y.H.; Li, P.; Cui, J.A.; Deng, Y.P.; Shi, T.L. Global protein interactome exploration through mining genome-scale data in Arabidopsis thaliana. BMC Genom. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, A.; Noble, W.S. Kernel methods for predicting protein–protein interactions. Bioinformatics 2005, 21, I38–I46. [Google Scholar] [CrossRef] [PubMed]
- Chatr-aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.C.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Lopez, D.; Gutierrez, M.A.; Lopes, K.P.; Prieto, C.; Santamaria, R.; de Las Rivas, J. APID interactomes: Providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acids Res. 2016, 44, W529–W535. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwinski, L.; Duan, X.Q.J.; Higney, P.; Kim, S.M.; Eisenberg, D. DIP, the database of interacting proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakai, K.; Nakamura, H. HitPredict: A database of quality assessed protein–protein interactions in nine species. Nucleic Acids Res. 2011, 39, D744–D749. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Reiser, L.; Berardini, T.Z.; Li, D.H.; Muller, R.; Strait, E.M.; Li, Q.; Mezheritsky, Y.; Vetushko, A.; Huala, E. Sustainable funding for biocuration: The Arabidopsis Information Resource (TAIR) as a case study of a subscription-based funding model. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.S.K.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human protein reference database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Pinese, M.; Kassahn, K.S.; Waddell, N.; Pearson, J.V.; Grimmond, S.M.; Biankin, A.V.; Hautaniemi, S.; Wu, J. PINA v2.0: Mining interactome modules. Nucleic Acids Res. 2012, 40, D862–D865. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Yu, H.Y. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.D.; Chang, J.W.; Guo, J.; Chen, D.; Li, S.; Xu, Q.; Deng, X.X.; Cheng, Y.J.; Chen, L.L. Prediction and functional analysis of the sweet orange protein–protein interaction network. BMC Plant Biol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Blohm, P.; Frishman, G.; Smialowski, P.; Goebels, F.; Wachinger, B.; Ruepp, A.; Frishman, D. Negatome 2.0: A database of non-interacting proteins derived by literature mining, manual annotation and protein structure analysis. Nucleic Acids Res. 2014, 42, D396–D400. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.; Lynn Fink, J.; Karunaratne, S.; Hanson, K.; Hamilton, N.A.; Teasdale, R.D. LOCATE: A mammalian protein subcellular localization database. Nucleic Acids Res. 2008, 36, D230–D233. [Google Scholar] [CrossRef] [PubMed]
- Peabody, M.A.; Laird, M.R.; Vlasschaert, C.; Lo, R.; Brinkman, F.S. PSORTdb: Expanding the bacteria and archaea protein subcellular localization database to better reflect diversity in cell envelope structures. Nucleic Acids Res. 2016, 44, D663–D668. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Rost, B. LocDB: Experimental annotations of localization for Homo sapiens and Arabidopsis thaliana. Nucleic Acids Res. 2011, 39, D230–D234. [Google Scholar] [CrossRef] [PubMed]
- Date, S.V.; Stoeckert, C.J. Computational modeling of the Plasmodium falciparum interactome reveals protein function on a genome-wide scale. Genome Res. 2006, 16, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, A.; Noble, W.S. Choosing negative examples for the prediction of protein–protein interactions. BMC Bioinform. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.S.; Chaudhuri, A.; Rothberg, J.M.; Chant, J. Gaining confidence in high-throughput protein interaction networks. Nat. Biotechnol. 2004, 22, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, B.; Meroueh, S.O. Structure-based computational approaches for small-molecule modulation of protein–protein interactions. Methods Mol. Biol. 2015, 1278, 77–92. [Google Scholar] [PubMed]
- Qi, Y.J.; Bar-Joseph, Z.; Klein-Seetharaman, J. Evaluation of different biological data and computational classification methods for use in protein interaction prediction. Proteins 2006, 63, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Yellaboina, S.; Tasneem, A.; Zaykin, D.V.; Raghavachari, B.; Jothi, R. DOMINE: A comprehensive collection of known and predicted domain–domain interactions. Nucleic Acids Res. 2011, 39, D730–D735. [Google Scholar] [CrossRef] [PubMed]
- Sprinzak, E.; Altuvia, Y.; Margalit, H. Characterization and prediction of protein–protein interactions within and between complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 14718–14723. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Deng, M.; Sun, F.; Chen, T. An integrated approach to the prediction of domain–domain interactions. BMC Bioinform. 2006, 7. [Google Scholar] [CrossRef]
- Izarzugaza, J.M.; Juan, D.; Pons, C.; Ranea, J.A.; Valencia, A.; Pazos, F. TSEMA: Interactive prediction of protein pairings between interacting families. Nucleic Acids Res. 2006, 34, W315–W319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Garzon, J.I.; Deng, L.; Honig, B. PrePPI: A structure-informed database of protein–protein interactions. Nucleic Acids Res. 2013, 41, D828–D833. [Google Scholar] [CrossRef] [PubMed]
- De Bodt, S.; Proost, S.; Vandepoele, K.; Rouze, P.; van de Peer, Y. Predicting protein–protein interactions in Arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genom. 2009, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.; Zhu, P.; Jiao, Y.; Meng, Y.; Chen, M. PRIN: A predicted rice interactome network. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Geisler-Lee, J.; O’Toole, N.; Ammar, R.; Provart, N.J.; Millar, A.H.; Geisler, M. A predicted interactome for Arabidopsis. Plant Physiol. 2007, 145, 317–329. [Google Scholar] [CrossRef] [PubMed]
- McDowall, M.D.; Scott, M.S.; Barton, G.J. PIPs: Human protein–protein interaction prediction database. Nucleic Acids Res. 2009, 37, D651–D656. [Google Scholar] [CrossRef] [PubMed]
- Hosur, R.; Peng, J.; Vinayagam, A.; Stelzl, U.; Xu, J.; Perrimon, N.; Bienkowska, J.; Berger, B. A computational framework for boosting confidence in high-throughput protein–protein interaction datasets. Genome Biol. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Nussinov, R.; Gursoy, A. PRISM: Protein–protein interaction prediction by structural matching. Methods Mol. Biol. 2008, 484, 505–521. [Google Scholar] [PubMed]
- Knisley, D.; Knisley, J. Predicting protein–protein interactions using graph invariants and a neural network. Comput. Biol. Chem. 2011, 35, 108–113. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Zhu, L.; Zheng, C.H.; Yu, H.J.; Deng, S.P.; Ji, Z. Prediction of protein–protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, B.; Huang, Z.; Shi, T.; Chen, Y.; Zhang, J. SPPS: A sequence-based method for predicting probability of protein–protein interaction partners. PLoS ONE 2012, 7, e30938. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.; Eskin, E.; Noble, W.S. The spectrum kernel: A string kernel for SVM protein classification. Pac. Symp. Biocomput. 2002, 7, 564–575. [Google Scholar]
- Martin, S.; Roe, D.; Faulon, J.L. Predicting protein–protein interactions using signature products. Bioinformatics 2005, 21, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, H.S.; Salavati, R. Sequence-based prediction of protein–protein interactions by means of codon usage. Genome Biol. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Dhole, K.; Singh, G.; Pai, P.P.; Mondal, S. Sequence-based prediction of protein–protein interaction sites with L1-logreg classifier. J. Theor. Biol. 2014, 348, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Mizuguchi, K. Applying the Naive Bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites. Bioinformatics 2010, 26, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.S.; Han, K.; Yang, J.Y.; Shen, H.B.; Yu, D.J. Protein–protein interaction sites prediction by ensembling SVM and sample-weighted random forests. Neurocomputing 2016, 193, 201–212. [Google Scholar] [CrossRef]
- Finn, R.D.; Miller, B.L.; Clements, J.; Bateman, A. iPfam: A database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 2014, 42, D364–D373. [Google Scholar] [CrossRef] [PubMed]
- Mosca, R.; Ceol, A.; Stein, A.; Olivella, R.; Aloy, P. 3Did: A catalog of domain-based interactions of known three-dimensional structure. Nucleic Acids Res. 2014, 42, D374–D379. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ainsworth, R.I.; Wu, M.X.; Ding, B.; Wang, W. MIEC-SVM: Automated pipeline for protein peptide/ligand interaction prediction. Bioinformatics 2016, 32, 940–942. [Google Scholar] [CrossRef] [PubMed]
- Tikk, D.; Thomas, P.; Palaga, P.; Hakenberg, J.; Leser, U. A comprehensive benchmark of Kernel methods to extract protein–protein interactions from literature. PLoS Comput. Biol. 2010, 6, e1000837. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Fonstein, M.; D’Souza, M.; Pusch, G.D.; Maltsev, N. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. USA 1999, 96, 2896–2901. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Iliopoulos, I.; Kyrpides, N.C.; Ouzounis, C.A. Protein interaction maps for complete genomes based on gene fusion events. Nature 1999, 402, 86–90. [Google Scholar] [PubMed]
- Sreenivasulu, N.; Usadel, B.; Winter, A.; Radchuk, V.; Scholz, U.; Stein, N.; Weschke, W.; Strickert, M.; Close, T.J.; Stitt, M.; et al. Barley grain maturation and germination: Metabolic pathway and regulatory network commonalities and differences highlighted by new MapMan/PageMan profiling tools. Plant Physiol. 2008, 146, 1738–1758. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.; Cheng, C.; Xuan, Z.; Sheng, H.; Ma, X. Understanding protein evolutionary rate by integrating gene co-expression with protein interactions. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.W.; Zhou, S.G.; Liu, X. Prediction of protein–protein interactions from primary sequences. Int. J. Data Min. Bioinform. 2010, 4, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.W.; Zhang, J.; Luo, X.M.; Zhu, W.L.; Yu, K.Q.; Chen, K.X.; Li, Y.X.; Jiang, H.L. Predictina protein–protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Z.; Yu, L.Z.; Wen, Z.N.; Li, M.L. Using support vector machine combined with auto covariance to predict proteinprotein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Aytuna, A.S.; Gursoy, A.; Keskin, O. Prediction of protein–protein interactions by combining structure and sequence conservation in protein interfaces. Bioinformatics 2005, 21, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.A.; Palmer, A.G.; Brunak, S.; Rost, B. Continuum secondary structure captures protein flexibility. Structure 2002, 10, 175–184. [Google Scholar] [CrossRef]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Raghavachari, B.; Tasneem, A.; Przytycka, T.M.; Jothi, R. DOMINE: A database of protein domain interactions. Nucleic Acids Res. 2008, 36, D656–D661. [Google Scholar] [CrossRef] [PubMed]
- Han, D.S.; Kim, H.S.; Jang, W.H.; Lee, S.D.; Suh, J.K. PreSPI: A domain combination based prediction system for protein–protein interaction. Nucleic Acids Res. 2004, 32, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Cusick, M.E.; Yu, H.Y.; Smolyar, A.; Venkatesan, K.; Carvunis, A.R.; Simonis, N.; Rual, J.F.; Borick, H.; Braun, P.; Dreze, M.; et al. Literature-curated protein interaction datasets. Nat. Methods 2009, 6, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Tudor, C.O.; Ross, K.E.; Li, G.; Vijay-Shanker, K.; Wu, C.H.; Arighi, C.N. Construction of phosphorylation interaction networks by text mining of full-length articles using the eFIP system. Database 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Lopez, Y.; Nakai, K.; Patil, A. HitPredict version 4: Comprehensive reliability scoring of physical protein–protein interactions from more than 100 species. Database 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Csank, C.; Costanzo, M.C.; Hirschman, J.; Hodges, P.; Kranz, J.E.; Mangan, M.; O’Neill, K.E.; Robertson, L.S.; Skrzypek, M.S.; Brooks, J.; et al. Three yeast proteome databases: YPD, PombePD, and CalPD (MycoPathPD). Method Enzymol. 2002, 350, 347–373. [Google Scholar]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Corney, D.P.A.; Buxton, B.F.; Langdon, W.B.; Jones, D.T. BioRAT: Extracting biological information from full-length papers. Bioinformatics 2004, 20, 3206–3213. [Google Scholar] [CrossRef] [PubMed]
- Tsuruoka, Y.; Tsujii, J.; Ananiadou, S. FACTA: A text search engine for finding associated biomedical concepts. Bioinformatics 2008, 24, 2559–2560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]

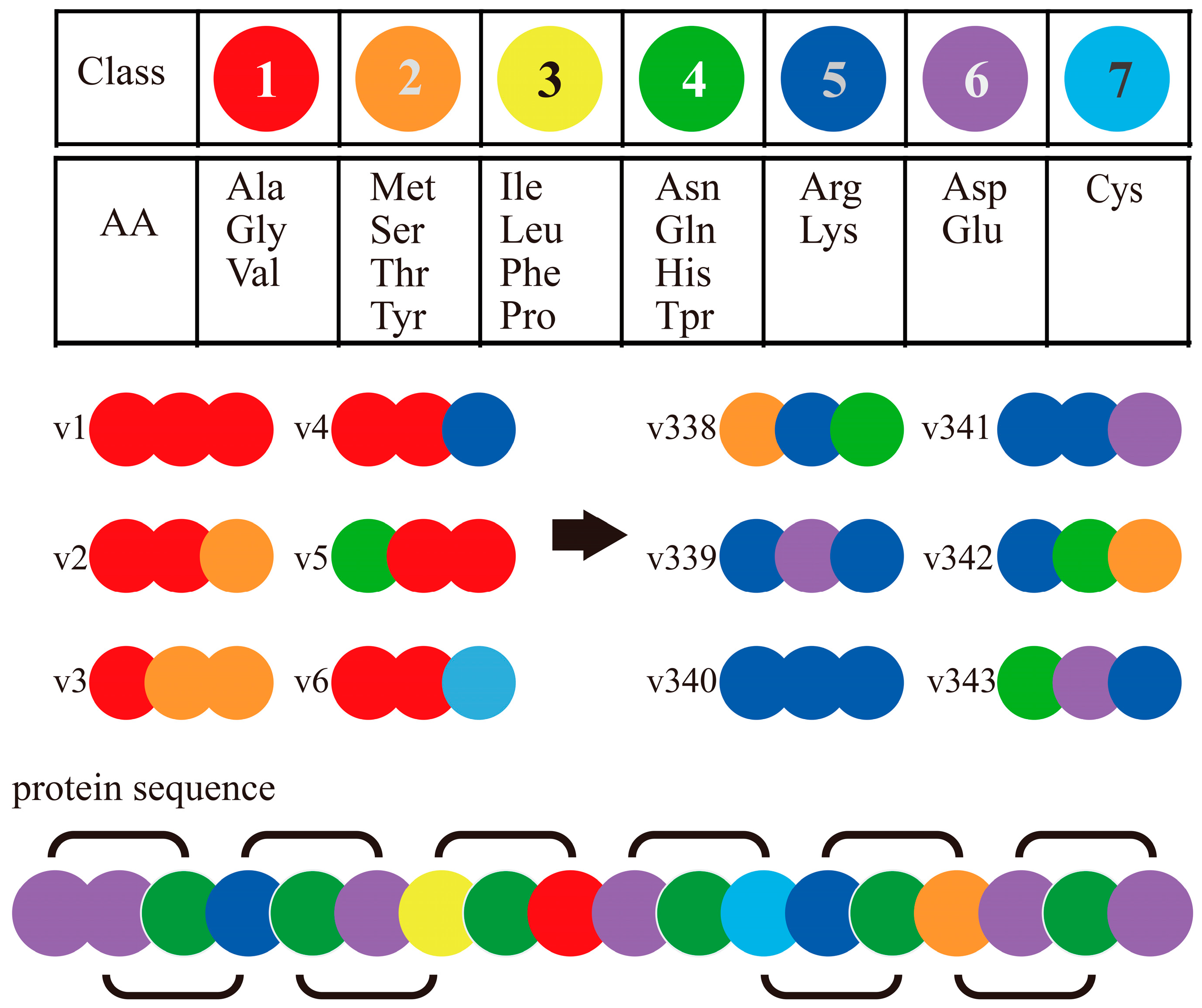

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Points/Edges | C | E | L | O | Last Update | Ref. I | Ref. II |
|---|---|---|---|---|---|---|---|---|---|
| BioGrid 3.4 | An interaction repository with data compiled through comprehensive curation efforts. | 65,099/836,212 | N | P | P | 61 | September 2016 | http://thebiogrid.org/ | [24] |
| IntAct 4.2.5 | Provides a strong, freely available, open source database system and analysis tools for molecular interaction data. | 93,856/653,104 | N | P | P | 8 | September 2016 | http://www.ebi.ac.uk/intact/ | [25] |
| PDB | A database containing experimentally determined three-dimensional structures of proteins. | 126,079/NA | N | P | P | NA | September 2016 | http://www.wwpdb.org/ | [36] |
| STRING | A database including protein interactions containing both physical and functional associations. | 9.6 million/184 million | P | P | P | 2031 | September 2016 | http://string-db.org/ | [26] |
| APID | Based on known experimentally validated PPIs and integrated interactomes with a methodological approach to report quality levels and coverage over the proteomes. | 90,379/678,441 | N | P | P | 25 | June 2016 | http://bioinfow.dep.usal.es/apid/ | [27] |
| DIP | A database combining experimental PPI information from a variety of sources. | 28,764/81,627 | N | P | P | 826 | Febrary 2014 | http://dip.doe-mbi.ucla.edu/dip/ | [28] |
| HitPredict 4 | A resource of experimentally determined PPI with reliability scores. | 70,808/398,696 | N | P | N | 105 | September 2015 | http://hintdb.hgc.jp/htp/ | [29] |
| MINT | Focuses on experimentally verified protein−protein interactions mined from the scientific literature by expert curators. | 25,530/125,464 | N | P | P | 611 | September 2013 | http://mint.bio.uniroma2.it/mint/ | [30] |
| TAIR-nbrowse | Provide Arabidopsis PPI data curated from the literature by TAIR curators. | 2452/8626 | N | P | P | 1 | September 2011 | http://www.arabidopsis.org/tools/nbrowse.jsp | [31] |
| HPRD Release 9 | A centralized platform to integrate interaction networks of human protein. | 30,047/41,327 | N | P | N | 1 | April 2010 | http://hprd.org/ | [32] |
| PINA2.0 | An integrated platform for protein interaction network construction, filtering, analysis, visualization and management. | 12,969/365,930 | N | P | N | 7 | May 2014 | http://cbg.garvan.unsw.edu.au/pina/ | [33] |
| Negatome 2.0 * | A collection containing experimentally supported non-interacting protein pairs and domain pairs which are unlikely engaged in direct physical interactions. | 3376/6532 | N | P | P | NA | 2014 | http://mips.helmholtz-muenchen.de/proj/ppi/negatome/ | [37] |
| Categories | Feature | Abbreviation | Ref. |
|---|---|---|---|
| EVO | Gene Fusion Event | FE | [21,22,45,46,47,48] |
| Gene Cluster | GCL | [21] | |
| Gene Neighborhood | GN | [21,22,45,47] | |
| Pylogenetic Profile | PP | [16,21,22,45,46,47,49,50] | |
| FF | GO Cellular Component | COM | [1,16,22,45,46,50,51,52] |
| Coessentiality | ESS | [45,50] | |
| Gene/Protein Coexpression | Exp | [1,16,18,22,45,46,47,50,52,53,54] | |
| GO Molecular Function | FUN | [1,16,22,45,46,48,50,52] | |
| Colocalization | Loc | [47,53,54] | |
| Ortholog/ Sequence Similar | ORT | [1,18,22,35,45,50,51,52,53,54,55,56] | |
| GO Biological Process | PRO | [1,16,18,22,45,46,47,48,50,51,52] | |
| Coregulation/Transcriptional Regulation | Reg | [45] | |
| TOP | Graphical Invariants | GI | [57] |
| Probabilistic Graphical Model | PGM | [57] | |
| Small-World Clustering Coefficients | SCC | [20,58] | |
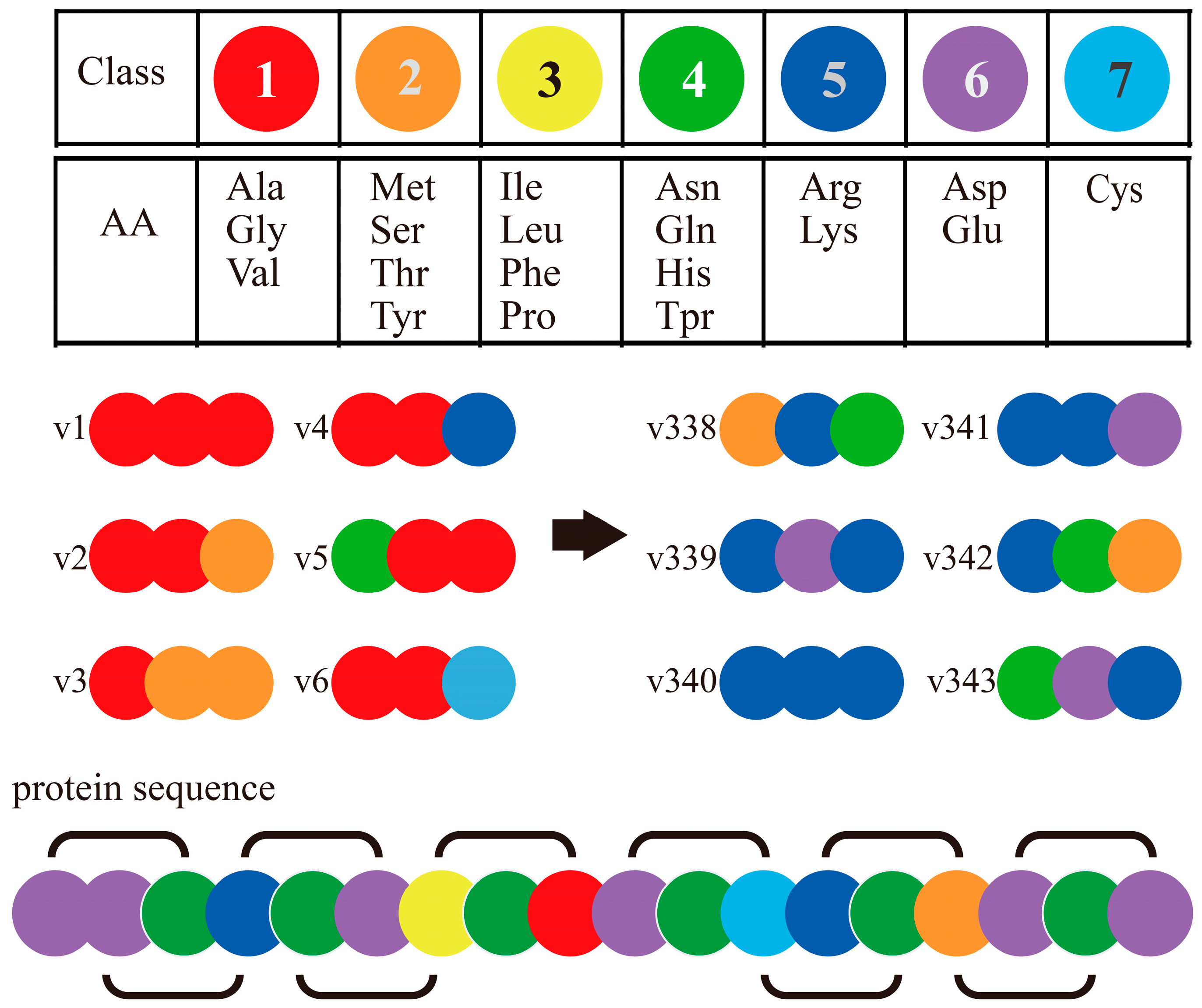
| SEQ | Conjoint Triad | COT | [35,58,59] |
| N-grams | NGR | [60,61] | |
| ORF Codon Usage | ORF | [62] | |
| Position-Specific Scoring Matrix | PSSM | [63,64,65] | |
| 2D Structure | 2DS | [20] | |
| STR | 3D Structure | 3DS | [46,66,67] |
| Average of the Cumulative Hydropathy Indices | ACH | [63,65] | |
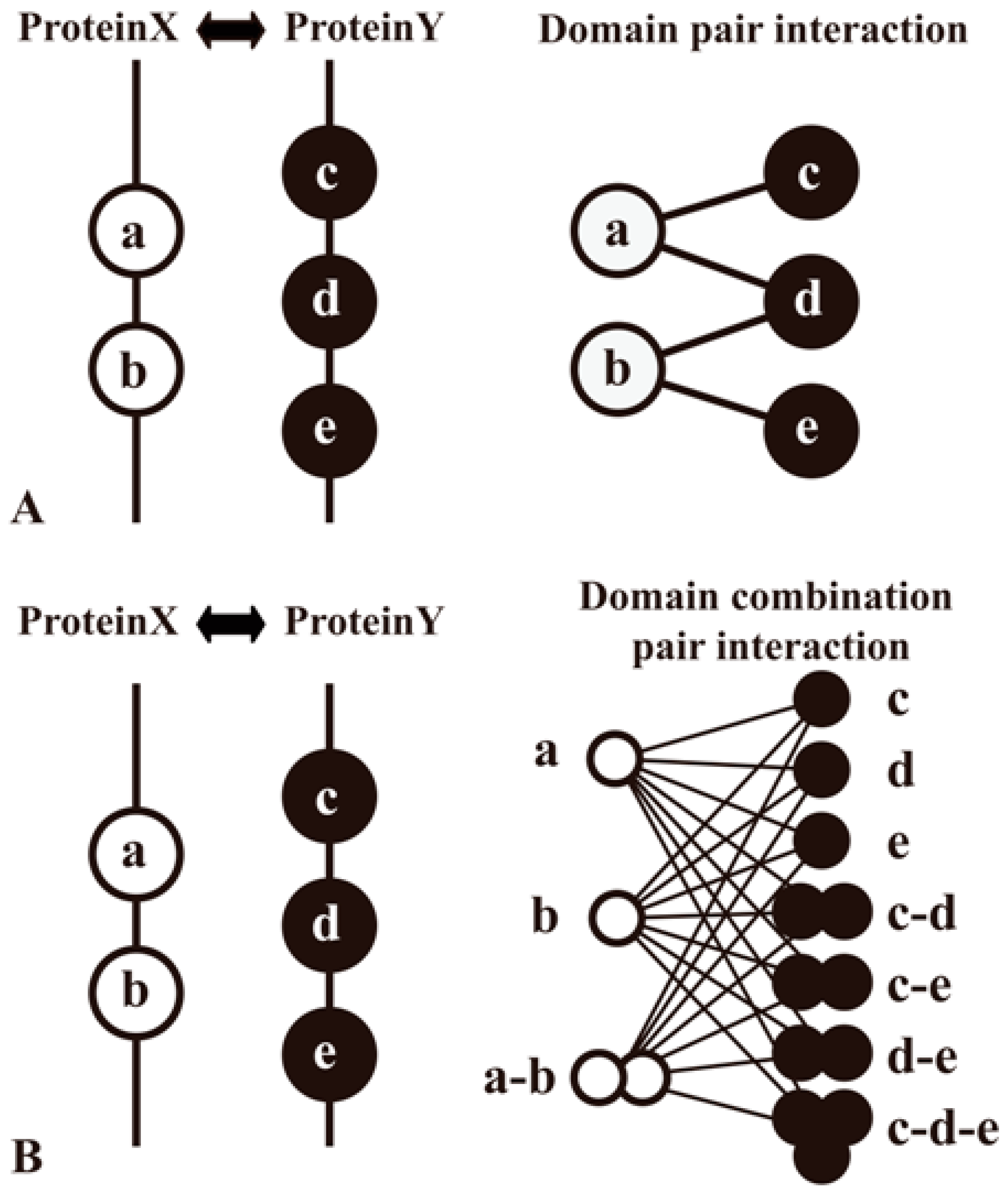
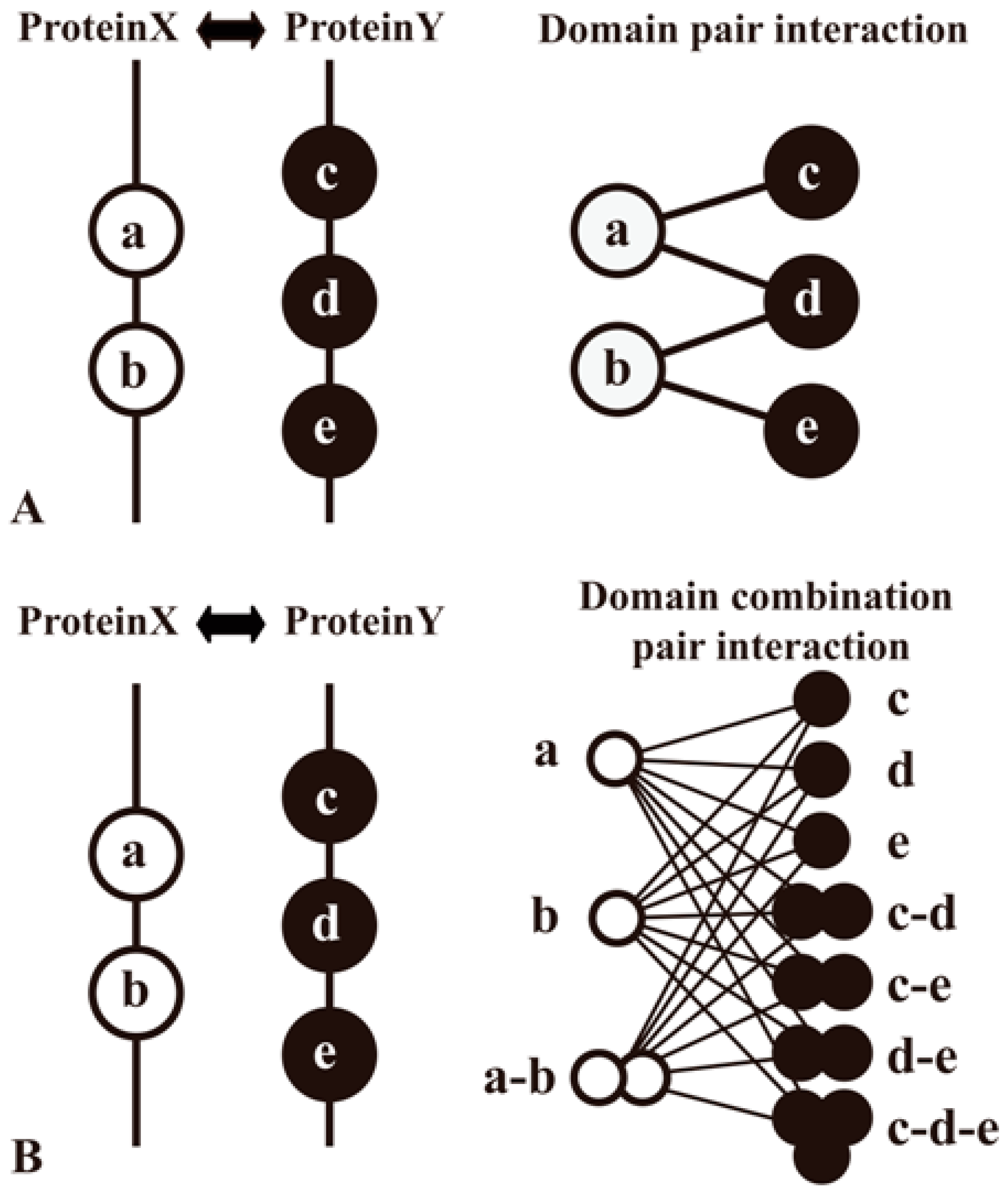
| Domain–Domain Interaction | DDI | [1,16,18,22,35,45,46,47,48,54] | |
| DSSP Structure in PDB | DSSP | [20] | |
| Electrostatics | ELE | [68] | |
| Protein Fold | Fold | [47] | |
| Generalized Born | GB | [68] | |
| High Quality AA Indices | HQI | [20] | |
| Predicted Accessibility | pA | [64] | |
| Physico-Chemical Properties | PHC | [20,57,66,67] | |
| PSIPRED Structure | PSIP | [1,20] | |
| Posttranslational Modifications | PTM | [1,54,68] | |
| Relative Solvent Accessibility | RSA | [56,63,65] | |
| Surface Area | SA | [68] | |
| Van Der Waals Forces | VDW | [68] | |
| TM | Literature-Curated | LC | [69] |
| Class | Description | Classifiers | Evidence | Organisms | Ref. | (URL) (Last Update) (Points/Edges) |
|---|---|---|---|---|---|---|
| DDI | iPfam: catalogs of protein family interactions, including domain and ligand interactions, calculated from known structures | NA | PHC, 3DS | NA | [66] | (http://ipfam.org/) (June 2013) (>9500/15,500) |
| DDI | 3did: database of three-dimensional interacting domains is a collection of DDIs in proteins for which high-resolution known 3D structures | NA | PHC, 3DS | NA | [67] | (http://3did.irbbarcelona.org/) (June 2016) (648/9952) |
| DDI | DOMINE is a database of known and predicted DDIs | POI | Exp, PP, FE, FUN, PRO, COM, DDI, 3DS | NA | [46,82] | (http://domine.utdallas.edu/cgi-bin/Domine) (September 2010) (5410/26,219) |
| DDI | Combine protein interaction datasets from multiple species to construct DDIs | NB, EC | FUN, PRO, GF, DDI, etc. | 4 (Hs, Dm, Sc, Ce) | [48] | NA |
| PPI | Predicting PPIs in Arabidopsis thaliana | EC | ORT, COM, PRO | 1 (At) | [51] | NA |
| PPI | CitrusNet: sweet orange PPI network | KNN | DDI, ORT, COT | 1 (Cs) | [35] | (http://citrus.hzau.edu.cn/orange/ppi/index.php) (June 2013) (8,195/124,491) |
| PPI | A predicted interactome for Arabidopsis. | EC | Exp, ORT, Loc | 1 (At) | [53] | NA |
| PPI | PRIN: a predicted rice interactome network | EC | FUN, PRO, COM, Exp, ORT | 1 (Os) | [52] | (http://bis.zju.edu.cn/prin/) (2010) (5049/76,585) |
| PPI | TSEMA: predicts the interaction between two families of proteins based on Monte Carlo approach | MC | PP | NA | [49] | (http://tsema.bioinfo.cnio.es/) |
| PPI | Predicting PPI using graph invariants and a neural network | NN | PGM, GI, PHC | NA | [57] | NA |
| PPI | IID: integrated interactions database providing tissue-specific PPIs for model organisms | EC | Exp, ORT, etc. | 6 (Sc, Ce, Dm, Mm, Hs, Rn) | [1] | (http://dcv.uhnres.utoronto.ca/iid/) (March 2016) (NA/1,741,568) |
| PPI | FpClass: interactions and properties of human proteins | association analysis | DDI, FUN, PRO, COM, PTM, Exp, ORT, PSIP | 1 (Hs) | [1] | (http://ophid.utoronto.ca/fpclass/) (NA) (10,531/250,498) |
| PPI | PAIR: the predicted Arabidopsis interactome resource | SVM | PP, PRO, FUN, COM, Exp, DDI | 1 (At) | [16] | (http://www.cls.zju.edu.cn/pair/) |
| PPI | SPPS: sequence-based protein partners search | SVM | COT | 5 (Sc, Ce, Dm, Ec, Hs) | [59] | (http://mdl.shsmu.edu.cn/SPPS/) (November 2011) (Hs = 23,719/39,191; Mm = 16,542/1225; Ce = 5348/4973; Dm = 8921/22,482; Sc = 16,506/25,064) |
| PPI | PIPs: human PPI prediction database | NB | Exp, ORT, DDI, Loc, PTM | 5 (Sc, Ce, Dm, Ec, Hs) | [54] | (http://www.compbio.dundee.ac.uk/www-pips/) (September 2008) (NA/79441) |
| PPI | Six classifiers and different biological data were used to predict interactions | RF, kRF, NB, DT, LR, SVM | Exp, FUN, PRO, COM, ESS, Reg, FE, GN, PP, ORT, DDI, etc. | NA | [45] | NA |
| PPI | SSWRF: an ensemble of SVM and SWRF method | SVM, SWRF | PSSM, ACH, RSA | NA | [65] | NA |
| PPI | Sequence-based approach is developed by combining MCD and SVM methods | MCD, SVM | COT, SeqS | 1 (Sc) | [58] | NA |
| PPI | PrePPI: predicts PPI using both structural and nonstructural information | LR | ORT, FUN, PRO, COM, ESS, Exp, PP, etc. | 2 (Sc, Hs) | [50,91] | (http://bhapp.c2b2.columbia.edu/PrePPI/) (August 2011) (Sc = NA/31,402; Hs = NA/317,813) |
| PPI | MLPPI: multi-level machine learning prediction of PPI in yeast | SVM | 2DS&PHC (PSIP, DSSP, HQI), SEQ, etc. | 1(Sc) | [20] | (http://zubekj.github.io/mlppi/) (NA) (NA) |
| PPI | Probabilistic model of the human PPI network | NB | PRO, Exp, ORT, DDI | 1 (Hs) | [18] | NA |
| PPI | Characterization and prediction of PPI in the yeast | LR | DDI, Fold, FE, PP, GN, Loc, PRO, Exp | 1 (Sc) | [47] | NA |
| PPI | InPrePPI method: an integrated method for prediction of PPI | AC | GCL, PP, FE, GN | 1 (Ec) | [21] | (http://inpreppi.biosino.org/InPrePPI/index.jsp) (NA) (6,429/17,855) |
| PPI | Global genome-scale PPI network in Arabidopsis thaliana. | NB | ORT, FE, GN, PP, FUN, PRO, COM, Exp, DDI | 1 (At) | [22] | NA |
| PPIS | LORIS method: sequence-based L1-logreg classifier proposed to identify PPIS | L1-logreg | PSSM, ACH, RSA | NA | [63] | NA |
| PPIS | Struct2Net, iWRAP & Coev2Net | PGM, LR, etc. | ORT | 3 (Hs, Sc, Dm) | [55] | (http://groups.csail.mit.edu/cb/struct2net/webserver) (2012) (NA) |
| PPIS | PRISM2: protein interactions by structural matching | EC | RSA, ORT | NA | [56] | (http://cosbi.ku.edu.tr/prism/) (NA) (NA) |
| PPIS | MIEC-SVM: structure-based method for predicting protein recognition specificity | SVM | VDW, ELE, GB, SA, PTM, etc. | NA | [68] | (http://wanglab.ucsd.edu/MIEC-SVM/) (NA) (NA) |
| PPIS | PSIVER method | NB, KDE | PSSM, pA | NA | [64] | NA |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-W.; Zhou, Y.-Q.; Ul Qamar, M.T.; Chen, L.-L.; Ding, Y.-D. Prediction of Protein–Protein Interactions by Evidence Combining Methods. Int. J. Mol. Sci. 2016, 17, 1946. https://doi.org/10.3390/ijms17111946
Chang J-W, Zhou Y-Q, Ul Qamar MT, Chen L-L, Ding Y-D. Prediction of Protein–Protein Interactions by Evidence Combining Methods. International Journal of Molecular Sciences. 2016; 17(11):1946. https://doi.org/10.3390/ijms17111946
Chicago/Turabian StyleChang, Ji-Wei, Yan-Qing Zhou, Muhammad Tahir Ul Qamar, Ling-Ling Chen, and Yu-Duan Ding. 2016. "Prediction of Protein–Protein Interactions by Evidence Combining Methods" International Journal of Molecular Sciences 17, no. 11: 1946. https://doi.org/10.3390/ijms17111946