
Genetic Heterogeneity of HER2 Amplification and Telomere Shortening in Papillary Thyroid Carcinoma


 ,
,
Abstract
:
1. Introduction
2. Results
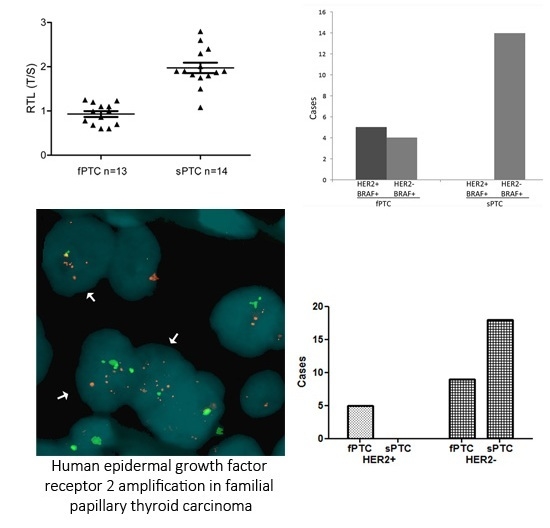
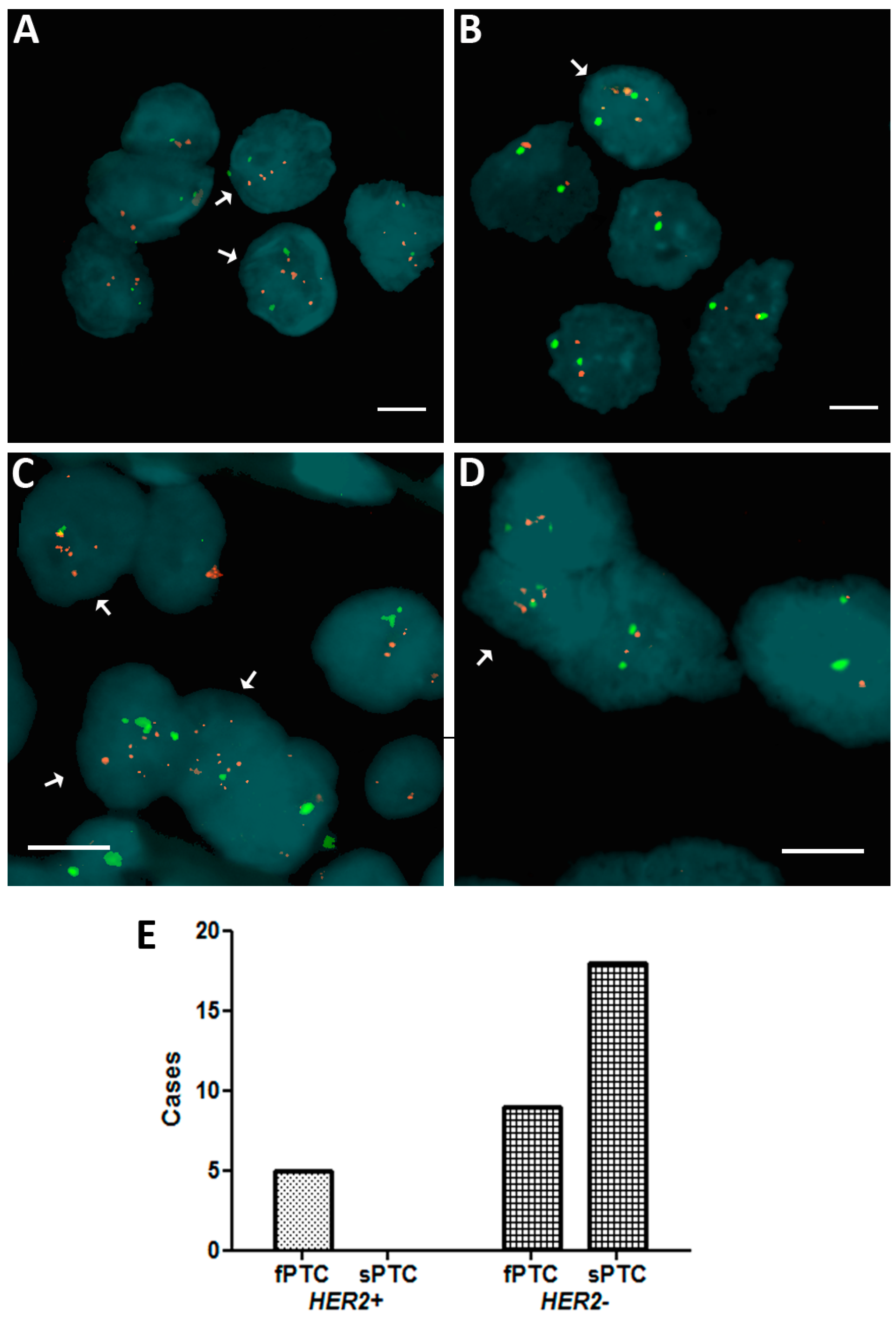
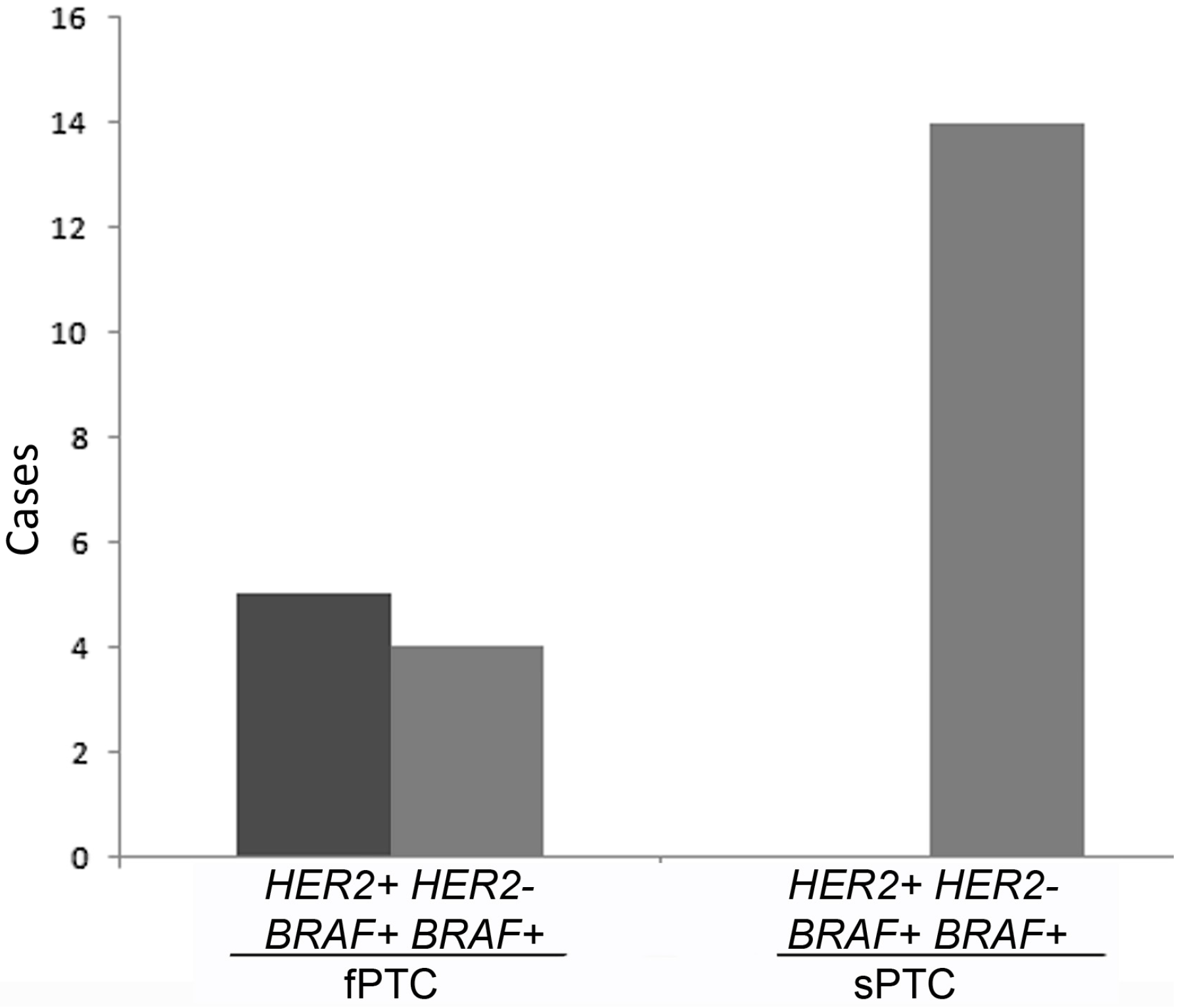
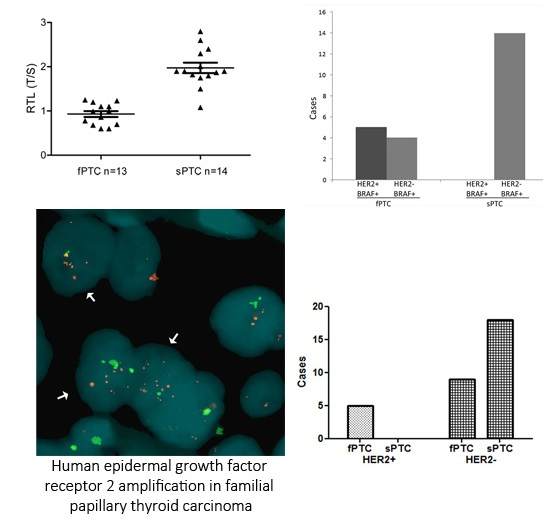
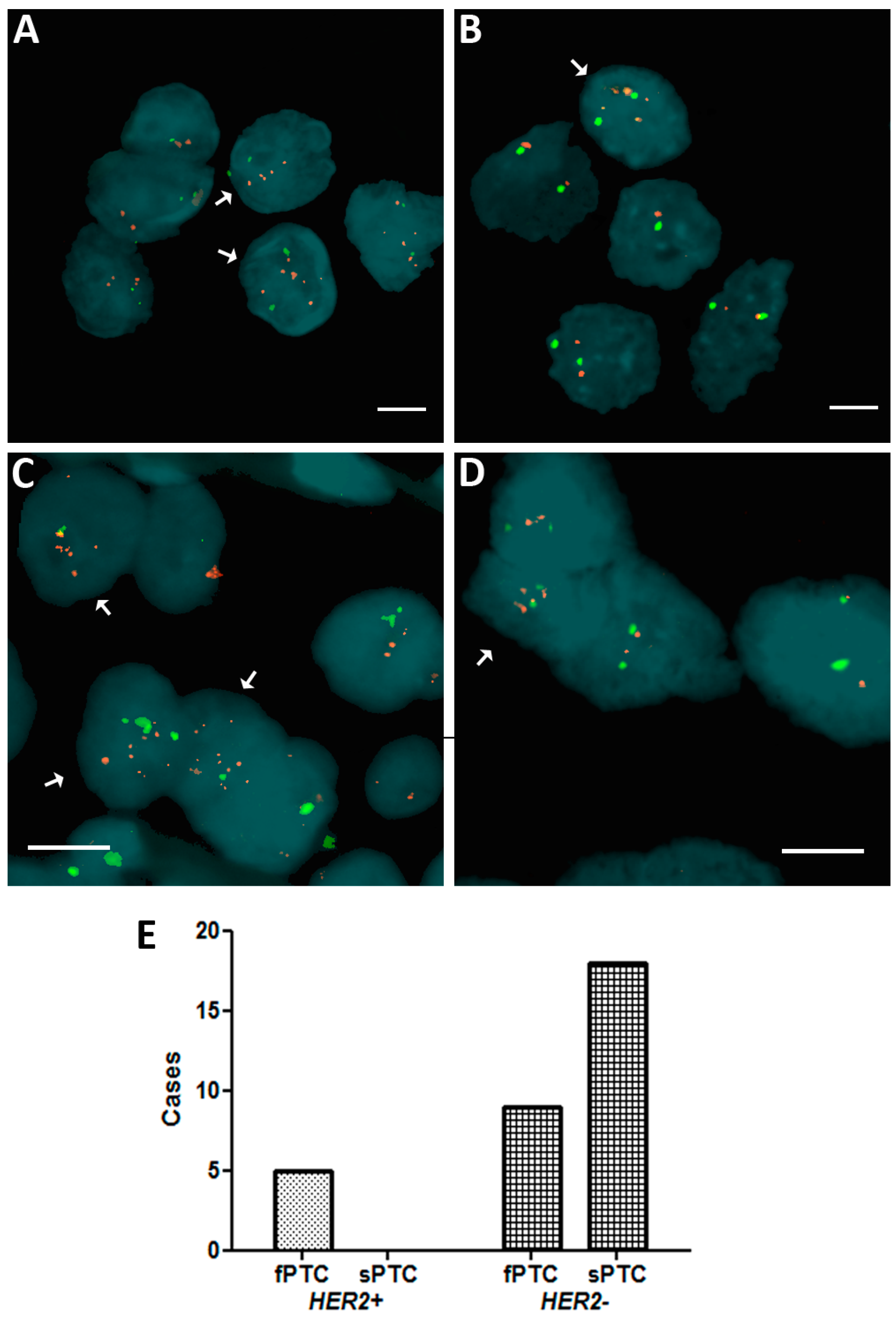
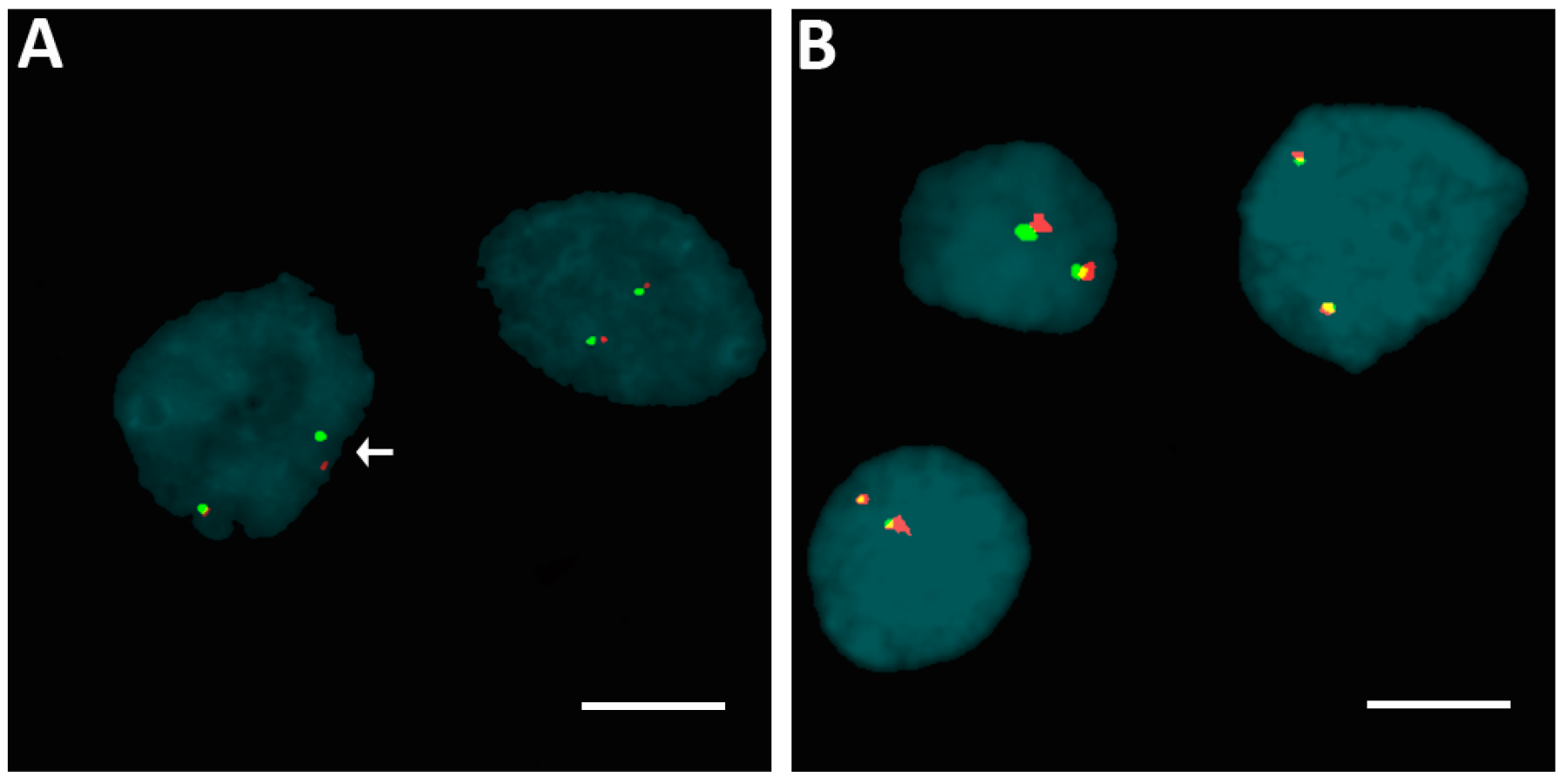
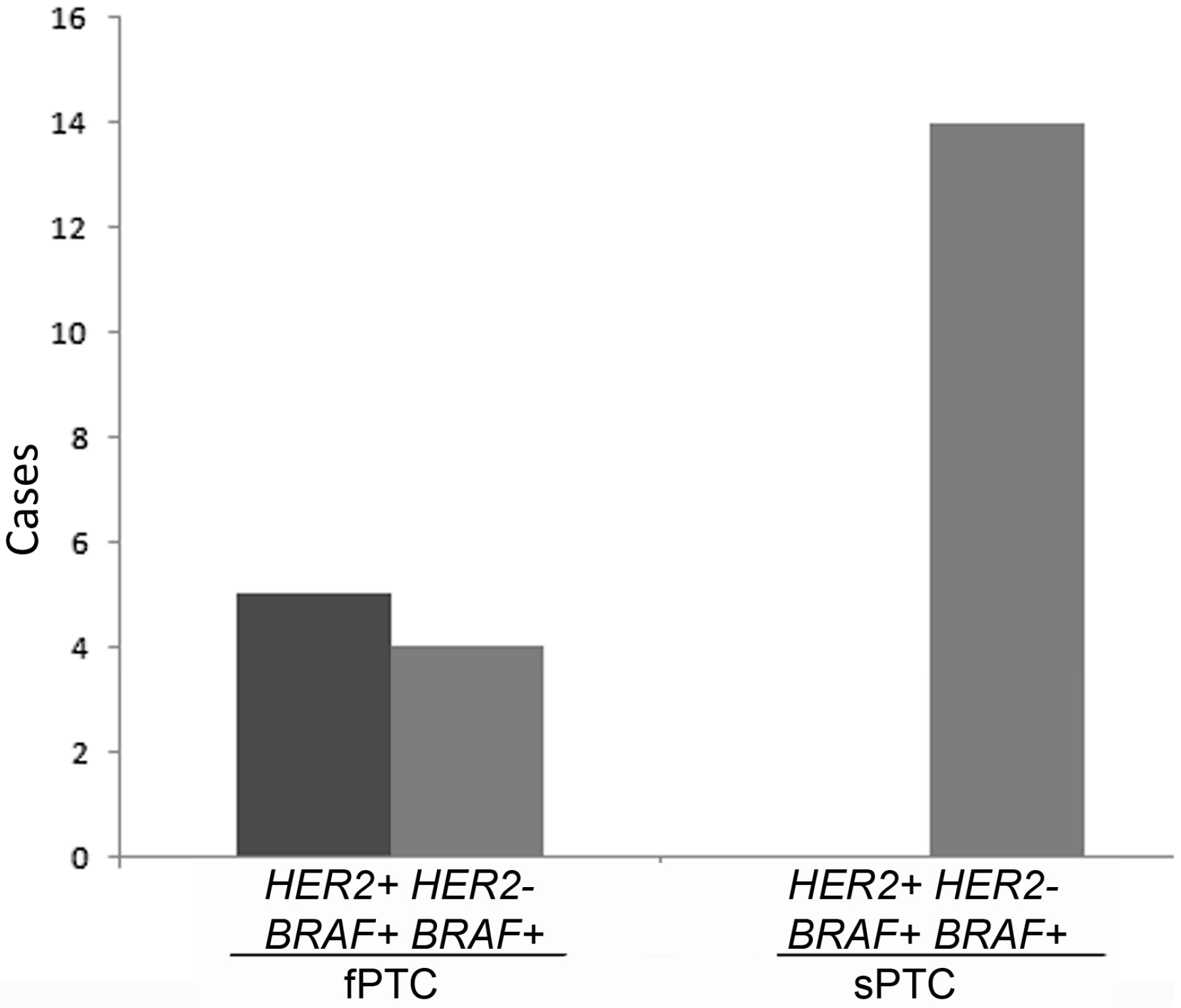
2.1. Human Epidermal Growth Factor Receptor 2 (HER2) Amplification in Familial Papillary Thyroid Carcinoma (fPTC) and Sporadic Papillary Thyroid Carcinoma (sPTC)
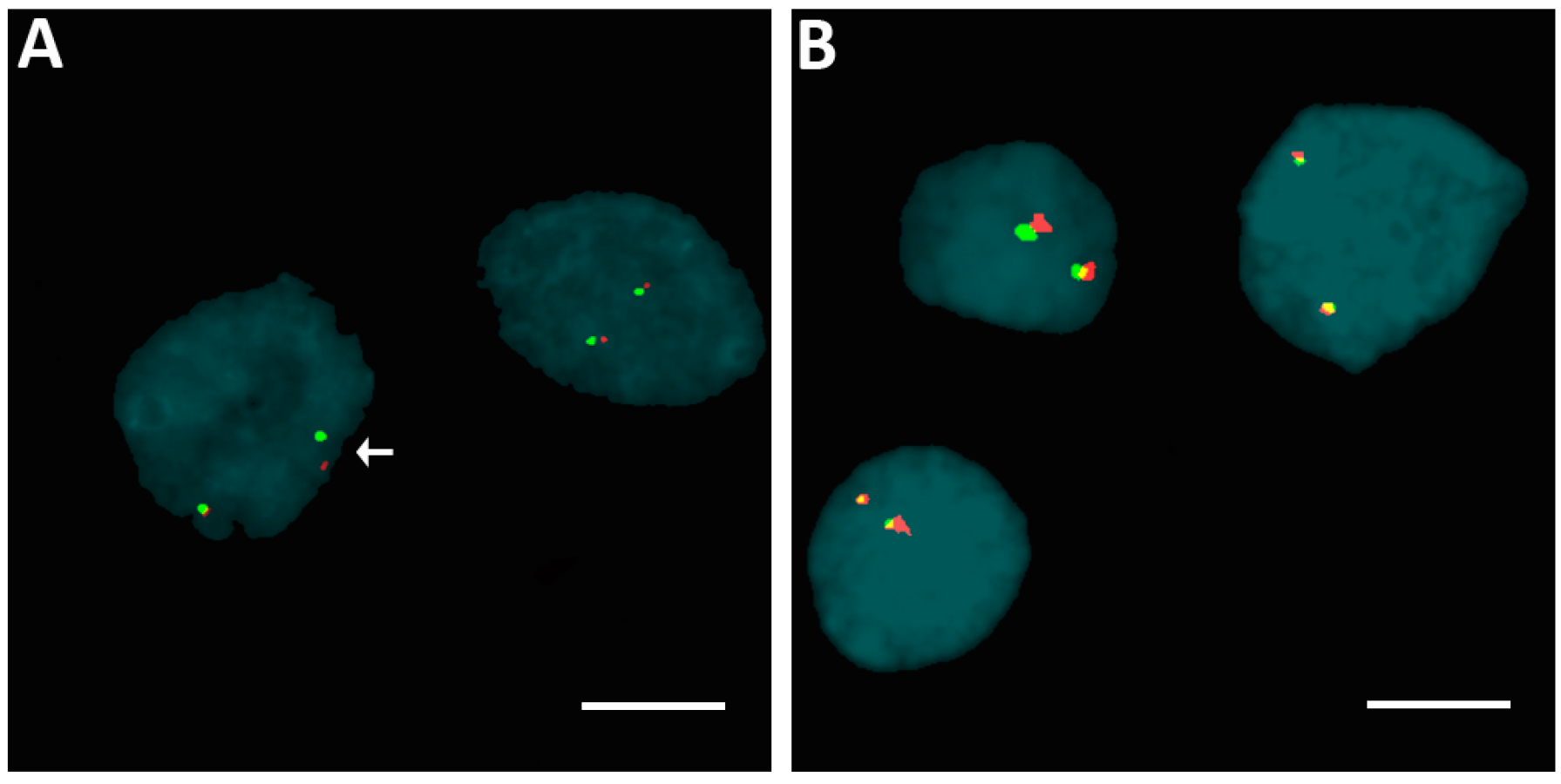
2.2. Rearrangements of ALK, BRAF, ETV6, and RET Genes
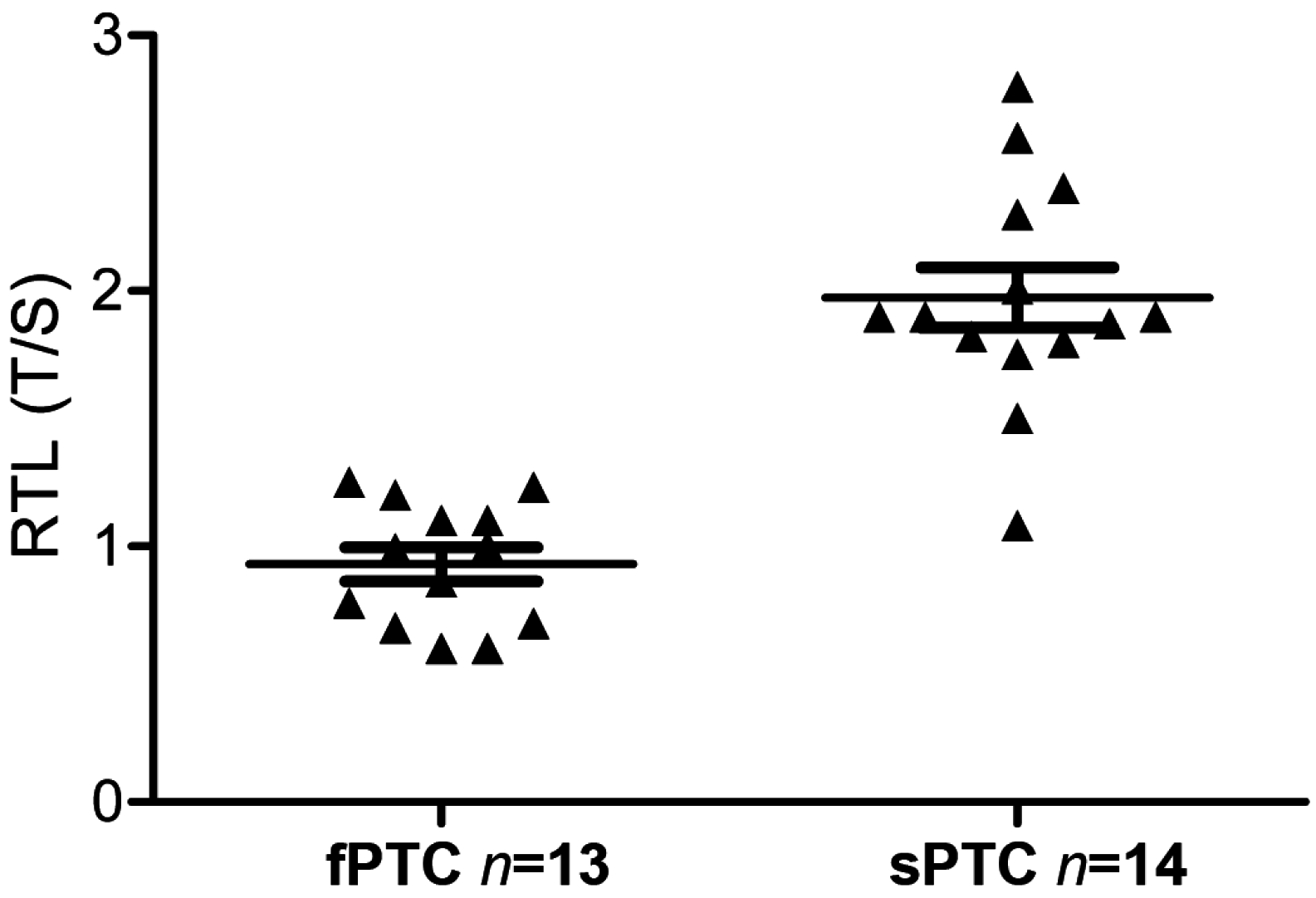
2.3. Telomere Length and BRAFV600E Mutation in fPTC and sPTC Patients
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Fluorescence In Situ Hybridization
4.3. Detection of HER2 Gene Copy Number Alterations or Amplification
4.4. Telomere Length and BRAFV600E Mutation
4.5. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thyroid Disease Manager. Available online: http://www.thyroidmanager.org/ (accessed on 31 July 2016).
- Bonora, E.; Tallini, G.; Romeo, G. Genetic predisposition to familial nonmedullary thyroid cancer: An update of molecular findings and state-of-the-art studies. J. Oncol. 2010, 385206. [Google Scholar] [CrossRef] [PubMed]
- Moses, W.; Weng, J.; Kebebew, E. Prevalence, clinicopathologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid 2011, 21, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Xing, M. Absence of germline mutations in genes within the MAP kinase pathway in familial non medullary thyroid cancer. Cell Cycle 2006, 5, 2036–2039. [Google Scholar] [CrossRef] [PubMed]
- Gara, S.K.; Jia, L.; Merino, M.J.; Agarwa, S.K.; Zhang, L.; Cam, M.; Patel, D.; Kebebew, E. Germline HABP2 mutation causing familial non medullary thyroid cancer. N. Engl. J. Med. 2015, 373, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Carmona, L.G.; Tomlinson, I.; Sahasrabudhe, R. Re: HABP2 G534E mutation in familial nonmedullary thyroid cancer. J. Natl. Cancer Inst. 2016, 108, djw108. [Google Scholar] [CrossRef] [PubMed]
- Weeks, A.L.; Wilson, S.G.; Ward, L.; Goldblatt, J.; Hui, J.; Walsh, J.P. HABP2 germline variants are uncommon in familial nonmedullary thyroid cancer. BMC Med. Genet. 2016, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Tomsic, J.; Fultz, R.; Liyanarachchi, S.; He, H.; Senter, L.; de la Chapelle, A. HABP2 G534E variant in papillary thyroid carcinoma. PLoS ONE 2016, 11, e0146315. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, P.; Mazzi, V.; Vita, R.; Ferrari, S.M.; Materazzi, G.; Galleri, D.; Benvenga, S.; Miccoli, P.; Antonelli, A. New therapies for dedifferentiated papillary thyroid cancer. Int. J. Mol. Sci. 2015, 16, 6153–6182. [Google Scholar] [CrossRef] [PubMed]
- Bikas, A.; Vachhani, S.; Jensen, K.; Vasko, V.; Burman, K.D. Targeted therapies in thyroid cancer: An extensive review of the literature. Expert Rev. Clin. Pharmacol. 2016, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E. Thyroid cancer in 2015: Molecular landscape of thyroid cancer continues to be deciphered. Nat. Rev. Endocrinol. 2016, 12, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.; Li, S.; Garcia-Rendueles, M.E.; Montero-Conde, C.; Voza, F.; Knauf, J.A.; Heguy, A.; Viale, A.; Bogdanova, T.; Thomas, G.A.; Mason, C.E.; et al. Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J. Clin. Investig. 2013, 123, 4935–4944. [Google Scholar] [CrossRef] [PubMed]
- Sugishita, Y.; Kammori, M.; Yamada, O.; Poon, S.S.; Kobayashi, M.; Onoda, N.; Yamazaki, K.; Fukumori, T.; Yoshikawa, K.; Onose, H.; et al. Amplification of the human epidermal growth factor receptor 2 gene in differentiated thyroid cancer correlates with telomere shortening. Int. J. Oncol. 2013, 42, 1589–1596. [Google Scholar] [PubMed]
- Capezzone, M.; Cantara, S.; Marchisotta, S.; Busonero, G.; Formichi, C.; Benigni, M.; Capuano, S.; Toti, P.; Pazaitou-Panayiotou, K.; Caruso, G.; et al. Telomere length in neoplastic and nonneoplastic tissues of patients with familial and sporadic papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2011, 96, E1852–E1856. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Haugen, B.R.; Schlumberger, M. Progress in molecular-based management of differentiated thyroid cancer. Lancet 2013, 381, 1058–1069. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef] [PubMed]
- Vance, G.H.; Barry, T.S.; Bloom, K.J.; Fitzgibbons, P.L.; Hicks, D.G.; Jenkins, R.B.; Persons, D.L.; Tubbs, R.R.; Hammond, M.E.H. Genetic heterogeneity in HER2 testing in breast cancer panel summary and guidelines. Arch. Pathol. Lab. Med. 2009, 133, 611–612. [Google Scholar] [PubMed]
- Wright, C.; Angus, B.; Nicholson, S.; Sainsbury, J.R.; Cairns, J.; Gullick, W.J.; Kelly, P.; Harris, A.L.; Horne, C.H. Expression of c-erbB-2 oncoprotein: A prognostic indicator in human breast cancer. Cancer Res. 1989, 49, 2087–2090. [Google Scholar] [PubMed]
- Caria, P.; Dettori, T.; Frau, D.V.; Borghero, A.; Cappai, A.; Riola, A.; Lai, M.L.; Boi, F.; Calò, P.; Nicolosi, A.; et al. Assessing RET/PTC in thyroid nodule fine-needle aspirates: The FISH point of view. Endocr. Relat. Cancer 2013, 20, 527–536. [Google Scholar] [CrossRef] [PubMed]
- De Lellis, R.A. WHO Classification of Tumours, Pathology and Genetics of Tumours of Endocrine Organs, 3rd ed.; Ronald, A., de Lellis, R.A., Riccardo, V.L., Philipp, U.H., Charis, E., Eds.; IARC Press: Lyon, France, 2004; Volume 8, p. 230. [Google Scholar]
- Ronckers, C.M.; McCarron, P.; Ron, E. Thyroid cancer and multiple primary tumors in the SEER cancer registries. Int. J. Cancer 2005, 117, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Omür, O.; Ozcan, Z.; Yazici, B.; Akgün, A.; Oral, A.; Ozkiliç, H. Multiple primary tumors in differentiated thyroid carcinoma and relationship to thyroid cancer outcome. Endocr. J. 2008, 55, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J. The distinctive nature of HER2-positive breast cancers. N. Engl. J. Med. 2005, 353, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Joseph, K.R.; Edirimanne, S.; Eslick, G.D. The association between breast cancer and thyroid cancer: A meta-analysis. Breast Cancer Res. Treat. 2015, 152, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, M.; Farkas, D.K.; Ehrenstein, V.; Jørgensen, J.O.; Dekkers, O.M.; Sorensen, H.T. Hypothyroidism and hyperthyroidism and breast cancer risk: A nationwide cohort study. Eur. J. Endocrinol. 2016, 174, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.P.; Chen, J.; Hitchcock, Y.J.; Szabo, A.; Shrieve, D.C.; Tward, J.D. The risk of second primary malignancies up to three decades after the treatment of differentiated thyroid cancer. J. Clin. Endocrinol. Metab. 2008, 93, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Verkooijen, R.B.; Smit, J.W.; Romijn, J.A.; Stokkel, M.P. The incidence of second primary tumors in thyroid cancer patients is increased, but not related to treatment of thyroid cancer. Eur. J. Endocrinol. 2006, 155, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, V.; Eslick, G.D. HER2 expression in gastric and oesophageal cancer: A metaanalytic review. J. Gastrointest. Cancer 2015, 6, 143–154. [Google Scholar]
- Verri, E.; Guglielmini, P.; Puntoni, M.; Perdelli, L.; Papadia, A.; Lorenzi, P.; Rubagotti, A.; Ragni, N.; Boccardo, F. HER2/neuoncoprotein overexpression in epithelial ovarian cancer: Evaluation of its prevalence and prognostic significance. Oncology 2005, 68, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.N.; Kwak, Y.; Kim, D.W.; Kang, S.B.; Choe, G.; Kim, W.H.; Lee, H.S. HER2 status in colorectal cancer: Its clinical significance and the relationship between HER2 gene amplification and expression. PLoS ONE 2014, 9, e98528. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Capezzone, M.; Marchisotta, S.; Cantara, S.; Busonero, G.; Brilli, L.; Pazaitou-Panayiotou, K.; Carli, A.F.; Caruso, G.; Toti, P.; Capitani, S.; et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr. Relat. Cancer 2008, 15, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Shong, Y.K.; Kim, T.Y.; Viola, D.; Elisei, R.; Bendlová, B.; Yip, L.; Mian, C.; et al. Association between BRAF V600E mutation and recurrence of papillary thyroid cancer. J. Clin. Oncol. 2015, 33, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Qiu, T.; Ling, Y.; Guo, L.; Zheng, B.; Wang, B.; Li, W.; Li, L.; Ying, J. Prevalence and clinicopathological characteristics of HER2 and BRAF mutation in Chinese patients with lung adenocarcinoma. PLoS ONE 2015, 10, e0130447. [Google Scholar] [CrossRef] [PubMed]
- Cantara, S.; Pisu, M.; Frau, D.V.; Caria, P.; Dettori, T.; Capezzone, M.; Capuano, S.; Vanni, R.; Pacini, F. Telomere abnormalities and chromosome fragility in patients affected by familial papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2012, 97, E1327–E1331. [Google Scholar] [CrossRef] [PubMed]
- Meeker, A.K.; Hicks, J.L.; Iacobuzio-Donahue, C.A.; Montgomery, E.A.; Westra, W.H.; Chan, T.Y.; Ronnett, B.M.; De Marzo, A.M. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin. Cancer Res. 2004, 10, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Jonson, T.; Petersen, A.; Strombeck, B.; Dal Cin, P.; Hoglund, M.; Mitelman, F.; Mertens, F.; Mandahl, N. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 12683–12688. [Google Scholar] [CrossRef] [PubMed]
- Gadji, M.; Vallente, R.; Klewes, L.; Righolt, C.; Wark, L.; Kongruttanachok, N.; Knecht, H.; Mai, S. Nuclear Remodeling as a Mechanism for Genomic Instability in Cancer; Gisselsson, D., Ed.; Academic Press: New York, NY, USA, 2011; Volume 112, pp. 77–126. [Google Scholar]
- Gadji, M.; Adebayo Awe, J.; Rodrigues, P.; Kumar, R.; Houston, D.S.; Klewes, L.; Dièye, T.N.; Rego, E.M.; Passetto, R.F.; de Oliveira, F.M.; et al. Profiling three-dimensional nuclear telomeric architecture of myelodysplastic syndromes and acute myeloid leukemia defines patient subgroups. Clin. Cancer Res. 2012, 18, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Gartner, J.; Mai, S. Identification of neuroblastoma subgroups based on three-dimensional telomere organization. Transl. Oncol. 2016, 9, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Wark, L.; Danescu, A.; Natarajan, S.; Zhu, X.; Cheng, S.Y.; Hombach-Klonisch, S.; Mai, S.; Klonisch, T. Three-dimensional telomere dynamics in follicular thyroid cancer. Thyroid 2014, 24, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [PubMed]
- Liu, R.; Bishop, J.; Zhu, G.; Zhang, T.; Ladenson, P.W.; Xing, M. Mortality risk stratification by combining BRAF V600E and TERT promoter mutations in papillary thyroid cancer: Genetic duet of BRAF and TERT promoter mutations in thyroid cancer mortality. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.L.; Sorensen, M.C.; Pedersen, S.; Rasmussen, M. Fluorescence in situ hybridization on formalin-fixed and paraffin-embedded tissue: Optimizing the method. Appl. Immunohistochem. Mol. Morphol. 2004, 12, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Caria, P.; Frau, D.V.; Dettori, T.; Boi, F.; Lai, M.L.; Mariotti, S.; Vanni, R. Optimizing detection of RET and PPARg rearrangements in thyroid neoplastic cells using a home-brew tetracolor probe. Cancer Cytopathol. 2014, 122, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, R.; Bown, N.; Tibiletti, M.G.; Debiec-Rychter, M.; Vanni, R.; Espinet, B.; van Roy, N.; Roberts, P.; van den Berg-de-Ruiter, E.; Bernheim, A.; et al. Guidelines for cytogenetic investigations in tumours. Eur. J. Hum. Genet. 2015, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cantara, S.; Capezzone, M.; Marchisotta, S.; Capuano, S.; Busonero, G.; Toti, P.; di Santo, A.; Caruso, G.; Carli, A.F.; Brilli, L.; et al. Impact of proto-oncogene mutation detection in cytological specimens from thyroid nodules improves the diagnostic accuracy of cytology. J. Clin. Endocrinol. Metab. 2010, 95, 1365–1369. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | Age at Diagnosis (Mean ± SD) | Sex (Males %) | PTC Size (Median/IQR) | TNM | Extrathyroidal Invasion N (%) | Multifocality N (%) | Lymphonode Metastases at Diagnosis N (%) | Final Outcome * N (%) | Follow-up (Mean Years) | Histology |
|---|---|---|---|---|---|---|---|---|---|---|
| fPTC (n = 13) | 51.5 ± 17.0 | 6 (46.1%) | 11/11.5 | pT1 8 (61.5%) pT2 2 (11.7%) pT3 3 (23.0%) | 5 (38.5%) | 7 (53.8%) | 3 (23%) | Remission 9 (69.2%) Persistent disease 4 (30.8%) | 7.59 ± 3.9 | 9 CV-PTC 4 FV-PTC |
| sPTC (n = 18) | 46.1 ± 8.5 | 3 (16.6%) | 9.5/8.5 | pT1 8 (44.4%) pT2 1 (5.5%) pT3 9 (50.%) | 9 (50%) | 6 (33.3%) | 5 (27.7%) | Remission 11 (61.1%) Persistent disease 2 (11.1%) | 5.5 ± 2.8 | 14 CV-PTC 2 FV-PTC 1 SV-PTC 1 TR-PTC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caria, P.; Cantara, S.; Frau, D.V.; Pacini, F.; Vanni, R.; Dettori, T. Genetic Heterogeneity of HER2 Amplification and Telomere Shortening in Papillary Thyroid Carcinoma. Int. J. Mol. Sci. 2016, 17, 1759. https://doi.org/10.3390/ijms17101759
Caria P, Cantara S, Frau DV, Pacini F, Vanni R, Dettori T. Genetic Heterogeneity of HER2 Amplification and Telomere Shortening in Papillary Thyroid Carcinoma. International Journal of Molecular Sciences. 2016; 17(10):1759. https://doi.org/10.3390/ijms17101759
Chicago/Turabian StyleCaria, Paola, Silvia Cantara, Daniela Virginia Frau, Furio Pacini, Roberta Vanni, and Tinuccia Dettori. 2016. "Genetic Heterogeneity of HER2 Amplification and Telomere Shortening in Papillary Thyroid Carcinoma" International Journal of Molecular Sciences 17, no. 10: 1759. https://doi.org/10.3390/ijms17101759