
The Inflammatory Role of Platelets: Translational Insights from Experimental Studies of Autoimmune Disorders


Abstract
:
1. Introduction
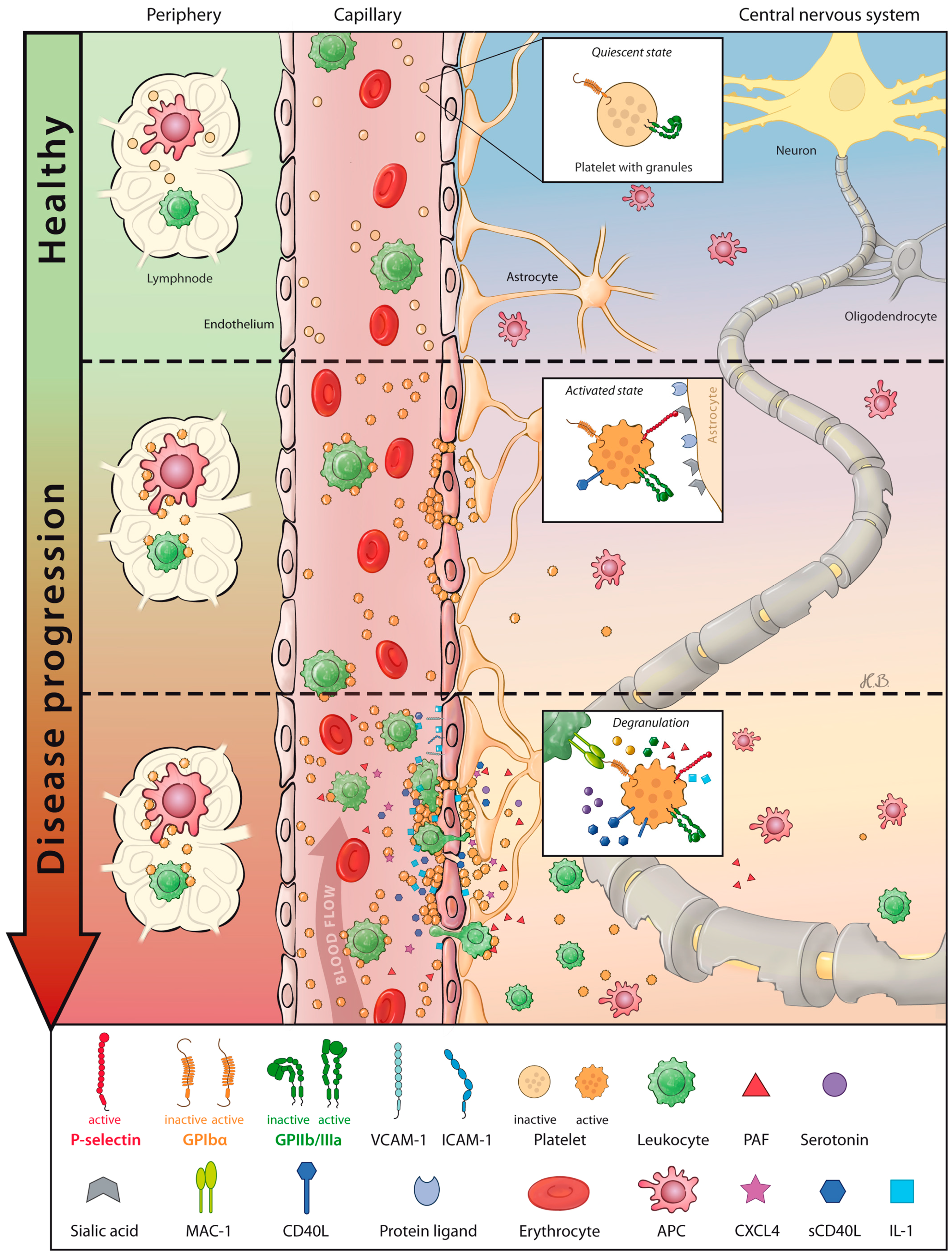
2. Platelets—Cellular Mediators of (Neuro-) Inflammation
3. Role of Platelet-Driven Neuroinflammation in Multiple Sclerosis
4. Role of Platelet-Driven Immune Responses in Other Non-Neurological Inflammatory Disorders
5. Rheumatoid Arthritis
Inflammatory Bowel Disease
6. Future Prospects/Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Ghoshal, K.; Bhattacharyya, M. Overview of platelet physiology: Its hemostatic and nonhemostatic role in disease pathogenesis. Sci. World J. 2014, 2014, 781857. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Gawaz, M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb. Haemost. 2008, 99, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet Activation: The Mechanisms and Potential Biomarkers. BioMed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Budnik, I.; Shenkman, B.; Savion, N. Synergistic effect of signaling from receptors of soluble platelet agonists and outside-in signaling in formation of a stable fibrinogen-integrin αIIbβ3-actin cytoskeleton complex. Thromb. Res. 2015, 135, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P. Arterial thrombosis—Insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Van Rooy, M.J.; Pretorius, E. Metabolic syndrome, platelet activation and the development of transient ischemic attack or thromboembolic stroke. Thromb. Res. 2015, 135, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Patzelt, J.; Verschoor, A.; Langer, H.F. Platelets and the complement cascade in atherosclerosis. Front. Physiol. 2015, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Nording, H.M.; Seizer, P.; Langer, H.F. Platelets in inflammation and atherogenesis. Front. Physiol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Chavakis, T. Leukocyte-endothelial interactions in inflammation. J. Cell. Mol. Med. 2009, 13, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Herter, J.M.; Rossaint, J.; Zarbock, A. Platelets in inflammation and immunity. J. Thromb. Haemost. 2014, 12, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [PubMed]
- Rondina, M.T.; Garraud, O. Emerging evidence for platelets as immune and inflammatory effector cells. Front. Immunol. 2014, 5, 653. [Google Scholar] [CrossRef] [PubMed]
- Horstman, L.L.; Jy, W.; Ahn, Y.S.; Zivadinov, R.; Maghzi, A.H.; Etemadifar, M.; Alexander, J.S.; Minagar, A. Role of platelets in neuroinflammation: A wide-angle perspective. J. Neuroinflamm. 2010, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Lannan, K.L.; Sahler, J.; Kim, N.; Spinelli, S.L.; Maggirwar, S.B.; Garraud, O.; Cognasse, F.; Blumberg, N.; Phipps, R.P. Breaking the mold: Transcription factors in the anucleate platelet and platelet-derived microparticles. Front. Immunol. 2015, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Horstman, L.L.; Ahn, Y.S. Platelet microparticles: A wide-angle perspective. Crit. Rev. Oncol. Hematol. 1999, 30, 111–142. [Google Scholar] [CrossRef]
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The Inflammatory Role of Platelets via Their TLRs and Siglec Receptors. Front. Immunol. 2015, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Oury, C.; Lecut, C.; Hego, A.; Wera, O.; Delierneux, C. Purinergic control of inflammation and thrombosis: Role of P2X1 receptors. Comput. Struct. Biotechnol. J. 2015, 13, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Mezger, M.; Gobel, K.; Kraft, P.; Meuth, S.G.; Kleinschnitz, C.; Langer, H.F. Platelets and vascular inflammation of the brain. Hamostaseologie 2015, 35, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Steinman, L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron 2009, 64, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Gobel, K.; Pankratz, S.; Schneider-Hohendorf, T.; Bittner, S.; Schuhmann, M.K.; Langer, H.F.; Stoll, G.; Wiendl, H.; Kleinschnitz, C.; Meuth, S.G. Blockade of the kinin receptor B1 protects from autoimmune CNS disease by reducing leukocyte trafficking. J. Autoimmun. 2011, 36, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Gobel, K.; Pankratz, S.; Asaridou, C.M.; Herrmann, A.M.; Bittner, S.; Merker, M.; Ruck, T.; Glumm, S.; Langhauser, F.; Kraft, P.; et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat. Commun. 2016, 7, 11626. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.A.; Bauer, J.; Flick, M.J.; Sikorski, S.L.; Nuriel, T.; Lassmann, H.; Degen, J.L.; Akassoglou, K. The fibrin-derived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J. Exp. Med. 2007, 204, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, M.; Savitsky, J.P. Platelet adhesive index studies in multiple sclerosis and other neurologic disorders. Bull. N. Y. Acad. Med. 1952, 28, 462–468. [Google Scholar] [PubMed]
- Millar, J.H.; Merrett, J.D.; Dalby, A.M. Platelet stickiness in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 1966, 29, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.H.; Hampton, J.R.; Phillipson, O.T. Platelet behaviour and plasma phospholipids in multiple sclerosis. Lancet 1968, 1, 99–104. [Google Scholar] [CrossRef]
- Andreoli, V.M.; Cazzullo, C.L. Platelet behaviour in multiple sclerosis. Lancet 1968, 1, 528–529. [Google Scholar] [CrossRef]
- Langer, H.F.; Choi, E.Y.; Zhou, H.; Schleicher, R.; Chung, K.J.; Tang, Z.; Gobel, K.; Bdeir, K.; Chatzigeorgiou, A.; Wong, C.; et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ. Res. 2012, 110, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Sotnikov, I.; Veremeyko, T.; Starossom, S.C.; Barteneva, N.; Weiner, H.L.; Ponomarev, E.D. Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation. PLoS ONE 2013, 8, e58979. [Google Scholar] [CrossRef] [PubMed]
- Starossom, S.C.; Veremeyko, T.; Yung, A.W.; Dukhinova, M.; Au, C.; Lau, A.Y.; Weiner, H.L.; Ponomarev, E.D. Platelets Play Differential Role During the Initiation and Progression of Autoimmune Neuroinflammation. Circ. Res. 2015, 117, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Callea, L.; Arese, M.; Orlandini, A.; Bargnani, C.; Priori, A.; Bussolino, F. Platelet activating factor is elevated in cerebral spinal fluid and plasma of patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol. 1999, 94, 212–221. [Google Scholar] [CrossRef]
- Sheremata, W.A.; Jy, W.; Horstman, L.L.; Ahn, Y.S.; Alexander, J.S.; Minagar, A. Evidence of platelet activation in multiple sclerosis. J. Neuroinflamm. 2008, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Cuesta, M.; Irizar, H.; Castillo-Trivino, T.; Munoz-Culla, M.; Osorio-Querejeta, I.; Prada, A.; Sepulveda, L.; Lopez-Mato, M.P.; de Munain, A.L.; Comabella, M.; et al. Circulating microparticles reflect treatment effects and clinical status in multiple sclerosis. Biomark. Med. 2014, 8, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Doring, A.; Wild, M.; Vestweber, D.; Deutsch, U.; Engelhardt, B. E- and P-selectin are not required for the development of experimental autoimmune encephalomyelitis in C57BL/6 and SJL mice. J. Immunol. 2007, 179, 8470–8479. [Google Scholar] [CrossRef] [PubMed]
- Kihara, Y.; Ishii, S.; Kita, Y.; Toda, A.; Shimada, A.; Shimizu, T. Dual phase regulation of experimental allergic encephalomyelitis by platelet-activating factor. J. Exp. Med. 2005, 202, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Duerschmied, D.; Suidan, G.L.; Demers, M.; Herr, N.; Carbo, C.; Brill, A.; Cifuni, S.M.; Mauler, M.; Cicko, S.; Bader, M.; et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 2013, 121, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, H.H.; Mossner, R.; Lesch, K.P.; Linker, R.A.; Toyka, K.V.; Gold, R. Absence of reuptake of serotonin influences susceptibility to clinical autoimmune disease and neuroantigen-specific interferon-gamma production in mouse EAE. Clin. Exp. Immunol. 2005, 142, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Mostert, J.P.; Admiraal-Behloul, F.; Hoogduin, J.M.; Luyendijk, J.; Heersema, D.J.; van Buchem, M.A.; de Keyser, J. Effects of fluoxetine on disease activity in relapsing multiple sclerosis: A double-blind, placebo-controlled, exploratory study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Sijens, P.E.; Mostert, J.P.; Irwan, R.; Potze, J.H.; Oudkerk, M.; de Keyser, J. Impact of fluoxetine on the human brain in multiple sclerosis as quantified by proton magnetic resonance spectroscopy and diffusion tensor imaging. Psychiatry Res. 2008, 164, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.; McColl, B.W.; Greenhalgh, A.; Denes, A.; Allan, S.M.; Rothwell, N.J. Platelet interleukin-1α drives cerebrovascular inflammation. Blood 2010, 115, 3632–3639. [Google Scholar] [CrossRef] [PubMed]
- Hawrylowicz, C.M.; Howells, G.L.; Feldmann, M. Platelet-derived interleukin 1 induces human endothelial adhesion molecule expression and cytokine production. J. Exp. Med. 1991, 174, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Henn, V.; Slupsky, J.R.; Grafe, M.; Anagnostopoulos, I.; Forster, R.; Muller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [PubMed]
- Graesser, D.; Solowiej, A.; Bruckner, M.; Osterweil, E.; Juedes, A.; Davis, S.; Ruddle, N.H.; Engelhardt, B.; Madri, J.A. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Investig. 2002, 109, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Rauch, B.H.; Braun, M.; Schror, K.; Weber, A.A. Platelet CD40 ligand (CD40L)—Subcellular localization, regulation of expression, and inhibition by clopidogrel. Platelets 2001, 12, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Sloka, S.; Metz, L.M.; Hader, W.; Starreveld, Y.; Yong, V.W. Reduction of microglial activity in a model of multiple sclerosis by dipyridamole. J. Neuroinflamm. 2013, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Starossom, S.C.; Veremeyko, T.; Dukhinova, M.; Yung, A.W.; Ponomarev, E.D. Glatiramer acetate (copaxone) modulates platelet activation and inhibits thrombin-induced calcium influx: Possible role of copaxone in targeting platelets during autoimmune neuroinflammation. PLoS ONE 2014, 9, e96256. [Google Scholar] [CrossRef] [PubMed]
- Gold, S.M.; Voskuhl, R.R. Pregnancy and multiple sclerosis: From molecular mechanisms to clinical application. Semin. Immunopathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.T.; Shah, M.; Lowe, G.D.; Belch, J.J.; Forbes, C.D.; Prentice, C.R. Plasma fibrinopeptide A and β-thromboglobulin in pre-eclampsia and pregnancy hypertension. Thromb. Haemost. 1982, 47, 54–55. [Google Scholar] [PubMed]
- Fitzgerald, D.J.; Mayo, G.; Catella, F.; Entman, S.S.; FitzGerald, G.A. Increased thromboxane biosynthesis in normal pregnancy is mainly derived from platelets. Am. J. Obstet. Gynecol. 1987, 157, 325–330. [Google Scholar] [CrossRef]
- Boilard, E.; Larabee, K.; Shnayder, R.; Jacobs, K.; Farndale, R.W.; Ware, J.; Lee, D.M. Platelets participate in synovitis via Cox-1-dependent synthesis of prostacyclin independently of microparticle generation. J. Immunol. 2011, 186, 4361–4366. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.G.; Hogg, N.; Revell, P.A. Lymphocytes, polymorphonuclear leukocytes, macrophages and platelets in synovium involved by rheumatoid arthritis. A study with monoclonal antibodies. Pathology 1986, 18, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, Y.T.; Bergroth, V.; Kulomaa, M.; Nordstrom, D.; Segerberg-Konttinen, M.; Keinanen, R.; Kemppinen, P.; Hukkanen, M.; Gronblad, M. Localisation of lysozyme mRNA in rheumatoid synovial membrane by in situ hybridisation. Ann. Rheum. Dis. 1989, 48, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, N.S.; Yan, C.G.; Li, J.H.; Tang, L.Q. The significance of platelet activation in rheumatoid arthritis. Clin. Rheumatol. 2007, 26, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Knijff-Dutmer, E.A.; Koerts, J.; Nieuwland, R.; Kalsbeek-Batenburg, E.M.; van de Laar, M.A. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum. 2002, 46, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Bunescu, A.; Seideman, P.; Lenkei, R.; Levin, K.; Egberg, N. Enhanced Fcgamma receptor I, αMβ2 integrin receptor expression by monocytes and neutrophils in rheumatoid arthritis: Interaction with platelets. J. Rheumatol. 2004, 31, 2347–2355. [Google Scholar] [PubMed]
- Gasparyan, A.Y.; Sandoo, A.; Stavropoulos-Kalinoglou, A.; Kitas, G.D. Mean platelet volume in patients with rheumatoid arthritis: The effect of anti-TNF-α therapy. Rheumatol. Int. 2010, 30, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Sellam, J.; Proulle, V.; Jungel, A.; Ittah, M.; Richard, C.M.; Gottenberg, J.E.; Toti, F.; Benessiano, J.; Gay, S.; Freyssinet, J.M.; et al. Increased levels of circulating microparticles in primary Sjogren’s syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res. Ther. 2009, 11, R156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goules, A.; Tzioufas, A.G.; Manousakis, M.N.; Kirou, K.A.; Crow, M.K.; Routsias, J.G. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J. Autoimmun. 2006, 26, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Pamuk, G.E.; Vural, O.; Turgut, B.; Demir, M.; Pamuk, O.N.; Cakir, N. Increased platelet activation markers in rheumatoid arthritis: Are they related with subclinical atherosclerosis? Platelets 2008, 19, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.E.; Harrison, P.; Mackie, I.J.; Isenberg, D.A.; Machin, S.J. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 2001, 115, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Endresen, G.K. Investigation of blood platelets in synovial fluid from patients with rheumatoid arthritis. Scand. J. Rheumatol. 1981, 10, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Endresen, G.K.; Forre, O. Human platelets in synovial fluid. A focus on the effects of growth factors on the inflammatory responses in rheumatoid arthritis. Clin. Exp. Rheumatol. 1992, 10, 181–187. [Google Scholar] [PubMed]
- Yaron, M.; Djaldetti, M. Platelets in synovial fluid. Arthritis Rheum. 1978, 21, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Farr, M.; Wainwright, A.; Salmon, M.; Hollywell, C.A.; Bacon, P.A. Platelets in the synovial fluid of patients with rheumatoid arthritis. Rheumatol. Int. 1984, 4, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.H.; Breth, G.; Skosey, J.L. Platelets in the synovial space. Arthritis Rheum. 1978, 21, 994–995. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Riddle, J.M.; Bluhm, G.B.; Pitchford, W.C.; McElroy, H.; Jimenea, C.; Leisen, J.; Venkatasubramanian, K. A comparative study of platelet reactivity in arthritis. Ann. N. Y. Acad. Sci. 1981, 370, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Mullan, P.A.M.; Peace, A.J.; Madigan, A.M.; Tedesco, A.F.; Kenny, D.; McCarthy, G.M. Platelet hyper-reactivity in active inflammatory arthritis is unique to the adenosine diphosphate pathway: A novel finding and potential therapeutic target. Rheumatology 2010, 49, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.E.; Mada, S.R.; Rico, M.C.; Cadena, R.A.D.; Kunapuli, S.P. Clopidogrel, a P2Y12 receptor antagonist, potentiates the inflammatory response in a rat model of peptidoglycan polysaccharide-induced arthritis. PLoS ONE 2011, 6, e26035. [Google Scholar] [CrossRef] [PubMed]
- Kawashiri, S.Y.; Taguchi, M.; Kawakami, A.; Eguchi, K. Clopidogrel-associated acute arthritis. Rheumatol. Int. 2012, 32, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Radvan, J.; Hopkinson, N. Clopidogrel associated with acute arthritis. BMJ 2000, 320, 483. [Google Scholar] [PubMed]
- Kanadiya, M.K.; Singhal, S.; Koshal, V.B. Prasugrel as a safe alternative for clopidogrel-associated arthritis. J. Invasive Cardiol. 2011, 23, E137–E138. [Google Scholar] [PubMed]
- Tayyareci, Y. Acute arthritis associated with loading dose of clopidogrel. J. Clin. Rheumatol. 2008, 14, 254–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.K.; Ginges, I.; Manolios, N. Clopidogrel-associated acute arthritis. Intern. Med. J. 2003, 33, 618–619. [Google Scholar] [CrossRef] [PubMed]
- Boulman, N.; Rozenbaum, M.; Slobodin, G.; Rosner, I. Acute polyarthritis associated with clopidogrel treatment. Israel Med. Assoc. J. 2005, 7, 670–671. [Google Scholar]
- Forrest, C.M.; Stone, T.W.; Mackay, G.M.; Oxford, L.; Stoy, N.; Harman, G.; Darlington, L.G. Purine metabolism and clinical status of patients with rheumatoid arthritis treated with dipyridamole. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Vowinkel, T.; Anthoni, C.; Wood, K.C.; Stokes, K.Y.; Russell, J.; Gray, L.; Bharwani, S.; Senninger, N.; Alexander, J.S.; Krieglstein, C.F.; et al. CD40-CD40 ligand mediates the recruitment of leukocytes and platelets in the inflamed murine colon. Gastroenterology 2007, 132, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Stokes, K.Y.; Vowinkel, T.; Watanabe, N.; Elrod, J.W.; Harris, N.R.; Lefer, D.J.; Hibi, T.; Granger, D.N. Colonic blood flow responses in experimental colitis: Time course and underlying mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1024–G1029. [Google Scholar] [CrossRef] [PubMed]
- Gironella, M.; Molla, M.; Salas, A.; Soriano, A.; Sans, M.; Closa, D.; Engel, P.; Salas, A.; Pique, J.M.; Panes, J. The role of P-selectin in experimental colitis as determined by antibody immunoblockade and genetically deficient mice. J. Leukocyte Biol. 2002, 72, 56–64. [Google Scholar] [PubMed]
- Nunez-Andrade, N.; Lamana, A.; Sancho, D.; Gisbert, J.P.; Gonzalez-Amaro, R.; Sanchez-Madrid, F.; Urzainqui, A. P-selectin glycoprotein ligand-1 modulates immune inflammatory responses in the enteric lamina propria. J. Pathol. 2011, 224, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B.; Cheresh, P.; Zhang, Z.; Ryu, H.; Managlia, E.; Barrett, T.A. P-selectin glycoprotein ligand-1 is needed for sequential recruitment of T-helper 1 (Th1) and local generation of Th17 T cells in dextran sodium sulfate (DSS) colitis. Inflamm. Bowel Dis. 2012, 18, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Rijcken, E.M.; Laukoetter, M.G.; Anthoni, C.; Meier, S.; Mennigen, R.; Spiegel, H.U.; Bruewer, M.; Senninger, N.; Vestweber, D.; Krieglstein, C.F. Immunoblockade of PSGL-1 attenuates established experimental murine colitis by reduction of leukocyte rolling. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G115–G124. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Granger, D.N. Inflammatory bowel disease: A paradigm for the link between coagulation and inflammation. Inflamm. Bowel Dis. 2009, 15, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Voudoukis, E.; Karmiris, K.; Koutroubakis, I.E. Multipotent role of platelets in inflammatory bowel diseases: A clinical approach. World J. Gastroenterol. 2014, 20, 3180–3190. [Google Scholar] [CrossRef] [PubMed]
- Harries, A.D.; Fitzsimons, E.; Fifield, R.; Dew, M.J.; Rhoades, J. Platelet count: A simple measure of activity in Crohn’s disease. Br. Med. J. 1983, 286, 1476. [Google Scholar] [CrossRef]
- Harries, A.D.; Beeching, N.J.; Rogerson, S.J.; Nye, F.J. The platelet count as a simple measure to distinguish inflammatory bowel disease from infective diarrhoea. J. Infect. 1991, 22, 247–250. [Google Scholar] [CrossRef]
- Larsen, T.B.; Nielsen, J.N.; Fredholm, L.; Lund, E.D.; Brandslund, I.; Munkholm, P.; Hey, H. Platelets and anticoagulant capacity in patients with inflammatory bowel disease. Pathophysiol. Haemost. Thromb. 2002, 32, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Morowitz, D.A.; Allen, L.W.; Kirsner, J.B. Thrombocytosis in chronic inflammatory bowel disease. Ann. Intern. Med. 1968, 68, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.E.; Rampton, D.S. Platelet dysfunction: A new dimension in inflammatory bowel disease. Gut 1995, 36, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.E.; Cahill, M.R.; Newland, A.C.; Rampton, D.S. Platelets circulate in an activated state in inflammatory bowel disease. Gastroenterology 1994, 106, 840–845. [Google Scholar] [CrossRef]
- Collins, C.E.; Rampton, D.S. Review article: Platelets in inflammatory bowel disease—Pathogenetic role and therapeutic implications. Aliment. Pharmacol. Ther. 1997, 11, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Katz, J.A.; Saibeni, S.; Papa, A.; Gasbarrini, A.; Vecchi, M.; Fiocchi, C. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut 2003, 52, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Irving, P.M.; Macey, M.G.; Shah, U.; Webb, L.; Langmead, L.; Rampton, D.S. Formation of platelet-leukocyte aggregates in inflammatory bowel disease. Inflamm. Bowel Dis. 2004, 10, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Rachchh, M.A.; Jadav, P.D. Evaluation of anti-inflammatory effect of anti-platelet agent-clopidogrel in experimentally induced inflammatory bowel disease. Indian J. Pharmacol. 2012, 44, 744–748. [Google Scholar] [PubMed]
- Kohnke, T.; Gomolka, B.; Bilal, S.; Zhou, X.; Sun, Y.; Rothe, M.; Baumgart, D.C.; Weylandt, K.H. Acetylsalicylic Acid reduces the severity of dextran sodium sulfate-induced colitis and increases the formation of anti-inflammatory lipid mediators. BioMed Res. Int. 2013, 2013, 748160. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Haubner, R.; Pichler, B.J.; Gawaz, M. Radionuclide imaging: A molecular key to the atherosclerotic plaque. J. Am. Coll. Cardiol. 2008, 52, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Platelets provide a bounty of potential targets for therapy in multiple sclerosis. Circ. Res. 2012, 110, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, R.I.; Reichenbach, F.; Kraft, P.; Kumar, A.; Lescan, M.; Todt, F.; Gobel, K.; Hilgendorf, I.; Geisler, T.; Bauer, A.; et al. Platelets induce apoptosis via membrane-bound FasL. Blood 2015, 126, 1483–1493. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Mouse Line (Genetic Background) | Model (Peptide) | Inflammatory Effect | Referene |
|---|---|---|---|
| ST3Gal-V−/− (C57BL/6) | EAE (MOG35–55) | Ameliorated disease course due to lack of brain-specific gangliosides that can recognize by platelets. Reduced CNS inflammation as determined by the infiltration of less lymphocytes, CD4 T cells and macrophages on day 21 after the EAE induction. | [31] |
| E-/P-selectin−/− (C57BL/6) | EAE (MOG35–55) | No effect on clinical symptoms. | [37] |
| E-/P-selectin−/− (SJL) | EAE (PLP139–151) | No effect on clinical symptoms. | [37] |
| 5-HTT−/−(C57BL/6) | EAE (MOG35–55) | Decreased disease severity. Reduced CNS inflammation. | [40] |
| 5-HTT−/− (C57BL/6) | EAE (rat MBP) | Decreased disease severity. | [40] |
| PAF receptor−/− (C57BL/6) | EAE (MOG35–55) | Decreased disease severity. Reduced CNS inflammation and demyelination. | [38] |
| FcR γ-chain −/− (C57BL/6J) | EAE (MOG35–55) | Reduced clinical symptoms. | [36] |
| PECAM-1−/− (C57BL/6) | EAE (MOG35–55) | Early onset of clinical symptoms associated with early leukocyte migration into CNS. | [46] |
| PECAM-1−/− (C57BL/6) | Adoptive transfer of EAE (MOG35–55) | Early onset of clinical symptoms regardless of whether KO mice were injected with MOG35–55-specific WT or PECAM-1−/− T cells. | [46] |
| Treatment | Model (Peptide) | Genetic Background/or Species | Inflammatory Effect | Reference(s) |
|---|---|---|---|---|
| Injection of brain lipid rafts on day 0 (platelet degranulation within brain) | EAE (MOG35–55) without PTx | C57BL/6 | EAE was induced. | [31] |
| Intracranial injection of platelet rich plasma on day 0 (systemic platelet degranulation) | EAE (MOG35–55) without PTx | C57BL/6 | EAE was induced. | [31] |
| Neuroaminidase (Prevention of platelet-lipid rafts interactions) | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [31] |
| LFA protein (Prevention of platelet-lipid rafts interactions) | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [31] |
| CTB (Prevention of platelet-lipid rafts interactions) | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [31] |
| Anti-GQ Ab (Prevention of platelet-lipid rafts interactions) | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [31] |
| Anti-M2 Ab on days 12, 14 & 16 (blockig Mac-1/GP1bα interaction) | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [30] |
| Anti-GPIIb/IIIα Fab on days 12, 14 & 16 | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [30] |
| Anti-GPIbα Fab on days 12, 14 & 16 | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [30] |
| Anti-GPIbα Fab on days 15, 17 & 19 | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. | [30] |
| Anti-thrombocyte serum (platelet depletion) on days 0, 2, 4 & 8 or 12 & 16 | EAE (MOG35–55) | C57BL/6 | Decreased disease severity. Reduced CNS inflammation. | [30,31] |
| Anti- thrombocyte serum (platelet depletion) on days 2 & 6 | EAE (MOG35–55) | C57BL/6 | No effect on clinical symptoms. | [30] |
| Dipyridamole | EAE (MOG35–55) | C57BL/6 | Reduced clinical symptoms, decerased microglial activity. | [48] |
| Mouse Line (Genetic Background) | Model (Peptide) | Inflammatory Effect | Reference |
|---|---|---|---|
| Cox1−/− | K/BxN serum transfer arthritis (K/BxN serum) | Ameliorated disease course due to platelet-derived COX-1 | [53] |
| Gp1ba−/− (C57BL/6) | K/BxN serum transfer arthritis (K/BxN serum) | No clinical effect | [70] |
| Gp6−/− (C57BL/6) | K/BxN serum transfer arthritis (K/BxN serum) | Decreased disease severity. Reduced inflammation, bone erosion, cartilage erosion | [70] |
| Tbxas1−/− (C57BL/6) | K/BxN serum transfer arthritis (K/BxN serum) | No effect on clinical symptoms | [70] |
| CD40−/− (C57BL/6) | Colitis (DSS) | Attenuated disease activity, reduced inflammation and MPO activity, reduced platelet and leukocyte adhesion | [81] |
| CD40L−/− (C57BL/6) | Colitis (DSS) | Attenuated disease activity, reduced inflammation and MPO activity, reduced platelet and leukocyte adhesion | [81] |
| P-Selectin−/− (C57BL/6) | Colitis (DSS) | Reduced platelet adhesion and rolling | [81] |
| Reduced platelet adhesion, decreased albumin extravasation | [82] | ||
| Enhanced disease activity, reduced MPO activity, inflammation, leukocyte rolling | [83] | ||
| PSGL-1−/− (C57BL/6) | Colitis (DSS) | Reduced platelet adhesion and rolling | [81] |
| Earlier disease onset, enhanced disease activity, enhanced infiltration, platelets not examined | [84] | ||
| Decreased disease activity, inflammation, reduced Th1 and Th17 infiltration, platelets not examined | [85] | ||
| PSGL-1−/− (Balb/c) | Colitis (DSS) | Reduced clinical disease activity, decreased leukocyte rolling, inflammation and MPO activity, platelets not examined | [86] |
| Treatment | Model (Peptide) | Genetic Background/or Species | Inflammatory Effect | Reference |
|---|---|---|---|---|
| Anti-GPIbα Ab (platelet depletion) | K/BxN serum transfer arthritis (K/BxN serum) | C57BL/6J | Reduced clinical symptoms, decreased inflammation, bone erosion and cartilage erosion | [70] |
| Anti-GPIIb/IIa Ab | Colitis (DSS) | C57BL/6 | No effect on adhesion of platelets and leukocytes | [82] |
| Anti-platelet serum | Colitis (DSS) | C57BL/6 | Decreased rolling and adhesion of leukocytes | [81] |
| Reactive arthritis (PG-PS) | Lewis rat | Exacerbated disease severity (increased joint diameter), increased synoviocyte hyperplasia (in both acute and chronic phases), blood vessel proliferation (chronic), inflammatory infiltration (acute and chronic) and fibrosis (chronic), increased IFNy, IL-1b, IL-6 plasma levels, reduced IL-10 plasma level, increased neutrophil and platelet count | [73] | |
| Crohn’s disease (TNBS) | Spargue Dawley rats | Attenuated disease course, reduced inflammation and MPO activity | [98] | |
| Ulcerative colitis (oxazolone) | Wistar rats | Attenuated disease course, reduced inflammation and MPO activity | [98] | |
| Prasugrel (P2Y12 receptor antagonist) | Reactive arthritis (PG-PS) | Lewis rat | Exacerbated disease severity (increased joint diameter), greater synoviocyte hyperplasia, leukocyte infiltration, fibrosis, bone destruction, and pannus formation, increased platelet and neutrophil count, decreased IL-10 plasma levels | [73] |
| SQ29548 (Thromboxane A2 receptor antagonist) | K/BxN serum transfer arthritis (K/BxN serum) | C57BL/6J | No effect on clinical symptoms | [70] |
| Anti-P-Selectin Ab | Colitis (DSS) | C57BL/6 | Decreased rolling and adhesion of platelets | [81] |
| Reduced adhesion of platelets and leukocytes | [82] | |||
| Reduced body weight loss, decreased disease activity, MPO activity, inflammation, leukocyte rolling in colon, enhanced MPO activity in lung | [83] | |||
| Anti-PSGL-1 Ab | Colitis (DSS) | C57BL/6 | Decreased rolling and adhesion of platelets | [81] |
| Reduced adhesion of platelets and leukocytes | [82] | |||
| Acetylsalicylic acid | Colitis (DSS) | C57BL/6 | Decrease in disease severity | [99] |
| Clopidogrel | Colitis (oxazolone) | Rats | Decrease in disease severity, protection against mucosal damage | [98] |
| Crohn’s disease (TNBS) | Rats | Decrease in disease severity, protection against mucosal damage | [98] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pankratz, S.; Bittner, S.; Kehrel, B.E.; Langer, H.F.; Kleinschnitz, C.; Meuth, S.G.; Göbel, K. The Inflammatory Role of Platelets: Translational Insights from Experimental Studies of Autoimmune Disorders. Int. J. Mol. Sci. 2016, 17, 1723. https://doi.org/10.3390/ijms17101723
Pankratz S, Bittner S, Kehrel BE, Langer HF, Kleinschnitz C, Meuth SG, Göbel K. The Inflammatory Role of Platelets: Translational Insights from Experimental Studies of Autoimmune Disorders. International Journal of Molecular Sciences. 2016; 17(10):1723. https://doi.org/10.3390/ijms17101723
Chicago/Turabian StylePankratz, Susann, Stefan Bittner, Beate E. Kehrel, Harald F. Langer, Christoph Kleinschnitz, Sven G. Meuth, and Kerstin Göbel. 2016. "The Inflammatory Role of Platelets: Translational Insights from Experimental Studies of Autoimmune Disorders" International Journal of Molecular Sciences 17, no. 10: 1723. https://doi.org/10.3390/ijms17101723