2.2. Parameter Selection
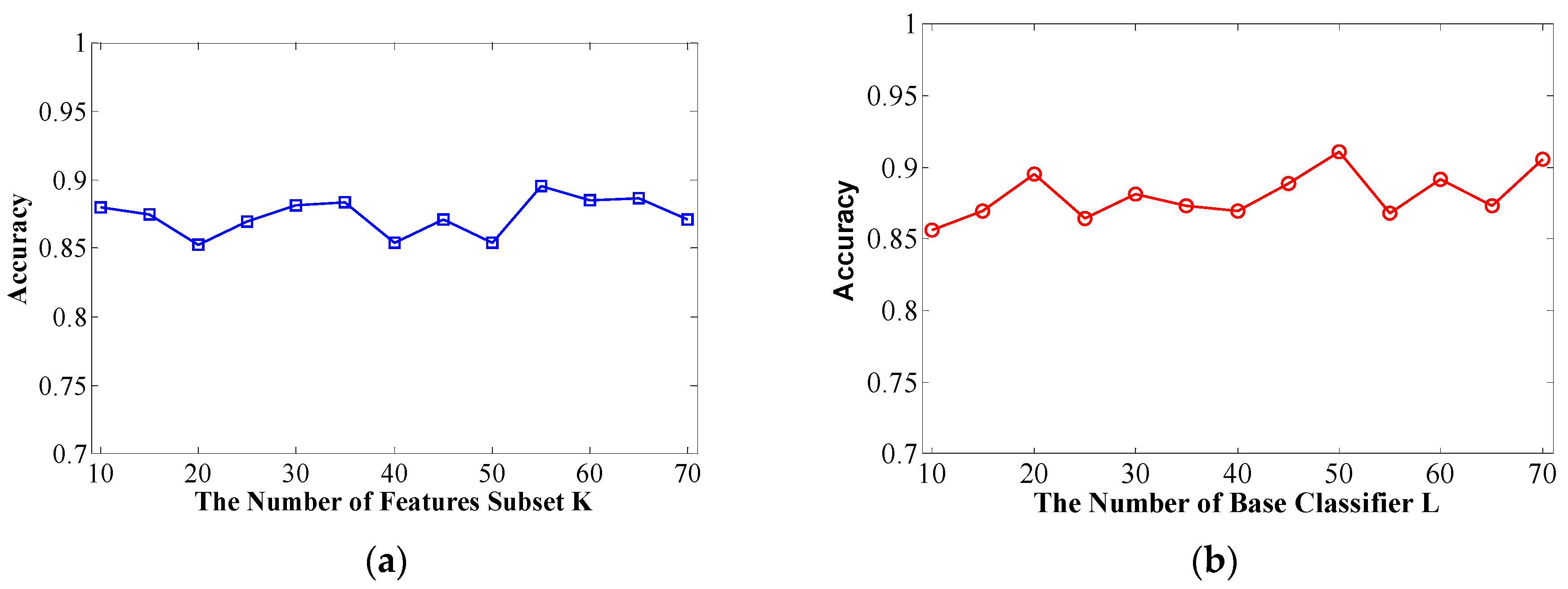
The number of feature subsets K and decision tree number L are crucial for the performance of the Rotation Forest classifier. Therefore, we need to set these two vital parameters in advance. It is quite complex to set the specific value and obtain the best performance for randomness and uncertainty. A higher value of K indicates more subsets, where each subset has fewer features, and a higher value of L indicates more basic classifiers in the ensemble classifier.
In this context, overall classification accuracy is evaluated on a Helicobacter pylori dataset using different K and L values in the first computational validation. Specifically, we adopt the parameter selection strategy that the first step is to fix L to 20 and tune K from 10 to 70 at intervals of 5. We then set K to the value obtained from the first step and tune L from 10 to 70 at intervals of 5.
The prediction results of
Helicobacter pylori are shown in
Figure 1. From
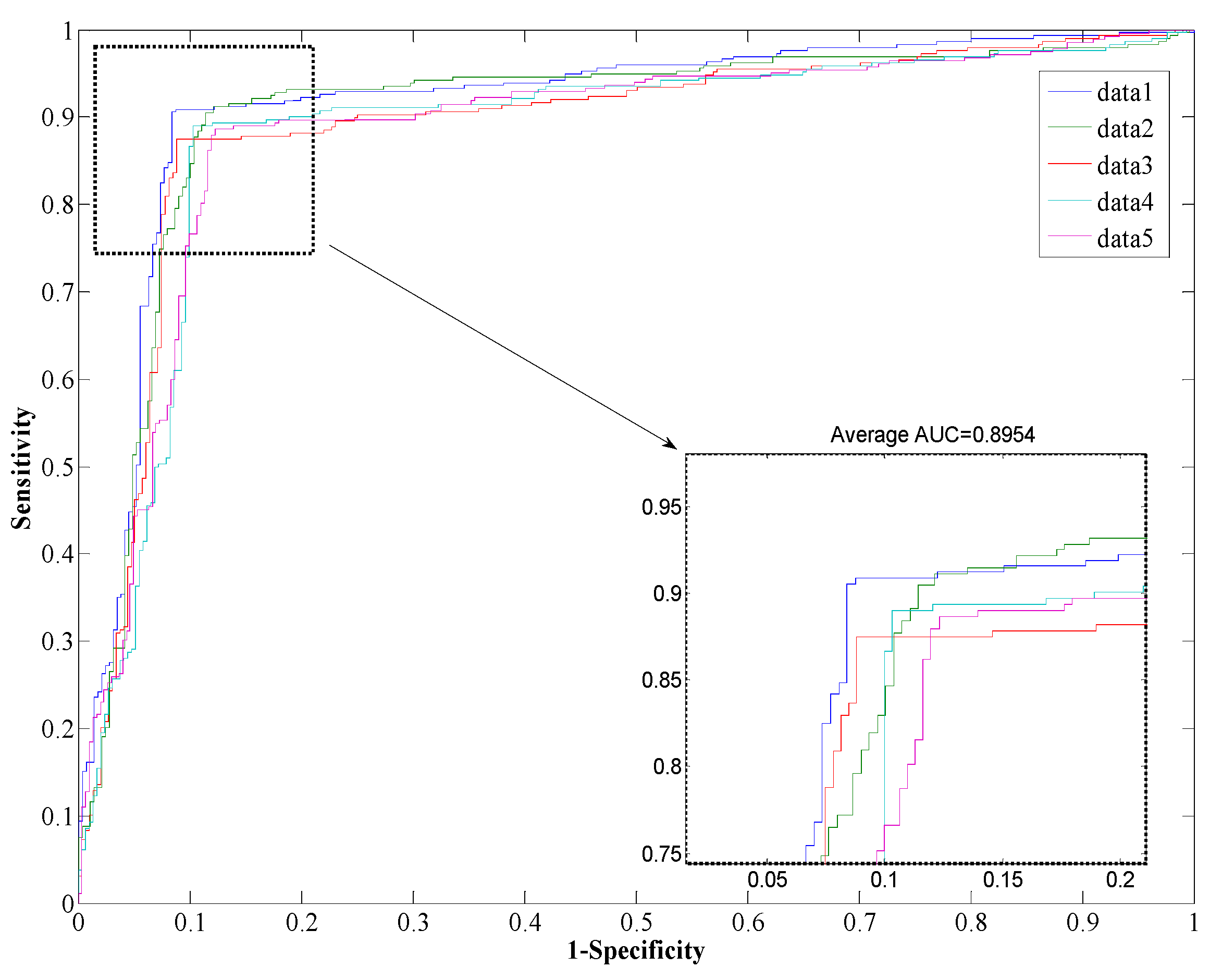
Figure 1a, we can see that setting the
K = 55 leads can obtain good result with an accuracy of 89.54% on the conditions of
L = 20. We then set
K to 55 and increase the value of
L from 10 to 70 at intervals of 5 to work out the results shown in
Figure 1b. We then determine that the optimal value of
L is 50.
The same parameter selection strategy is adopted when exploring the other two datasets. The proposed method on the Human dataset yields an accuracy of 97.91% with the optimized settings (K = 25; L = 40). For the Saccharomyces cerevisiae dataset, it achieves the best accuracy of 94.32% with the optimized settings (K = 65, L = 40).
Figure 1.
(a) Overall prediction accuracy rate with increasing K of feature subsets; (b) Overall prediction accuracy rate with increasing L of decision trees.
Figure 1.
(a) Overall prediction accuracy rate with increasing K of feature subsets; (b) Overall prediction accuracy rate with increasing L of decision trees.
2.3. Prediction Performance of Proposed Model
To validate the proposed model, we apply it to three prevalent PPIs datasets, including the Helicobacter pylori dataset, Homo sapiens dataset, and Saccharomyces cerevisiae dataset. To avoid the problem of over-fitting, five-fold cross-validation is used for performance evaluation. We also operate the support vector machine (SVM) to compare its performance with the proposed model.
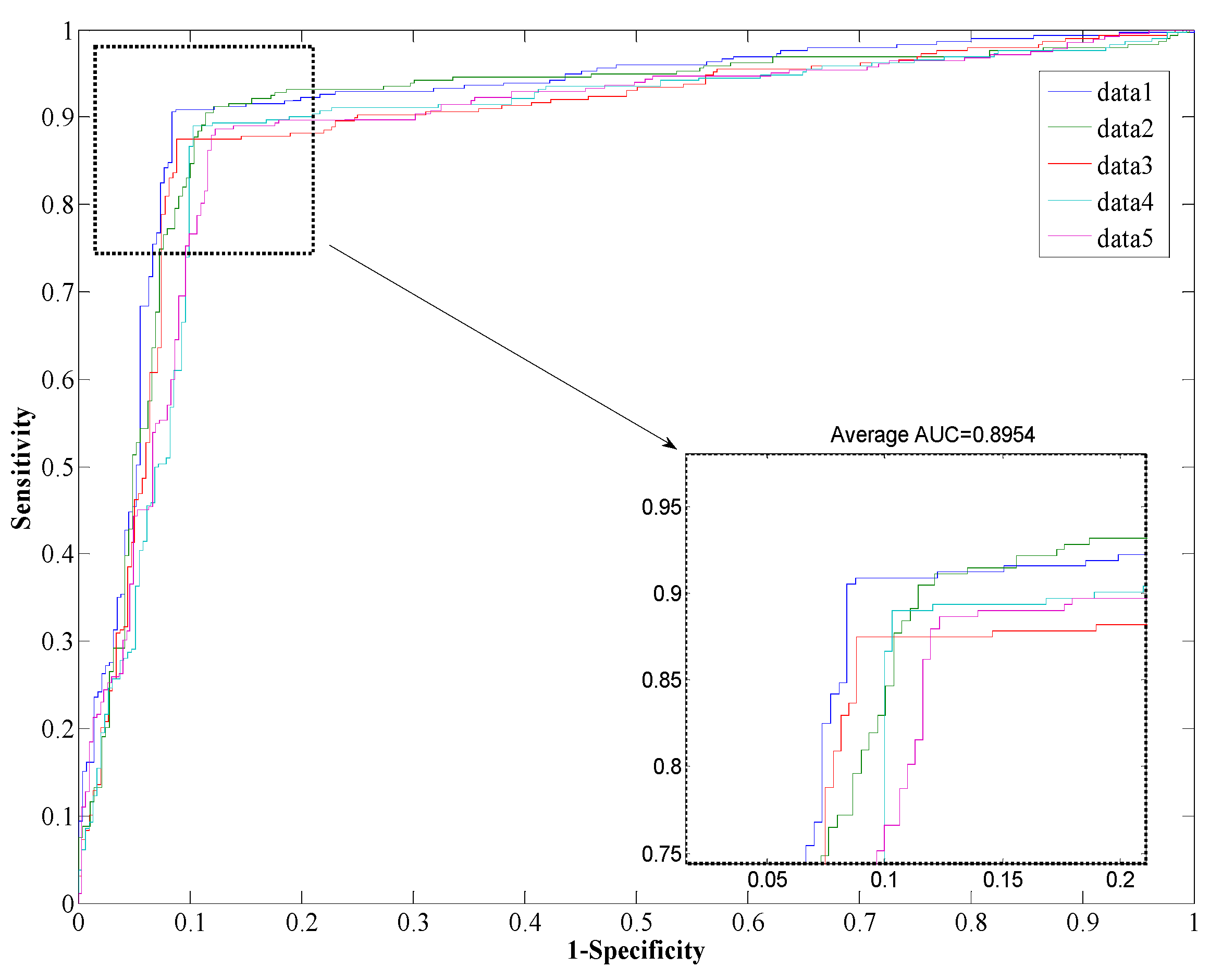
The performance of the
Helicobacter pylori and
Saccharomyces cerevisiae dataset are shown in
Table 1 and
Table 2, which list the overall accuracy, sensitivity, precision, MCC, and AUC. And the ROC curves are plotted in
Figure 2 and
Figure 3. We can see from
Table 1 that the proposed method yields a high accuracy of 89.47% on average on the
Helicobacter pylori dataset. The average value of the AUC is close to 0.90, which indicates the method has high precision in predicting PPIs. The standard deviation of the accuracy, precision, sensitivity, MCC, and AUC are 1.05%, 1.77%, 1.41%, 0.0167, and 0.0145, respectively. When employed on the
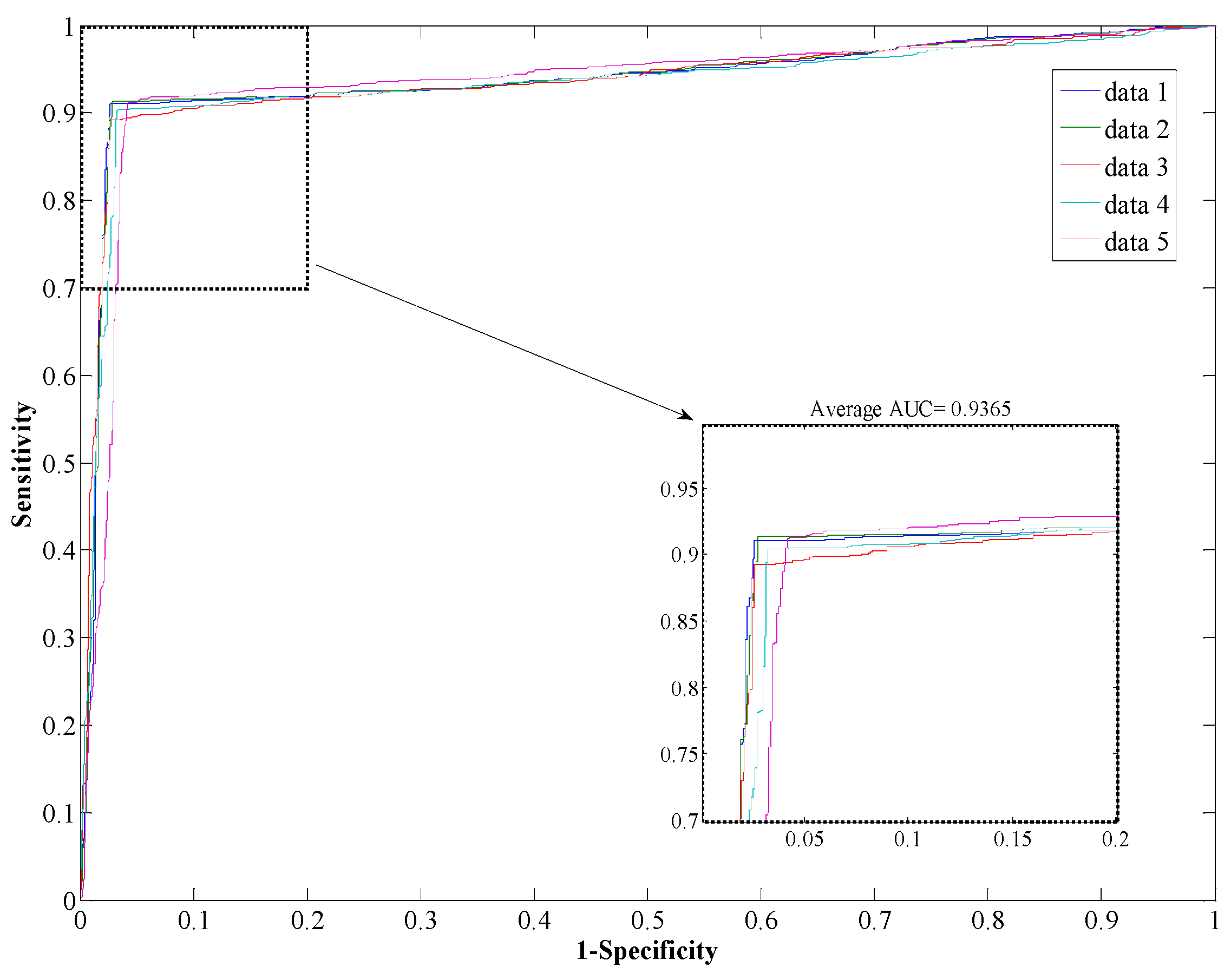
Saccharomyces cerevisiae dataset, our proposed method yields an AUC of 0.93 with a high accuracy of 93.80%, and the values of precision and sensitivity are 96.66% and 90.64%, respectively. The standard deviations of accuracy, precision, sensitivity, MCC, and AUC are 0.50%, 0.62%, 0.87%, 0.009, and 0.002, respectively.
Table 1.
The prediction results of the H. pylori dataset using the proposed method.
Table 1.
The prediction results of the H. pylori dataset using the proposed method.
| Test Set | Sensitivity (%) | Precision (%) | Accuracy (%) | MCC | AUC |
|---|
| 1 | 90.57 | 91.81 | 91.08 | 0.8375 | 0.9158 |
| 2 | 90.48 | 88.96 | 89.54 | 0.8126 | 0.9048 |
| 3 | 87.15 | 90.61 | 89.19 | 0.8070 | 0.8896 |
| 4 | 89.04 | 89.66 | 89.37 | 0.8099 | 0.8823 |
| 5 | 88.65 | 87.11 | 88.16 | 0.7912 | 0.8842 |
| Average | 89.18 ± 1.42 | 89.63 ± 1.77 | 89.47 ± 1.05 | 0.81 ± 0.0167 | 0.90 ± 0.0145 |
Table 2.
The prediction results of the S. cerevisiae dataset using the proposed method.
Table 2.
The prediction results of the S. cerevisiae dataset using the proposed method.
| Test Set | Sensitivity (%) | Precision (%) | Accuracy (%) | MCC | AUC |
|---|
| 1 | 89.22 | 97.16 | 93.34 | 0.8752 | 0.9381 |
| 2 | 91.18 | 95.61 | 93.47 | 0.8779 | 0.9387 |
| 3 | 90.40 | 96.61 | 93.52 | 0.8786 | 0.9368 |
| 4 | 91.07 | 97.06 | 94.32 | 0.8924 | 0.9331 |
| 5 | 91.34 | 96.88 | 94.37 | 0.8933 | 0.9358 |
| Average | 90.64 ± 0.87 | 96.66 ± 0.62 | 93.80 ± 0.50 | 0.88 ± 0.009 | 0.94 ± 0.002 |
The Support Vector Machine (SVM) is a state-of-the-art classification model. Therefore, we compare the Rotation Forest classifier with the SVM model on the
Human dataset. The experimental results are shown in
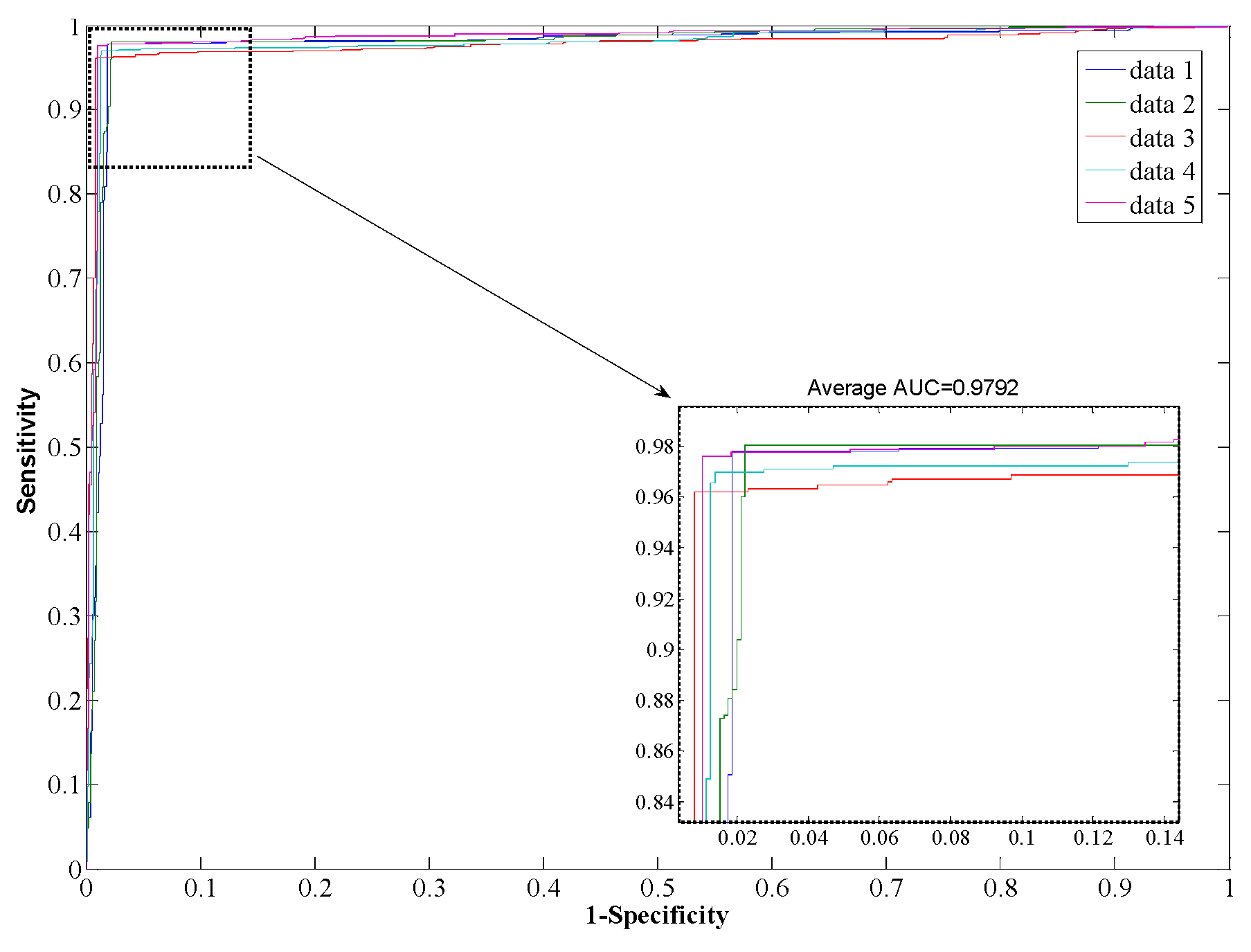
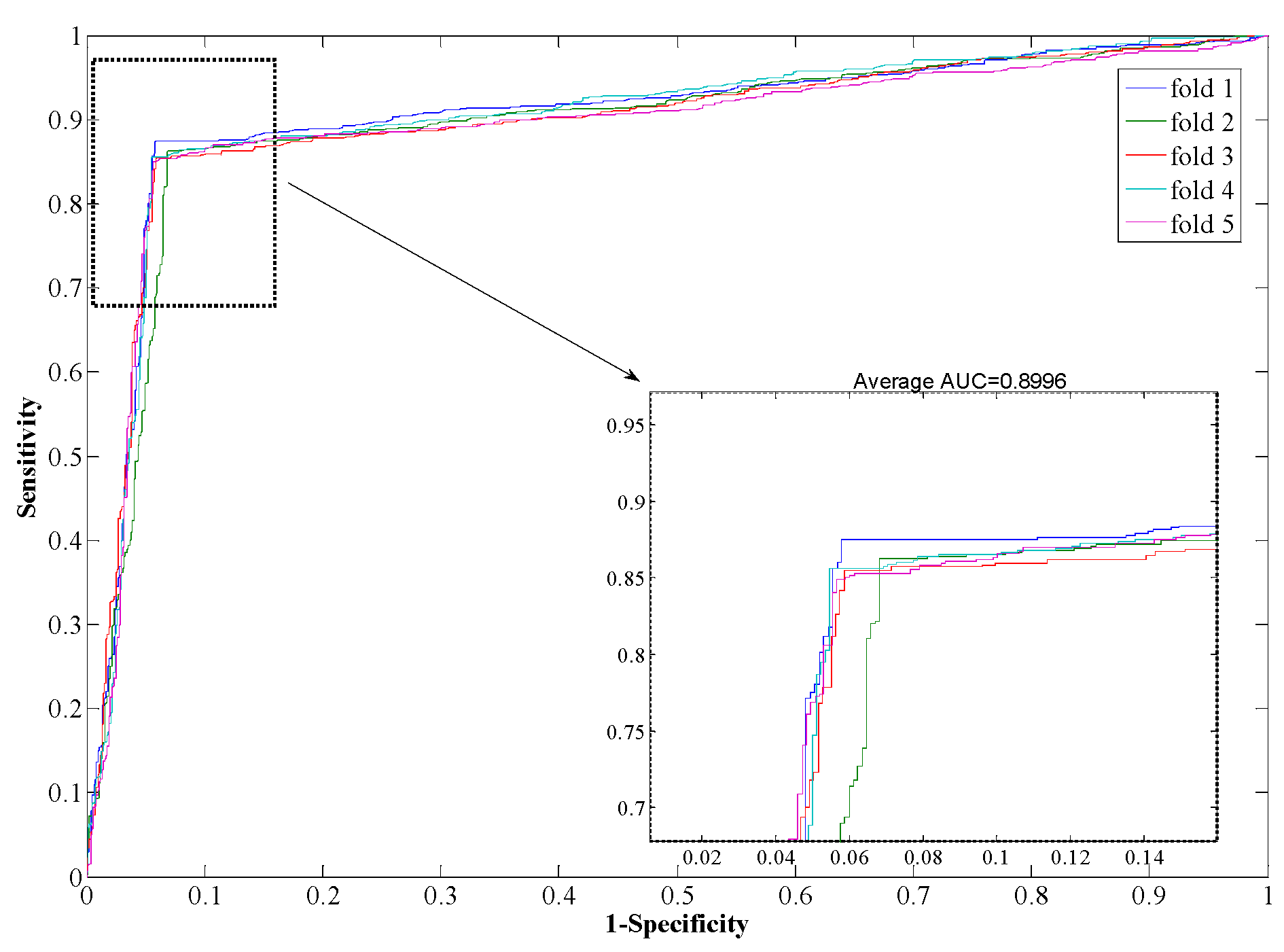
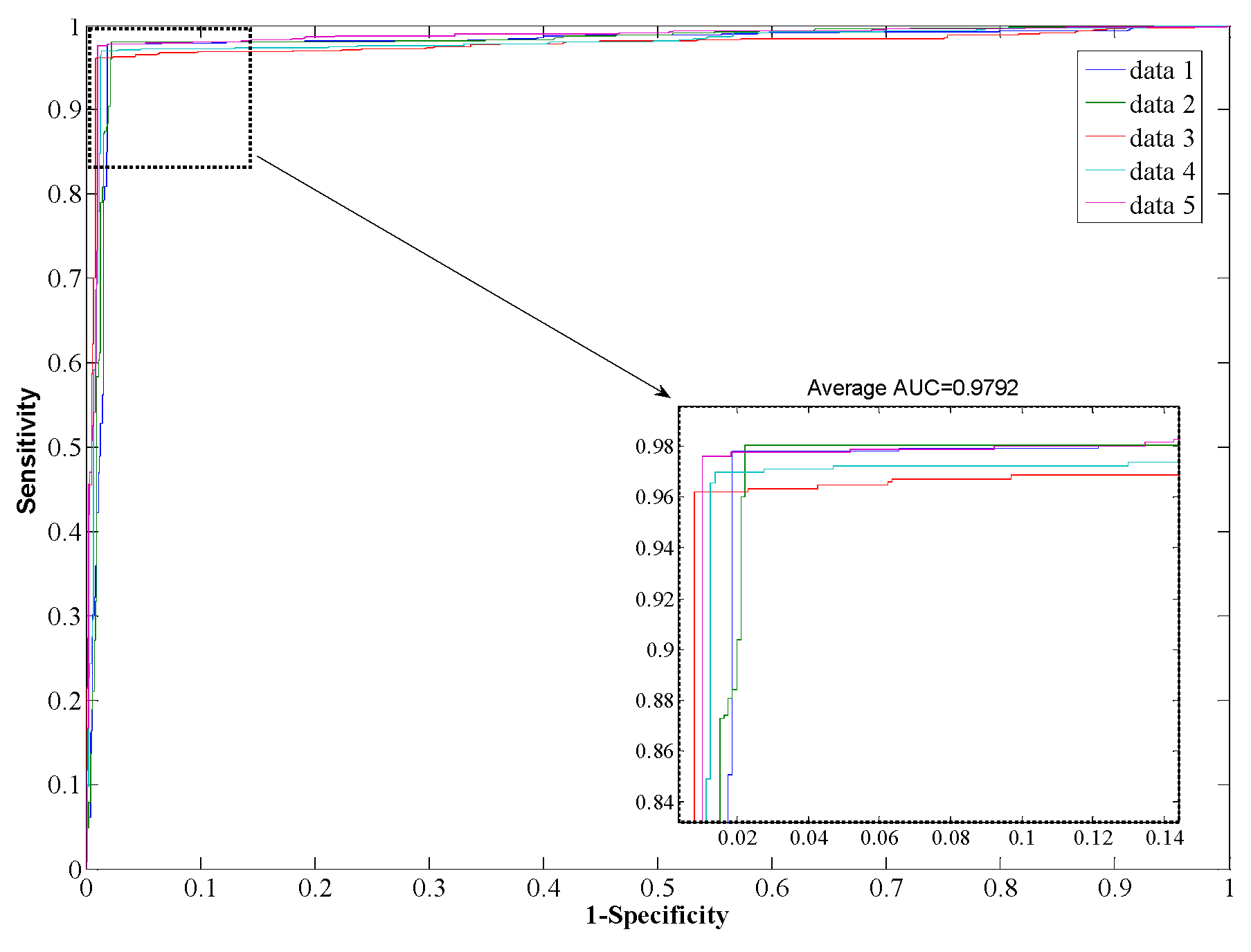
Table 3, from which it can be seen that our proposed method yields good results reflected in average values of accuracy, precision, sensitivity, and MCC as high as 97.96%, 98.35%, 97.32%, and 0.96, respectively. When employing the SVM model for prediction, the average values of accuracy, precision, sensitivity, and MCC are 90.21%, 93.00%, 85.96%, and 0.82, respectively. From the ROC curves of
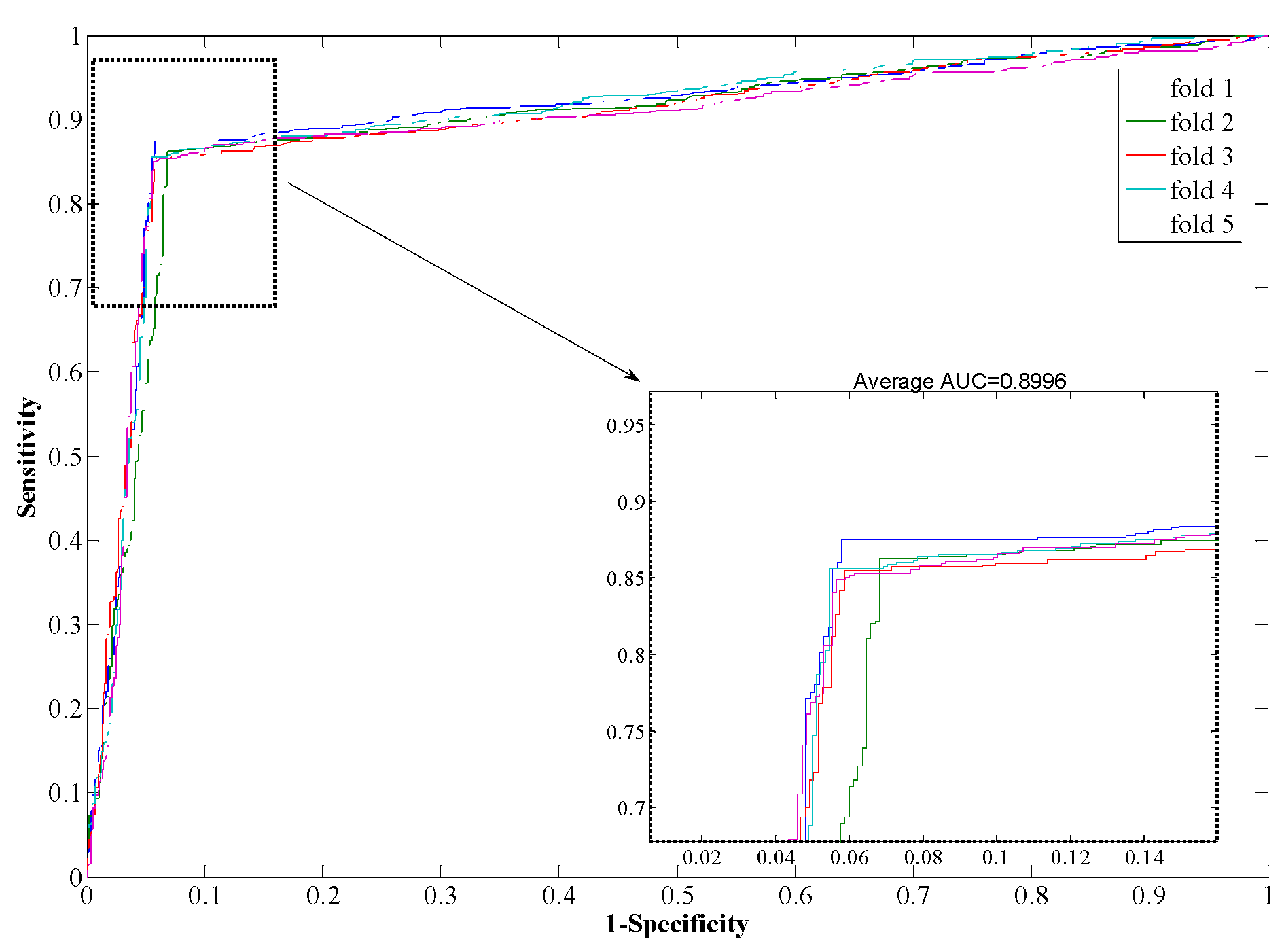
Figure 4 and
Figure 5, it can also be seen that the average AUC score of the proposed method was 0.9792, and the value of SVM was 0.8996. In addition, the standard deviations of accuracy, sensitivity, and MCC yielded by the proposed method were as low as 0.22%, 0.73%, and 0.0042, respectively, which are lower than the values obtained by the SVM model of 0.46%, 0.99%, and 0.0077, respectively. In conclusion, the experimental results above suggested that our proposed method is much better than the SVM-based method.
Table 3.
The prediction results of the Human dataset using the proposed method compared with SVM.
Table 3.
The prediction results of the Human dataset using the proposed method compared with SVM.
| Model | Test Set | Sensitivity (%) | Precision (%) | Accuracy (%) | MCC | AUC |
|---|
| Rotation Forest | 1 | 97.68 | 97.93 | 97.91 | 0.9590 | 0.97.68 |
| 2 | 98.07 | 97.57 | 97.91 | 0.9591 | 0.97.93 |
| 3 | 96.21 | 99.06 | 97.79 | 0.9566 | 0.97.65 |
| 4 | 96.98 | 98.40 | 97.85 | 0.9578 | 0.97.79 |
| 5 | 97.64 | 98.80 | 98.34 | 0.9673 | 0.98.53 |
| Average | 97.32 ± 0.73 | 98.35 ± 0.61 | 97.96 ± 0.22 | 0.96 ± 0.004 | 0.98 ± 0.004 |
| SVM | 1 | 87.52 | 93.59 | 90.92 | 0.8343 | 0.9055 |
| 2 | 86.28 | 92.07 | 89.88 | 0.8170 | 0.8959 |
| 3 | 85.46 | 93.00 | 90.01 | 0.8185 | 0.8985 |
| 4 | 85.62 | 93.05 | 90.44 | 0.8244 | 0.9047 |
| 5 | 84.93 | 93.27 | 89.82 | 0.8156 | 0.8935 |
| Average | 85.96 ± 0.99 | 93.00 ± 0.57 | 90.21 ± 0.46 | 0.82 ± 0.008 | 0.90 ± 0.005 |
Figure 2.
Receiver Operating Characteristic (ROC) from proposed method result for H. pylori protein-protein interaction (PPI) dataset.
Figure 2.
Receiver Operating Characteristic (ROC) from proposed method result for H. pylori protein-protein interaction (PPI) dataset.
Figure 3.
ROC from proposed method result for S. cerevisiae PPI dataset.
Figure 3.
ROC from proposed method result for S. cerevisiae PPI dataset.
Figure 4.
ROC from proposed method result for Human PPI dataset.
Figure 4.
ROC from proposed method result for Human PPI dataset.
Figure 5.
ROC from SVM-based method result for Human PPI dataset.
Figure 5.
ROC from SVM-based method result for Human PPI dataset.
2.4. Comparison with Other Methods
Many methods have been proposed for predicting PPIs. Here, we compare the prediction performance of the proposed method with the existing approaches. All the results yielded by different methods on the
Saccharomyces cerevisiae dataset are shown in
Table 4. We can observe from
Table 4 that Zhou’s work performs well with the lowest standard deviation of 0.33% for accuracy, and Guo’s work has a higher accuracy of 89.33%. In addition, Yang’s work makes a higher precision value of 90.24%. It should be noticed that the proposed method yields the best performance in terms of sensitivity, precision, accuracy and MCC at 90.64%, 96.66%, 93.80%, and 88.35%, respectively. The corresponding standard deviations are 0.87%, 0.62%, 0.50%, and 0.87%, respectively. The above results show that the performance of our proposed method is superior.
We also compare our proposed method with other methods on the
Helicobacter pylori dataset and the results are shown in
Table 5. Compared with the other methods, the proposed method achieves outstanding performance for its high sensitivity, precision, accuracy, and MCC. In detail, the performances of the classifiers are quite disparate. The worst result, yielded by the phylogenetic bootstrap, has an accuracy of 75.80%, precision of 80.20%, and sensitivity of 69.80%. HKNN achieves 84.00% accuracy, 84% precision, and 86% sensitivity. In contrast, the proposed method achieves an accuracy of 89.47%, precision of 89.63%, sensitivity of 89.18%, and an MCC of 81.16%, respectively. The above results indicate that our proposed method is promising and exhibits good performance for PPIs prediction.
Table 4.
Comparison of other methods on the S. cerevisiae dataset.
Table 4.
Comparison of other methods on the S. cerevisiae dataset.
| Model | Test Set | Sensitivity (%) | Precision (%) | Accuracy (%) | MCC (%) |
|---|
| Zhou’s work | SVM + LD | 87.37 ± 0.22 | 89.50 ± 0.60 | 88.56 ± 0.33 | 77.15 ± 0.68 |
| Guo’s work | ACC | 89.93 ± 3.68 | 88.87 ± 6.16 | 89.33 ± 2.67 | N/A |
| AC | 87.30 ± 0.22 | 87.82 ± 4.33 | 87.36 ± 1.38 | N/A |
| Yang’s work | Cod1 | 75.81 ± 1.20 | 74.75 ± 1.23 | 75.08 ± 1.13 | N/A |
| Cod2 | 76.77 ± 0.69 | 82.17 ± 1.35 | 80.04 ± 1.06 | N/A |
| Cod3 | 78.14 ± 0.90 | 81.86 ± 0.99 | 80.41 ± 0.47 | N/A |
| Cod4 | 81.03 ± 1.74 | 90.24 ± 1.34 | 86.15 ± 1.17 | N/A |
| Proposed Method | Average | 90.64 ± 0.87 | 96.66 ± 0.62 | 93.80 ± 0.50 | 88.35 ± 0.87 |
Table 5.
Comparison of other methods on the H. pylori dataset.
Table 5.
Comparison of other methods on the H. pylori dataset.
| Model | Sensitivity (%) | Precision (%) | Accuracy (%) | MCC (%) |
|---|
| Phylogenetic bootstrap | 69.80 | 80.20 | 75.80 | N/A |
| Boosting | 80.37 | 81.69 | 79.52 | 70.64 |
| Signature products | 79.90 | 85.70 | 83.40 | N/A |
| HKNN | 86.00 | 84.00 | 84.00 | N/A |
| Proposed Method | 89.18 | 89.63 | 89.47 | 81.16 |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}