
Multiple Factors Drive Replicating Strand Composition Bias in Bacterial Genomes

Abstract
:1. Introduction
2. Results and Discussion
2.1. Composition Bias in Obligate Intracellular Bacteria
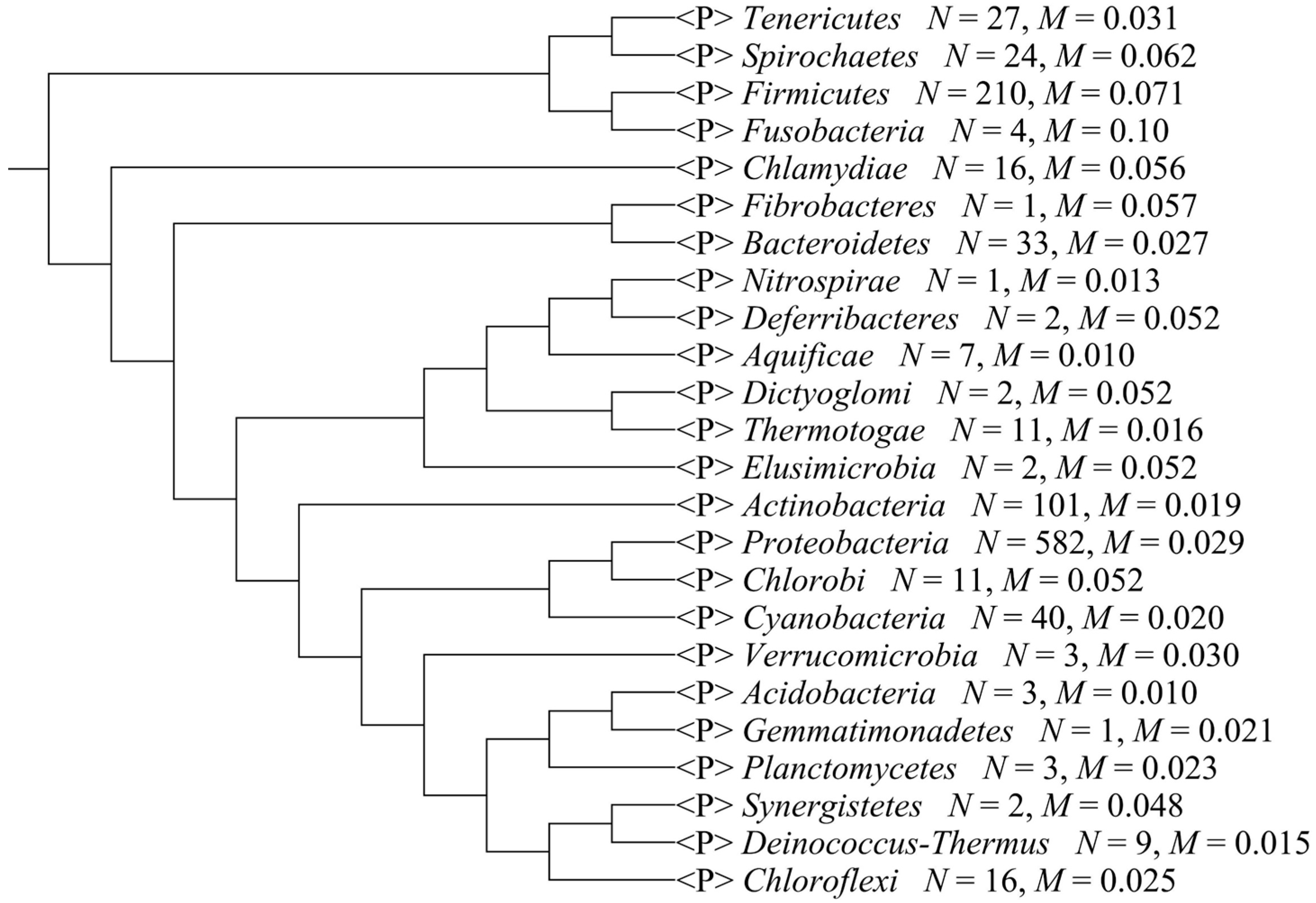
2.2. Composition Bias in Different Bacterial Phyla
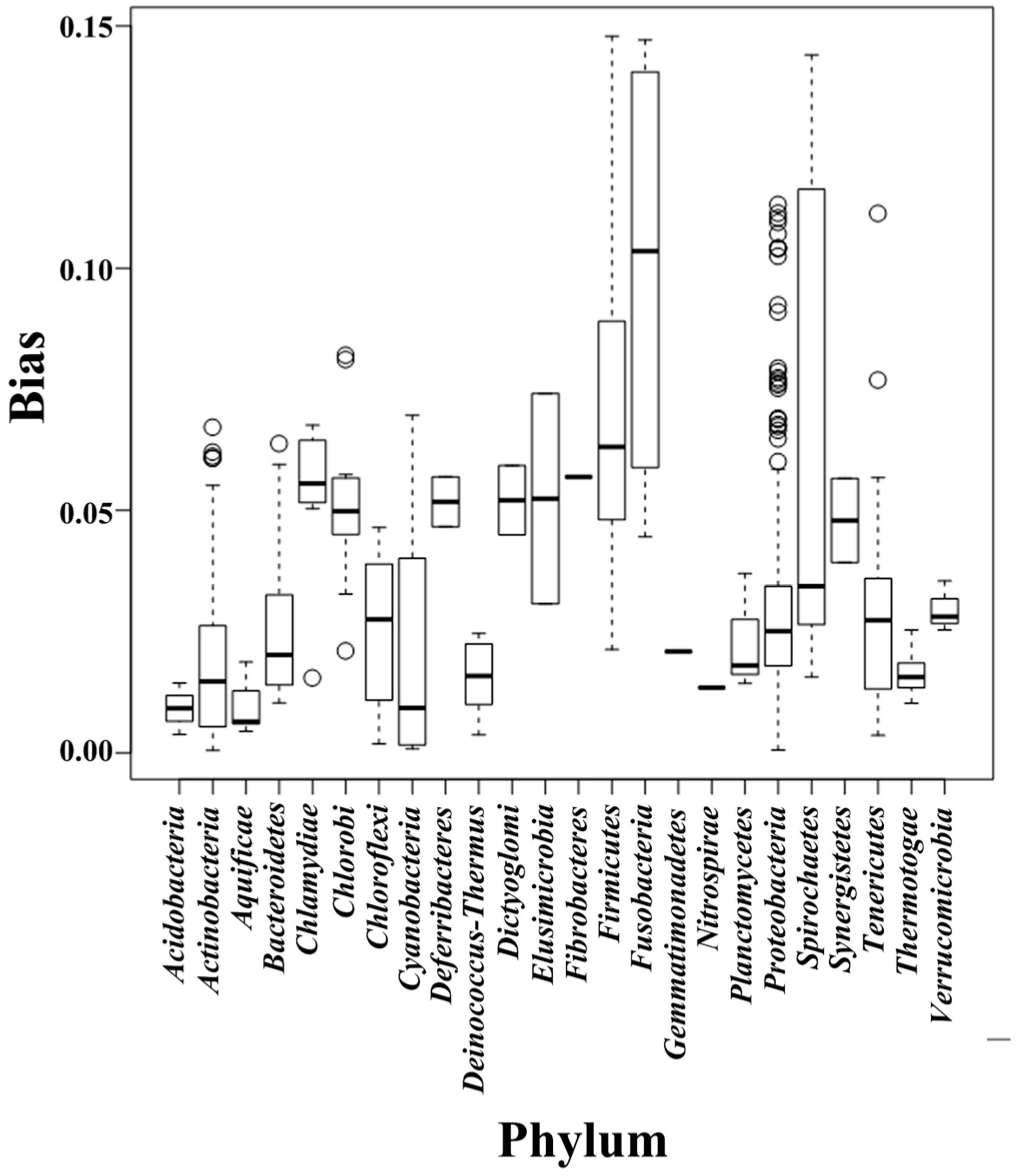

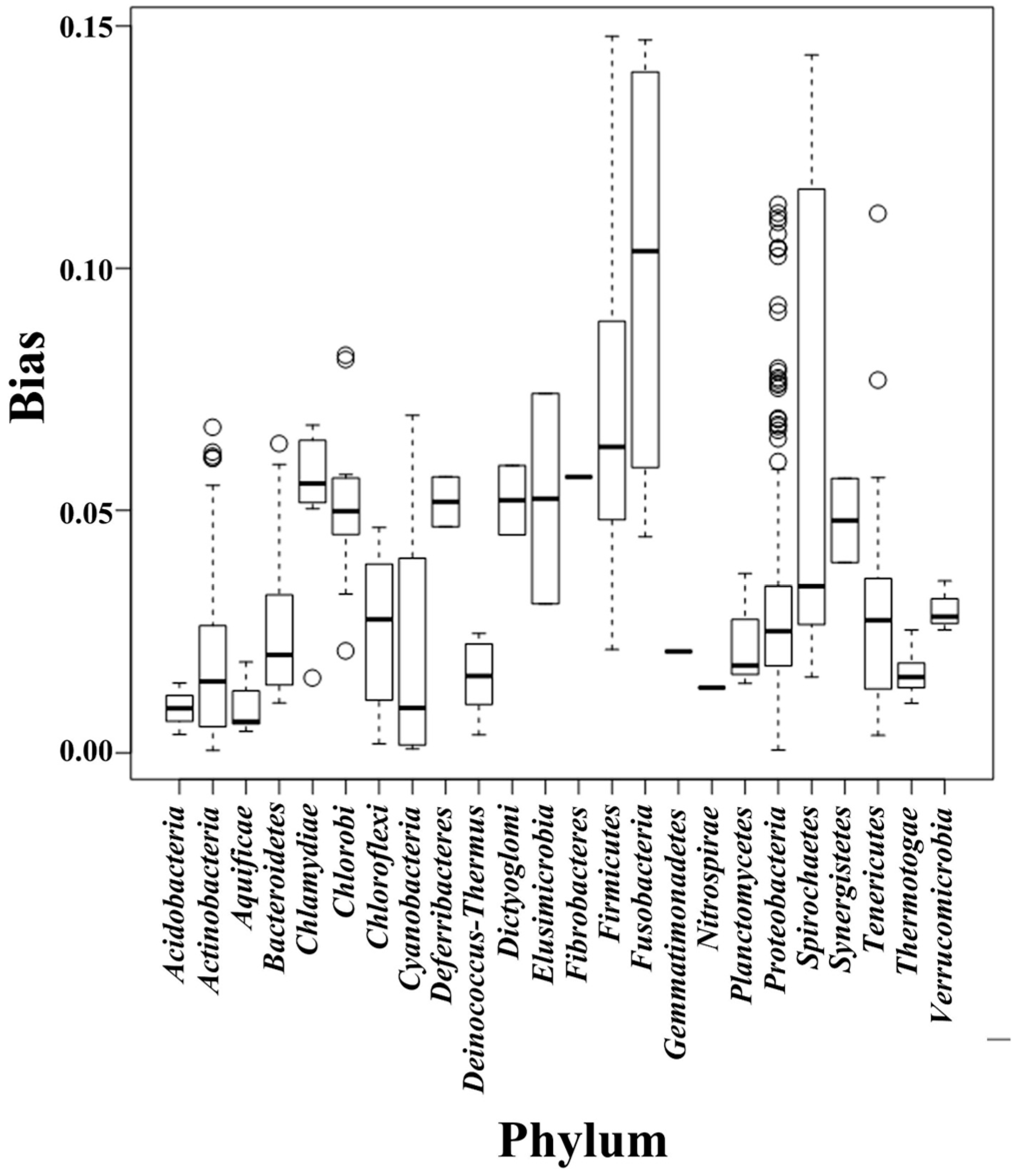{kind=link}
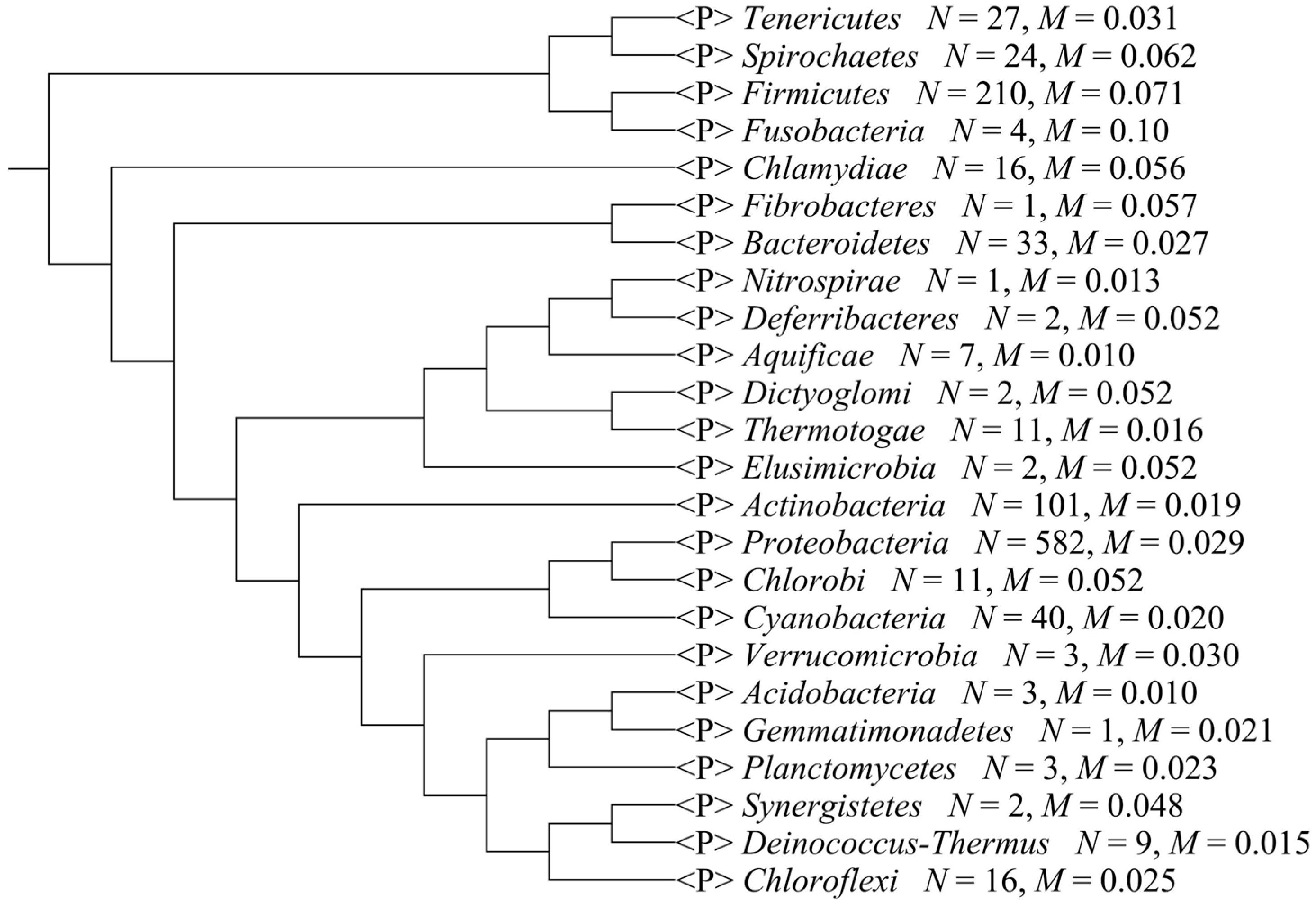{kind=link}
| Phylum | Standard Deviation | Variance | Mean |
|---|---|---|---|
| Acidobacteria | 0.005309 | 2.82 × 10−5 | 0.009124 |
| Actinobacteria | 0.015749 | 0.000248 | 0.018728 |
| Aquificae | 0.005263 | 2.77 × 10−5 | 0.00957 |
| Bacteroidetes | 0.016805 | 0.000282 | 0.027048 |
| Chlamydiae | 0.012521 | 0.000157 | 0.055526 |
| Chlorobi | 0.018046 | 0.000326 | 0.051947 |
| Chloroflexi | 0.015056 | 0.000227 | 0.024993 |
| Cyanobacteria | 0.021638 | 0.000468 | 0.019847 |
| Deferribacteres | 0.007318 | 5.36 × 10−5 | 0.051752 |
| Deinococcus-Thermus | 0.007668 | 5.88 × 10−5 | 0.015442 |
| Dictyoglomi | 0.010132 | 0.000103 | 0.052093 |
| Elusimicrobia | 0.030697 | 0.000942 | 0.052418 |
| Fibrobacteres | NA | NA | 0.056901 |
| Firmicutes | 0.028571 | 0.000816 | 0.071236 |
| Fusobacteria | 0.048886 | 0.00239 | 0.099682 |
| Gemmatimonadetes | NA | NA | 0.020857 |
| Nitrospirae | NA | NA | 0.013445 |
| Planctomycetes | 0.012161 | 0.000148 | 0.023082 |
| Proteobacteria | 0.017163 | 0.000295 | 0.028607 |
| Spirochaetes | 0.046978 | 0.002207 | 0.062153 |
| Synergistetes | 0.012306 | 0.000151 | 0.047907 |
| Tenericutes | 0.023255 | 0.000541 | 0.030599 |
| Thermotogae | 0.004126 | 1.70 × 10−5 | 0.016197 |
| Verrucomicrobia | 0.005228 | 2.73 × 10−5 | 0.029585 |
| Phylum | Genome Size | GC Content | Gene Density | gcRF | taRF |
|---|---|---|---|---|---|
| Acidobacteria | 6,581,121.33 | 0.602611 | 0.524179 | 0.546299 | 0.239179 |
| Actinobacteria | 4,434,386.26 | 0.647473 | 0.591745 | 0.655926 | 0.5707 |
| Aquificae | 1,680,594.86 | 0.3874153 | 0.514286 | 0.026764 | 0.090473 |
| Bacteroidetes | 3,688,038.52 | 0.4246355 | 0.553854 | 0.035009 | 0.101365 |
| Chlamydiae | 1,265,852.44 | 0.4046721 | 0.544713 | 0.022567 | 0.081014 |
| Chlorobi | 2,618,734.27 | 0.5079388 | 0.583907 | 0.061015 | 0.114787 |
| Chloroflexi | 2,435,937.54 | 0.5531583 | 0.519221 | 0.044977 | 0.063278 |
| Cyanobacteria | 3,397,176.98 | 0.4460103 | 0.508569 | −0.33356 | −0.55797 |
| Deferribacteres | 2,728,233 | 0.3682745 | 0.642415 | 0.012609 | 0.057666 |
| Deinococcus-Thermus | 2,411,100.11 | 0.66285 | 0.517812 | −0.10793 | −0.12243 |
| Dictyoglomi | 1,907,773.5 | 0.3384917 | 0.681195 | 0.01941 | 0.055101 |
| Elusimicrobia | 1,384,709.5 | 0.3757977 | 0.726988 | 0.014904 | 0.078649 |
| Fibrobacteres | 3,842,635 | 0.4805184 | 0.580603 | 0.047916 | 0.088216 |
| Firmicutes | 3,077,249.49 | 0.3853 | 0.786812 | 0.020021 | 0.081354 |
| Fusobacteria | 2,680,383 | 0.29141 | 0.72341 | 0.01046 | 0.05595 |
| Gemmatimonadetes | 4,636,964 | 0.6427436 | 0.566455 | 0.043068 | 0.055612 |
| Nitrospirae | 2,003,803 | 0.341289 | 0.552386 | 0.019141 | 0.07548 |
| Planctomycetes | 6,254,950 | 0.5550987 | 0.502151 | 0.116125 | 0.138471 |
| Proteobacteria | 3,506,416.55 | 0.5337785 | 0.569934 | 0.067462 | 0.135439 |
| Spirochaetes | 1,702,653.17 | 0.3721947 | 0.600467 | 0.021591 | 0.121083 |
| Synergistetes | 1,914,533 | 0.5454971 | 0.75006 | 0.023406 | 0.050368 |
| Tenericutes | 892,007.889 | 0.2794737 | 0.665323 | −0.02018 | −0.08702 |
| Thermotogae | 1,976,742.36 | 0.4028872 | 0.54724 | 0.024232 | 0.083806 |
| Verrucomicrobia | 3,998,507 | 0.5480856 | 0.51413 | 0.093882 | 0.10771 |
| Mean | 3,329,265.48 | 0.4952767 | 0.612158 | 0.092191 | 0.127667 |
2.3. Composition Bias in Genomes with Different S Values
2.4. Composition Bias in Genomes with Different Generation Times
2.5. Composition Bias in Genomes with Different Genome Sizes
2.6. Composition Bias in Genomes with Different Gene Densities of the Leading Strand
2.7. Composition Bias in Genomes with Different GC Contents
2.8. Composition Bias in Genomes with Different Recombination Rates
2.9. Composition Bias in Different COG Functional Categories
2.9.1. Percentage of Gene Number for Each COG Functional Subcategory
| COG Functional Category | p Value | Correlation | |
|---|---|---|---|
| Information Storage and Processing | |||
| J | Translation, ribosomal structure and biogenesis P | 8.11 × 10−32 | 0.341886 |
| A | RNA processing and modification N | 2.44 × 10−13 | −0.21728 |
| K | Transcription | 0.099239 | −0.04948 |
| L | Replication, recombination and repair P | 1.01 × 10−8 | 0.170797 |
| B | Chromatin structure and dynamics | 0.002404 | −0.09097 |
| Cellular Processes and Signaling | |||
| D | Cell cycle control, cell division, chromosome partitioning P | 1.05 × 10−45 | 0.407564 |
| Y | Nuclear structure | 0.222949 | 0.036592 |
| V | Defense mechanisms P | 3.93 × 10−14 | 0.224269 |
| T | Signal transduction mechanisms | 1.77 × 10−7 | −0.15589 |
| M | Cell wall/membrane/envelope biogenesis | 0.609835 | −0.01533 |
| Cellular Processes and Signaling | |||
| N | Cell motility | 0.198305 | 0.038623 |
| Z | Cytoskeleton | 0.006632 | −0.0814 |
| W | Extracellular structures | 0.901043 | −0.00373 |
| U | Intracellular trafficking, secretion, and vesicular transport | 0.908091 | 0.003467 |
| O | Posttranslational modification, protein turnover, chaperones | 0.188347 | −0.0395 |
| Metabolism | |||
| C | Energy production and conversion N | 4.51 × 10−11 | −0.1959 |
| G | Carbohydrate transport and metabolism | 0.193919 | 0.039003 |
| E | Amino acid transport and metabolism | 0.417676 | −0.02434 |
| F | Nucleotide transport and metabolism P | 5.99 × 10−39 | 0.377498 |
| H | Coenzyme transport and metabolism | 0.01405 | 0.073666 |
| I | Lipid transport and metabolism N | 1.22 × 10−19 | −0.26737 |
| P | Inorganic ion transport and metabolism | 0.081681 | −0.05226 |
| Q | Secondary metabolites biosynthesis, transport and catabolism N | 6.65 × 10−40 | −0.38194 |
2.9.2. Proportion of Replication and Repair Genes
2.9.3. Average Value of Times between Strong-Biased Group and Weak-Biased Group for Each Functional Subcategory
| COG | AVDT | COG | AVDT |
|---|---|---|---|
| D | 5.709197 | C | 1.086021 |
| K | 3.415376 | H | 1.053758 |
| N | 2.848684 | F | 1.046122 |
| T | 2.229241 | V | 1.02066 |
| M | 2.181872 | E | 0.99786 |
| O | 2.089135 | I | 0.936222 |
| U | 2.013089 | P | 0.914553 |
| G | 1.472415 | A | 0.864394 |
| L | 1.363586 | Z | 0.775298 |
| B | 1.266486 | Q | 0.64794 |
| J | 1.23429 | W | 0.6 |
| Pathway | Function | p Value | Correlation |
|---|---|---|---|
| ko03030 | DNA replication | 3.69 × 10−10 | 0.18656 |
| ko03032 | DNA replication proteins | 6.70 × 10−9 | 0.172841 |
| ko03036 | Chromosome and associated proteins | 3.28 × 10−7 | 0.152472 |
| ko03400 | DNA repair and recombination proteins | 6.73 × 10−10 | 0.183808 |
| ko03410 | Base excision repair | 2.11 × 10−6 | 0.141724 |
| ko03420 | Nucleotide excision repair | 4.15 × 10−12 | 0.2059713 |
| ko03430 | Mismatch repair | 9.39 × 10−12 | 0.2025802 |
| ko03440 | Homologous recombination | 1.16 × 10−10 | 0.191753 |
| ko03450 | Non-homologous end-joining | 0.926821 | 0.002759 |
| ko03460 | Fanconi anemia pathway | 0.002531 | 0.090509 |
2.10. Conjoint Analysis of Multiple Factors and Composition Bias by Principal Component Regression
| Genomic Features | Genome Size | Gene Density | GC Content | gcRF | taRF | SCOGs | WCOGs | A |
|---|---|---|---|---|---|---|---|---|
| R2 | 0.0558 | 0.0648 | 0.0391 | 0.0004 | 0.0003 | 0.0332 | 0.0326 | 0.0122 |
| Genomic features | C | D | F | I | J | L | Q | V |
| R2 | 0.0634 | 0.0348 | 0.0272 | 0.0238 | 0.0299 | 0.0371 | 0.0262 | 0.0297 |
3. Experimental Section
3.1. Data Source
3.2. Computation of Strand Composition Bias
3.3. Computation of Counteracting Effect of Recombination
3.4. Computation of the Percentage of Each COG Functional Subcategory
3.5. Computation of Average Value of Differences between Strong-Biased Group and Weak-Biased Group for Each Functional Subcategory
3.6. Proportion of SCOGs and WCOGs
3.7. Proportion of Replication and Repair Genes
3.8. Statistical Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rocha, E.P. The replication-related organization of bacterial genomes. Microbiology 2004, 150, 1609–1627. [Google Scholar] [PubMed]
- Frank, A.C.; Lobry, J.R. Asymmetric substitution patterns: A review of possible underlying mutational or selective mechanisms. Gene 1999, 238, 65–77. [Google Scholar] [CrossRef]
- Asakawa, S.; Kumazawa, Y.; Araki, T.; Himeno, H.; Miura, K.; Watanabe, K. Strand-specific nucleotide composition bias in echinoderm and vertebrate mitochondrial genomes. J. Mol. Evol. 1991, 32, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Lobry, J.R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996, 13, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DNA replication and strand asymmetry in prokaryotic and mitochondrial genomes. Curr. Genom. 2012, 13, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Necsulea, A.; Lobry, J.R. A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 2007, 24, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-B.; Ning, L.-W. Strand-Specific Composition Bias in Bacterial Genomes; INTECH Open Access Publisher: Rijeka, Croatia, 2011. [Google Scholar]
- Guo, F.B. Replicating strand asymmetry in bacterial and eukaryotic genomes. Curr. Genom. 2012, 13, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, K.; Tomita, M. Measures of compositional strand bias related to replication machinery and its applications. Curr. Genom. 2012, 13, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Cui, P.; Ding, F.; Hu, S.; Yu, J. Replication-associated mutational pressure (RMP) governs strand-biased compositional asymmetry (SCA) and gene organization in animal mitochondrial genomes. Curr. Genom. 2012, 13, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.V.; Barkovsky, E.V. A blueprint for a mutationist theory of replicative strand asymmetries formation. Curr. Genom. 2012, 13, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, K.; Suzuki, H.; Tomita, M. Quantitative analysis of replication-related mutation and selection pressures in bacterial chromosomes and plasmids using generalised GC skew index. BMC Genom. 2009, 10, 640. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.V.; Barkovsky, E.V. Study of completed archaeal genomes and proteomes: Hypothesis of strong mutational at pressure existed in their common predecessor. Genom. Proteom. Bioinform. 2010, 8, 22–32. [Google Scholar] [CrossRef]
- Lobry, J.R.; Sueoka, N. Asymmetric directional mutation pressures in bacteria. Genome Biol. 2002, 3, RESEARCH0058. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.V.; Barkovsky, E.V. “Protoisochores” in certain archaeal species are formed by replication-associated mutational pressure. Biochimie 2011, 93, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.V.; Barkovsky, E.V. The probability of nonsense mutation caused by replication-associated mutational pressure is much higher for bacterial genes from lagging than from leading strands. Genomics 2010, 96, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Beletskii, A.; Bhagwat, A.S. Transcription-induced mutations: Increase in c to t mutations in the nontranscribed strand during transcription in escherichia coli. Proc. Natl. Acad. Sci. USA 1996, 93, 13919–13924. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro-Stone, M.; Nikolaishvili-Feinberg, N. Asymmetry of DNA replication and translesion synthesis of UV-induced thymine dimers. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2002, 510, 91–106. [Google Scholar] [CrossRef]
- Khrustalev, V.V.; Barkovsky, E.V. The level of cytosine is usually much higher than the level of guanine in two-fold degenerated sites from third codon positions of genes from simplex- and varicelloviruses with G plus C higher than 50%. J. Theor. Biol. 2010, 266, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.; Barkovsky, E. Bioinformatical approaches for studies on replication-associated and transcription-associated mutational pressure, interpretations and applications. Adv. Genet. Res. 2011, 6, 1–108. [Google Scholar]
- Francino, M.P.; Ochman, H. Strand asymmetries in DNA evolution. Trends Genet. 1997, 13, 240–245. [Google Scholar] [CrossRef]
- Nikolaou, C.; Almirantis, Y. A study on the correlation of nucleotide skews and the positioning of the origin of replication: Different modes of replication in bacterial species. Nucleic Acids Res. 2005, 33, 6816–6822. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P. The organization of the bacterial genome. Annu. Rev. Genet. 2008, 42, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Guo, F.B. Strong strand composition bias in the genome of ehrlichia canis revealed by multiple methods. Open Microbiol. J. 2010, 4, 98–102. [Google Scholar] [PubMed]
- Guo, F.B.; Yu, X.J. Separate base usages of genes located on the leading and lagging strands in chlamydia muridarum revealed by the Z curve method. BMC Genom. 2007, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.B.; Yuan, J.B. Codon usages of genes on chromosome, and surprisingly, genes in plasmid are primarily affected by strand-specific mutational biases in lawsonia intracellularis. DNA Res. 2009, 16, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Bennett, K.W.; Eley, A. Fusobacteria: New taxonomy and related diseases. J. Med. Microbiol. 1993, 39, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhao, X.; Yu, J. Replication-associated purine asymmetry may contribute to strand-biased gene distribution. Genomics 2007, 90, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Bailes, E.; Grocock, R.J.; Peden, J.F.; Sockett, R.E. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005, 33, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Silva, S.; Rocha, E. The systemic imprint of growth and its uses in ecological (meta) genomics. PLoS Genet. 2010, 6, e1000808. [Google Scholar] [CrossRef] [PubMed]
- McLean, M.J.; Wolfe, K.H.; Devine, K.M. Base composition skews, replication orientation, and gene orientation in 12 prokaryote genomes. J. Mol. Evol. 1998, 47, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P. Is there a role for replication fork asymmetry in the distribution of genes in bacterial genomes? Trends Microbiol. 2002, 10, 393–395. [Google Scholar] [CrossRef]
- Karlin, S. Bacterial DNA strand compositional asymmetry. Trends Microbiol. 1999, 7, 305–308. [Google Scholar] [CrossRef]
- Hildebrand, F.; Meyer, A.; Eyre-Walker, A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010, 6, e1001107. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Million-Weaver, S.; Chattopadhyay, S.; Sokurenko, E.; Merrikh, H. Accelerated gene evolution through replication-transcription conflicts. Nature 2013, 495, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Klasson, L.; Andersson, S.G. Strong asymmetric mutation bias in endosymbiont genomes coincide with loss of genes for replication restart pathways. Mol. Biol. Evol. 2006, 23, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.; Danchin, A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002, 18, 291–294. [Google Scholar] [CrossRef]
- Lin, Y.; Gao, F.; Zhang, C.T. Functionality of essential genes drives gene strand-bias in bacterial genomes. Biochem. Biophys. Res. Commun. 2010, 396, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Luo, H.; Zhang, C.T. Doric 5.0: An updated database of oric regions in both bacterial and archaeal genomes. Nucleic Acids Res. 2013, 41, D90–D93. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.-L.; Xia, Z.-K.; Zhang, F.-Z.; Ye, Y.-N.; Guo, F.-B. Multiple Factors Drive Replicating Strand Composition Bias in Bacterial Genomes. Int. J. Mol. Sci. 2015, 16, 23111-23126. https://doi.org/10.3390/ijms160923111
Zhao H-L, Xia Z-K, Zhang F-Z, Ye Y-N, Guo F-B. Multiple Factors Drive Replicating Strand Composition Bias in Bacterial Genomes. International Journal of Molecular Sciences. 2015; 16(9):23111-23126. https://doi.org/10.3390/ijms160923111
Chicago/Turabian StyleZhao, Hai-Long, Zhong-Kui Xia, Fa-Zhan Zhang, Yuan-Nong Ye, and Feng-Biao Guo. 2015. "Multiple Factors Drive Replicating Strand Composition Bias in Bacterial Genomes" International Journal of Molecular Sciences 16, no. 9: 23111-23126. https://doi.org/10.3390/ijms160923111