
Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury

Abstract
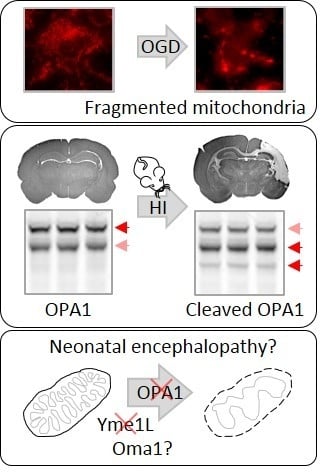
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
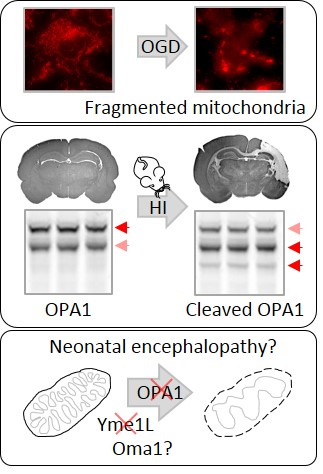
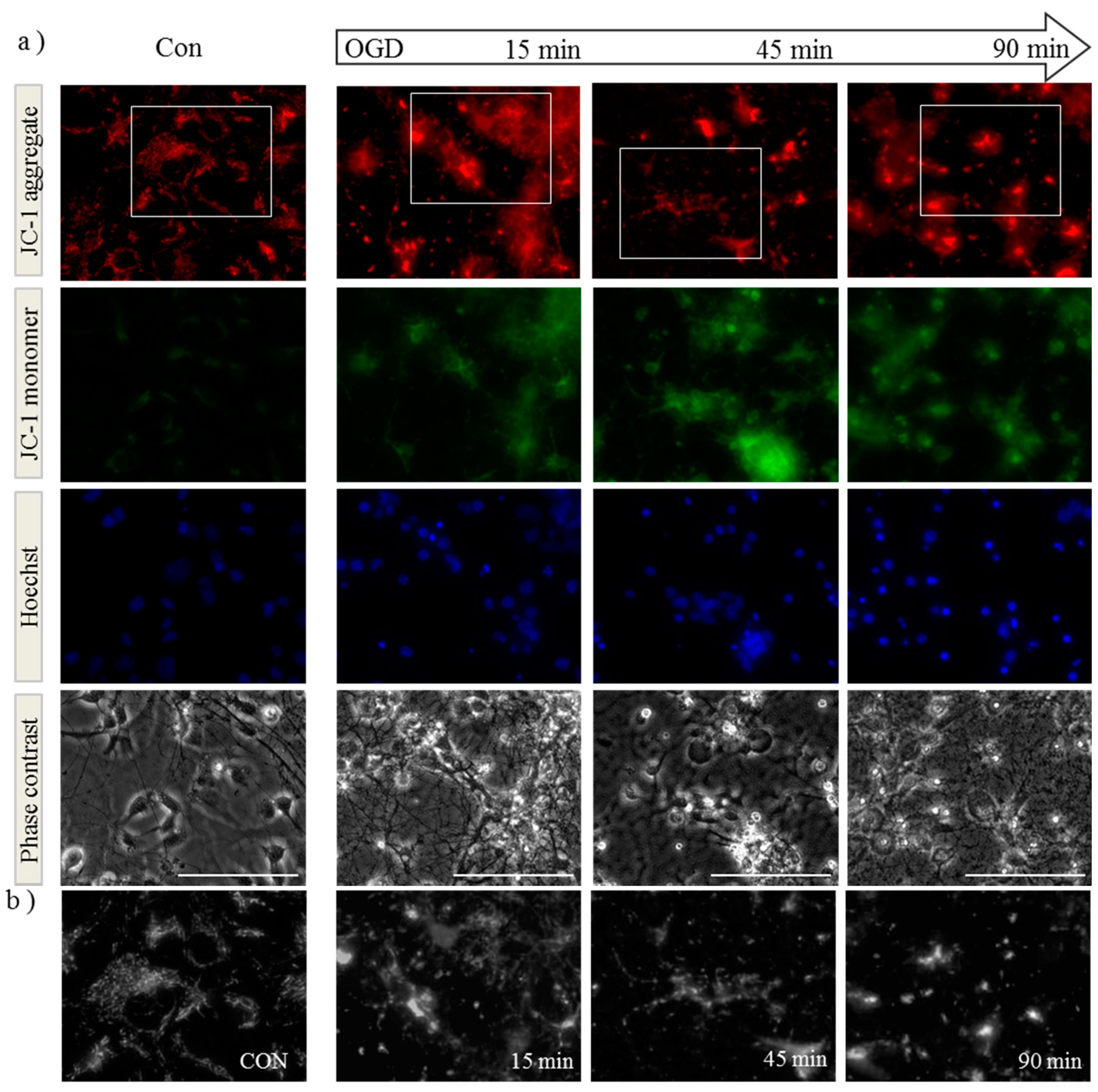
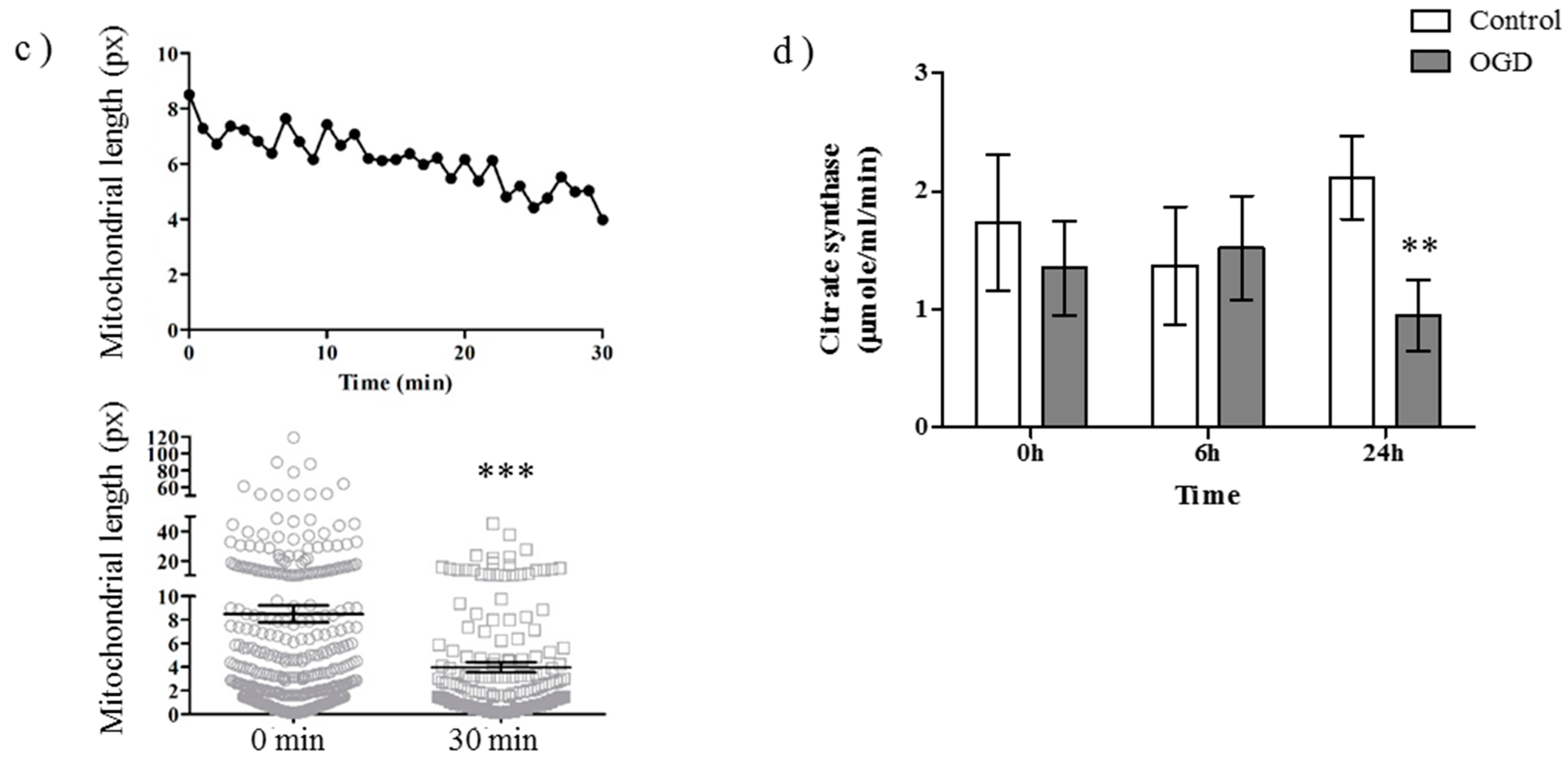
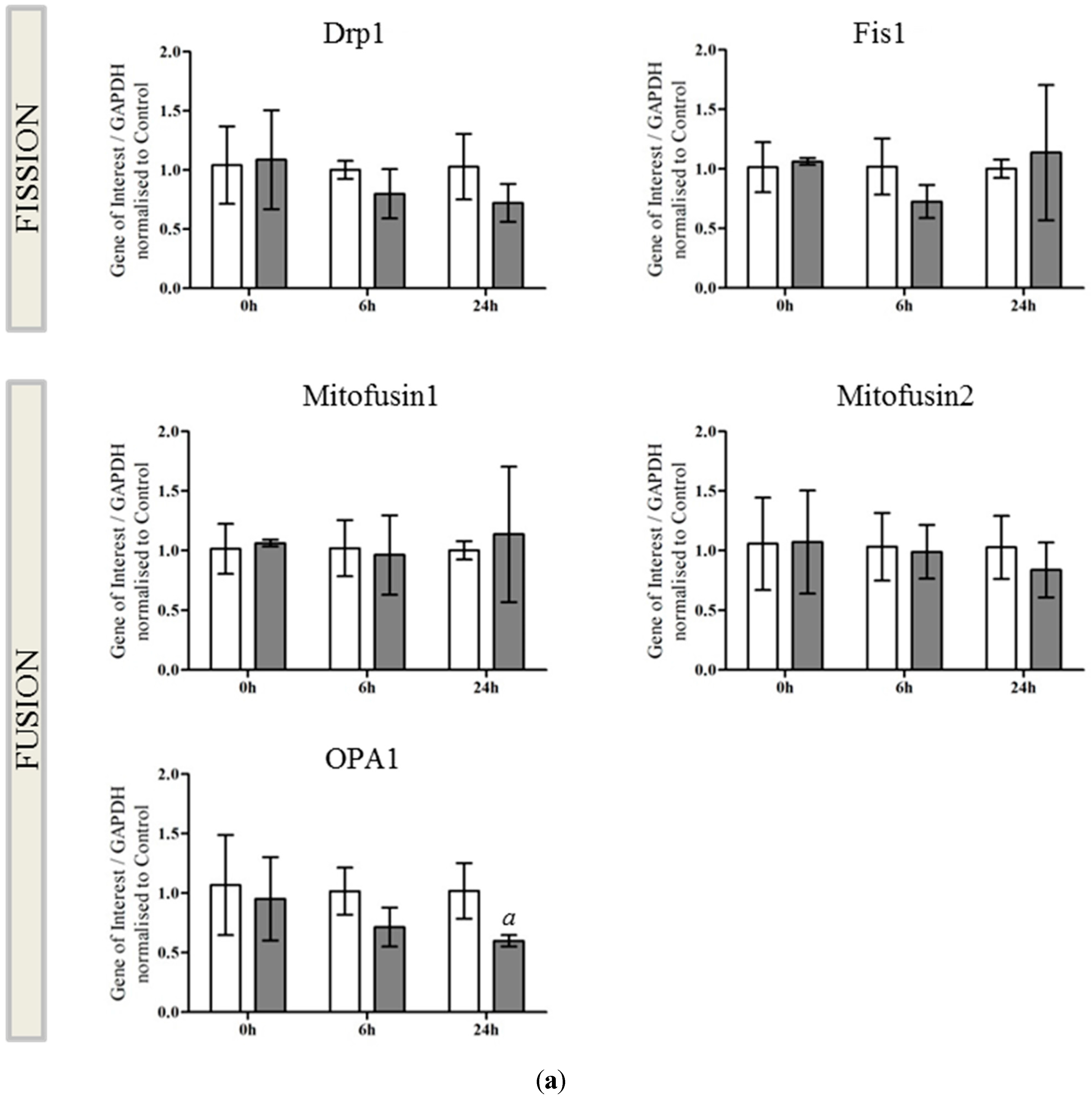
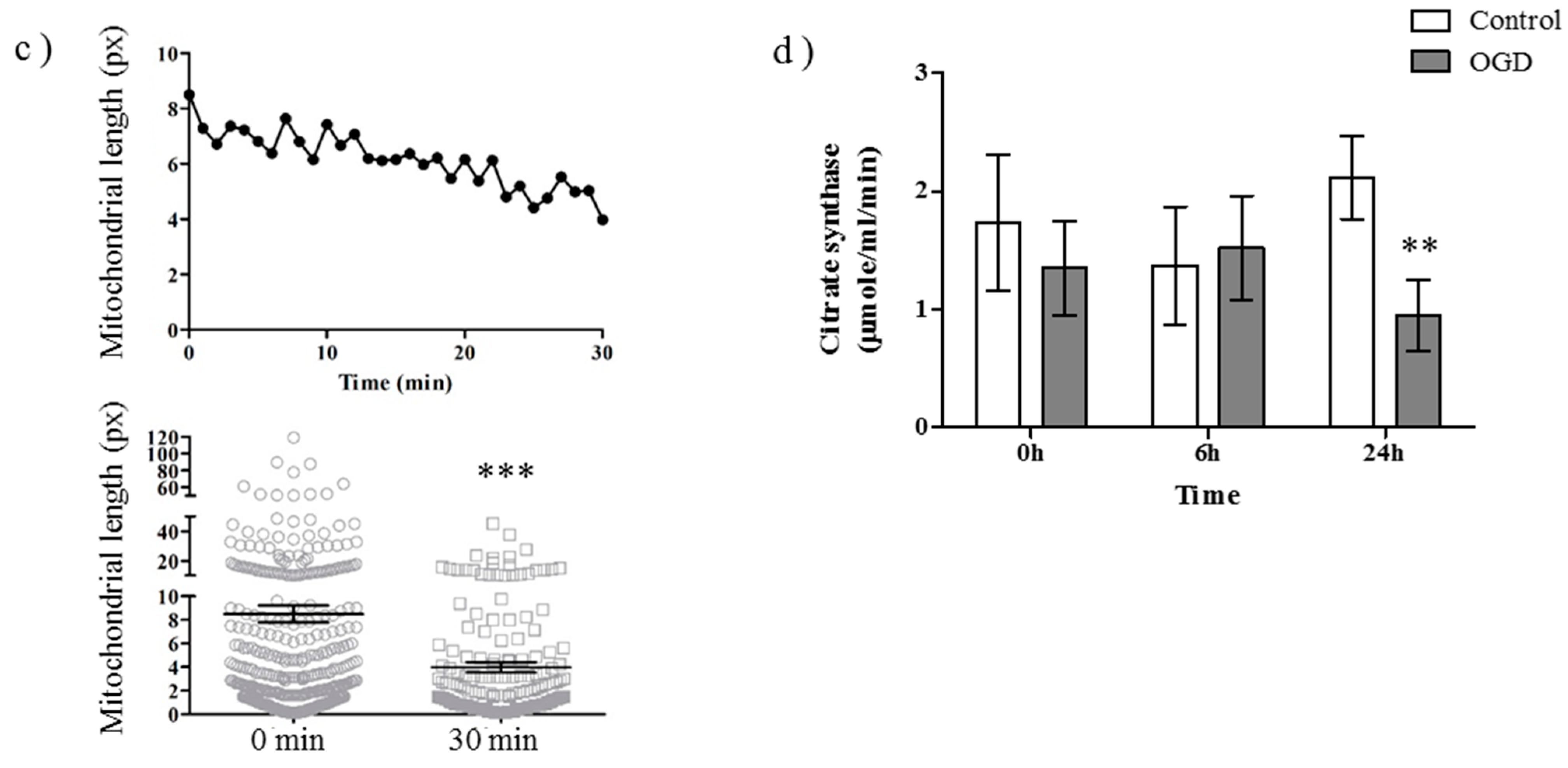
2.1. OGD in C17.2 Cells Alters Mitochondrial Function and Morphology
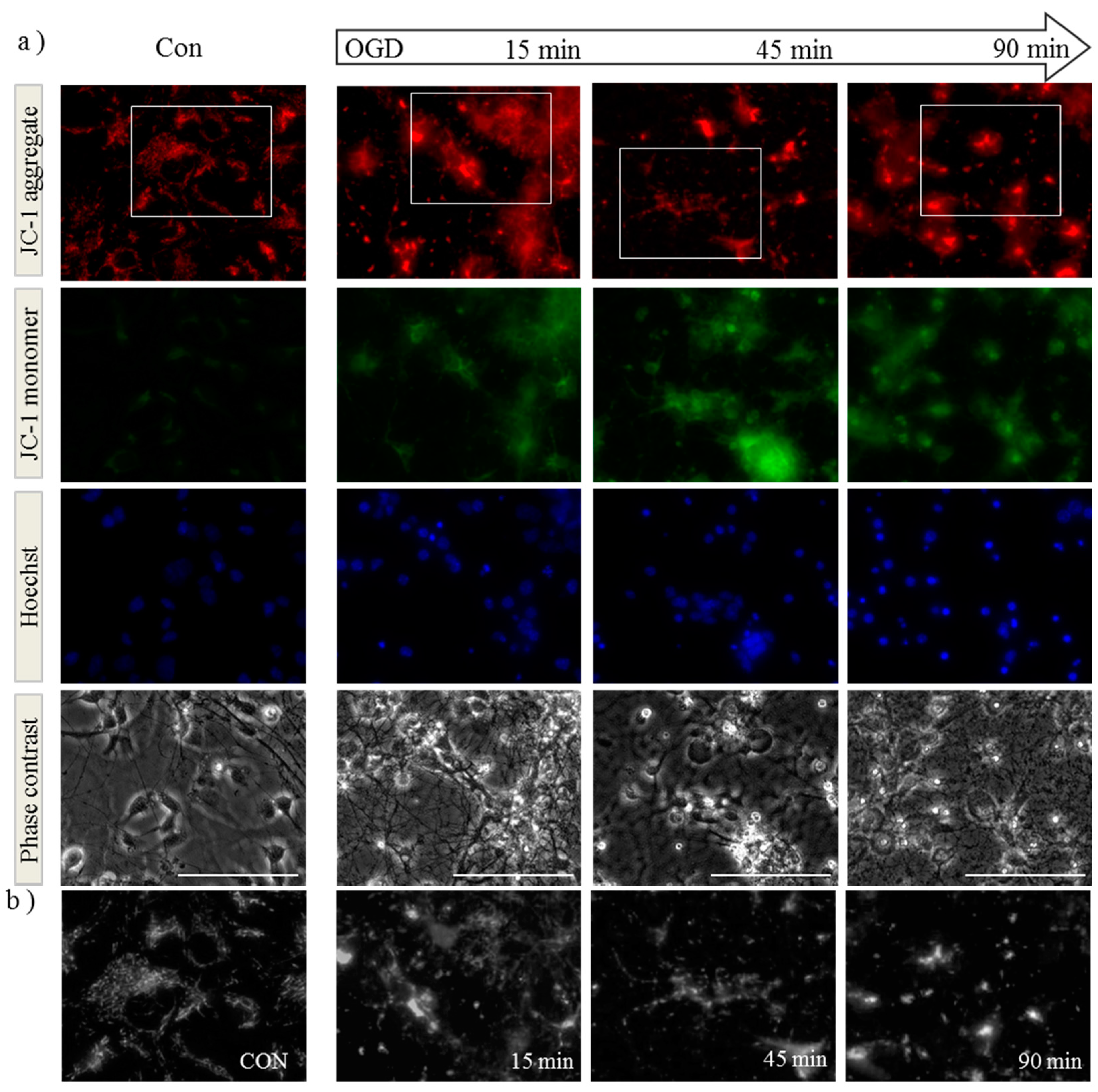
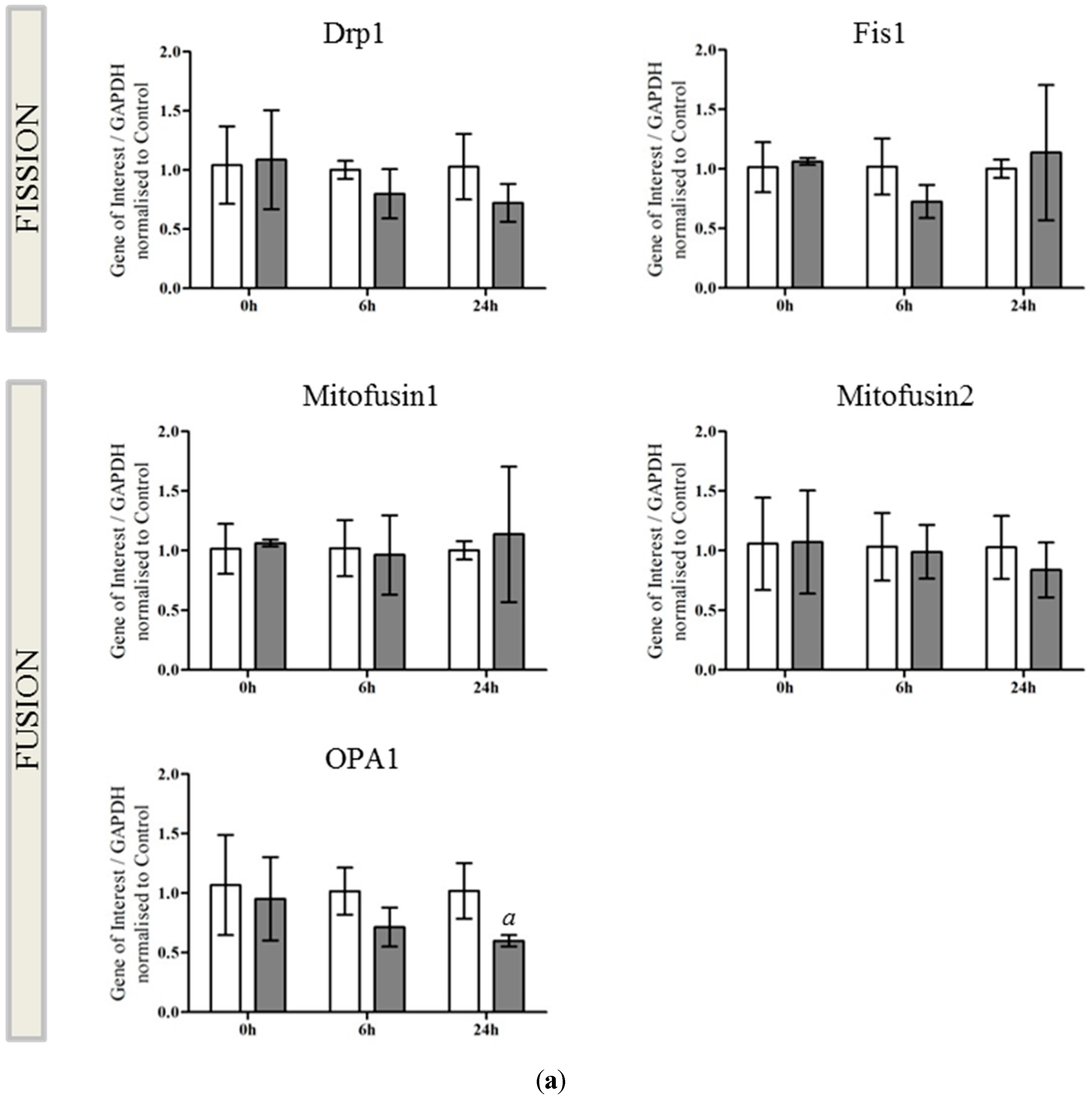
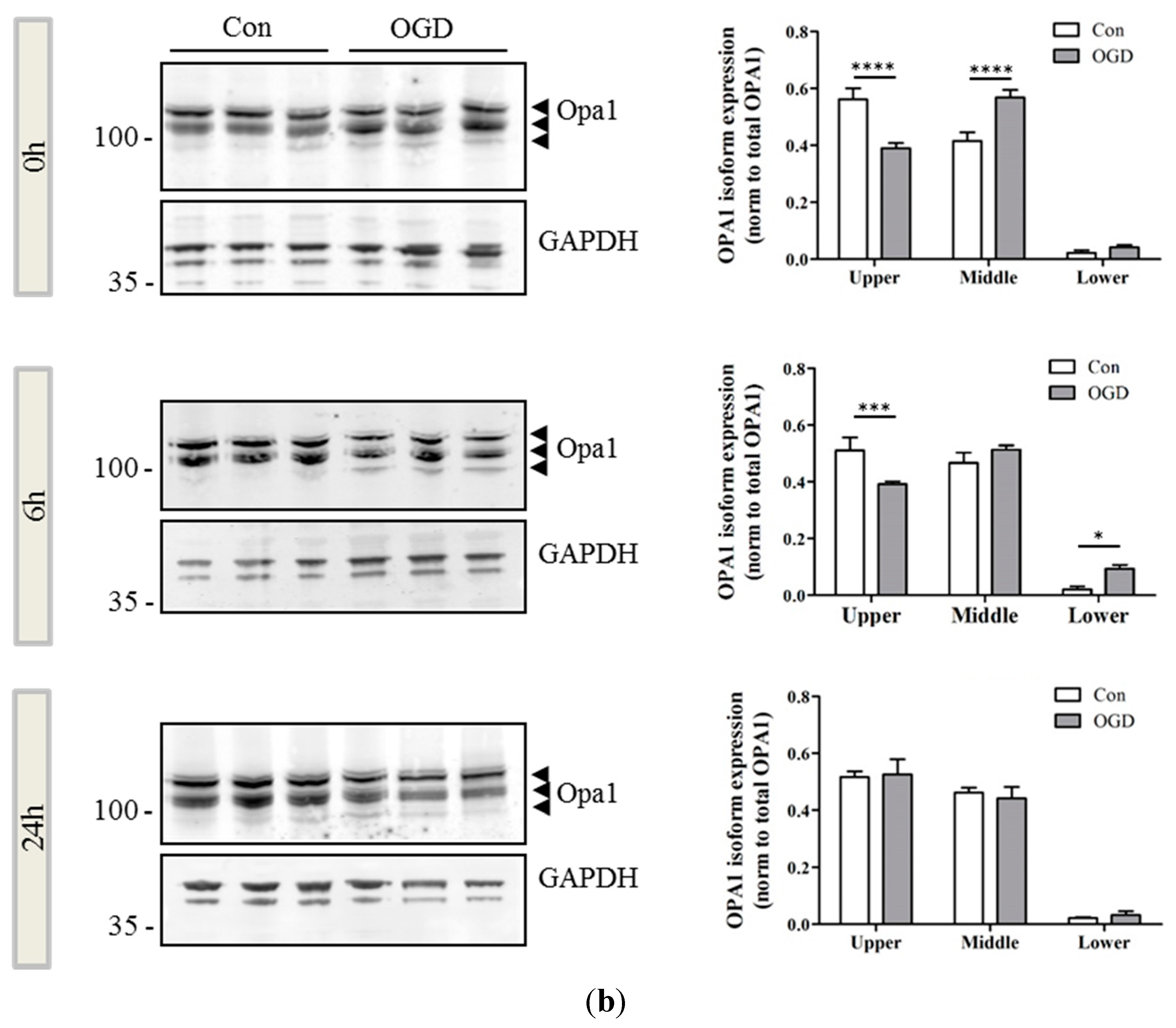
2.2. OPA1 Processing Is Altered after OGD
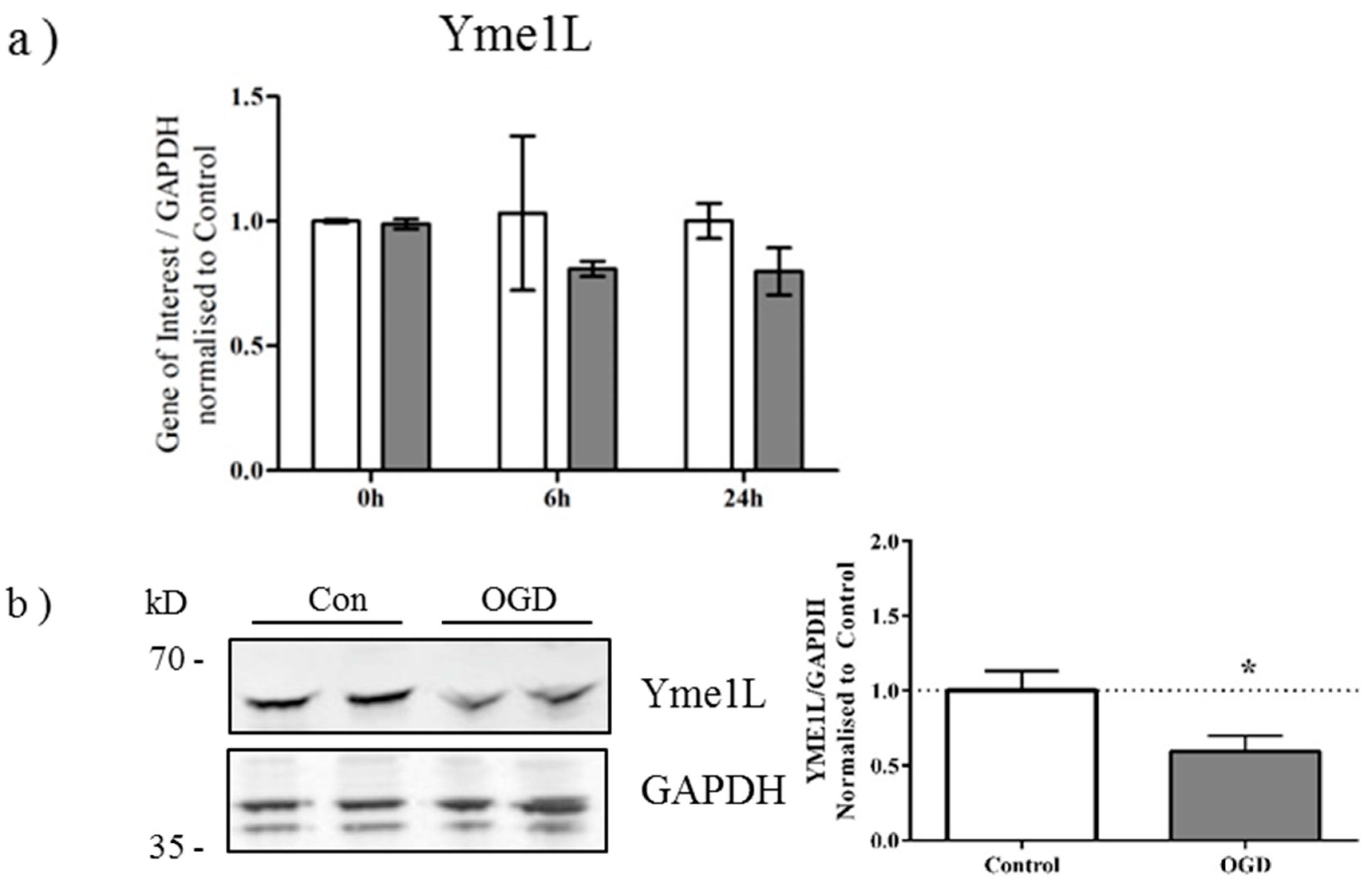
2.3. OGD Reduces Yme1L Protein Expression in Primary Neurons
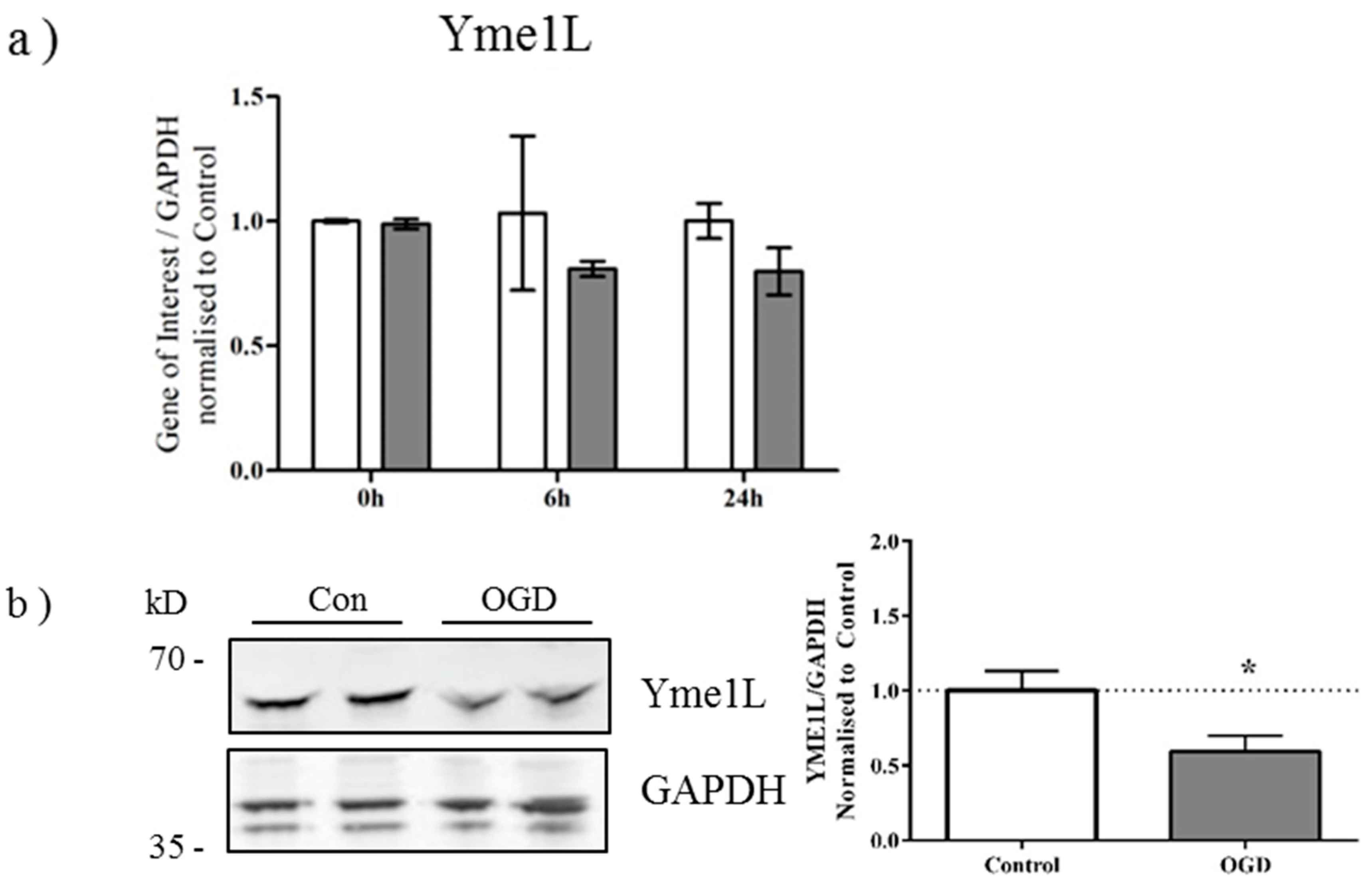
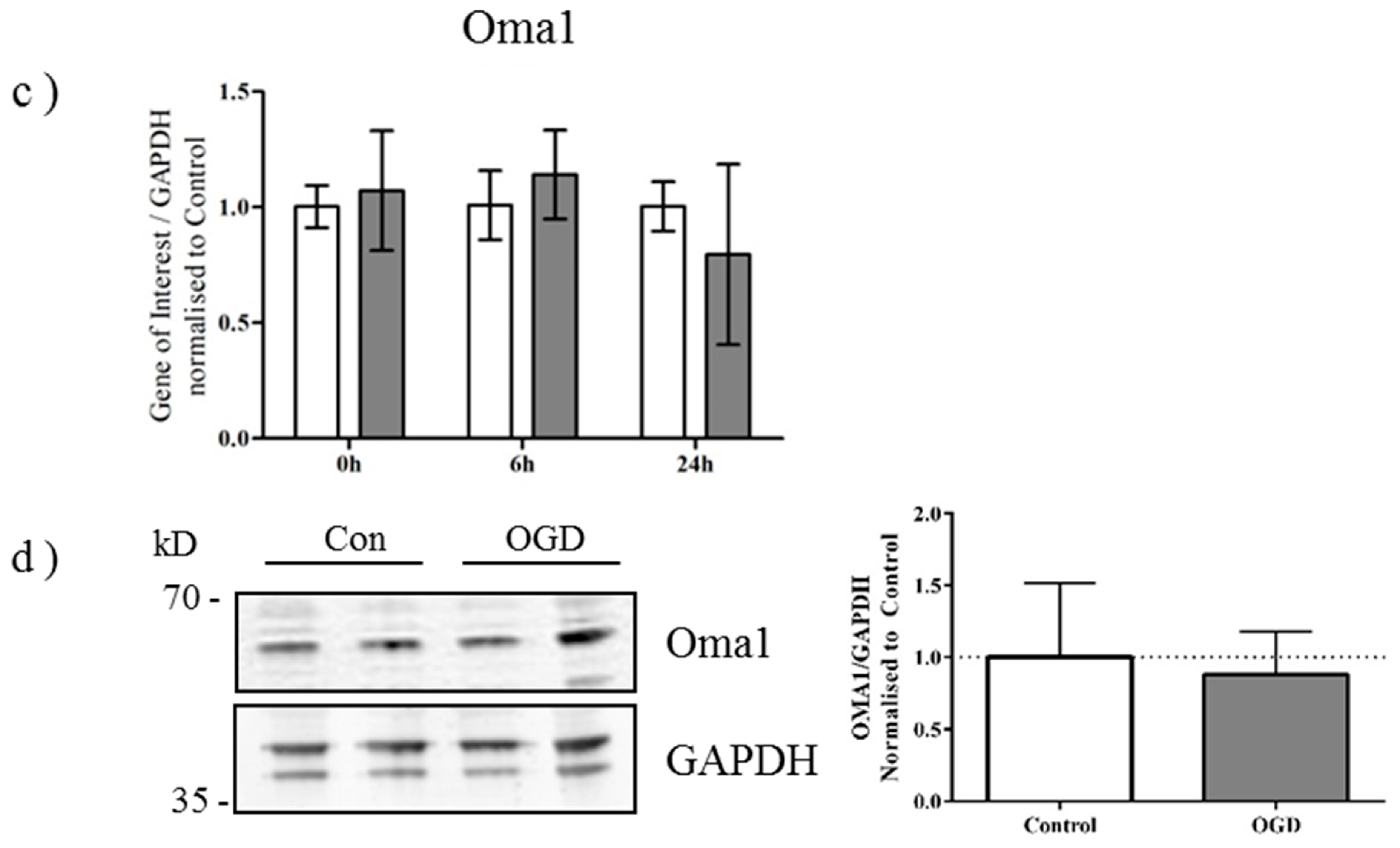
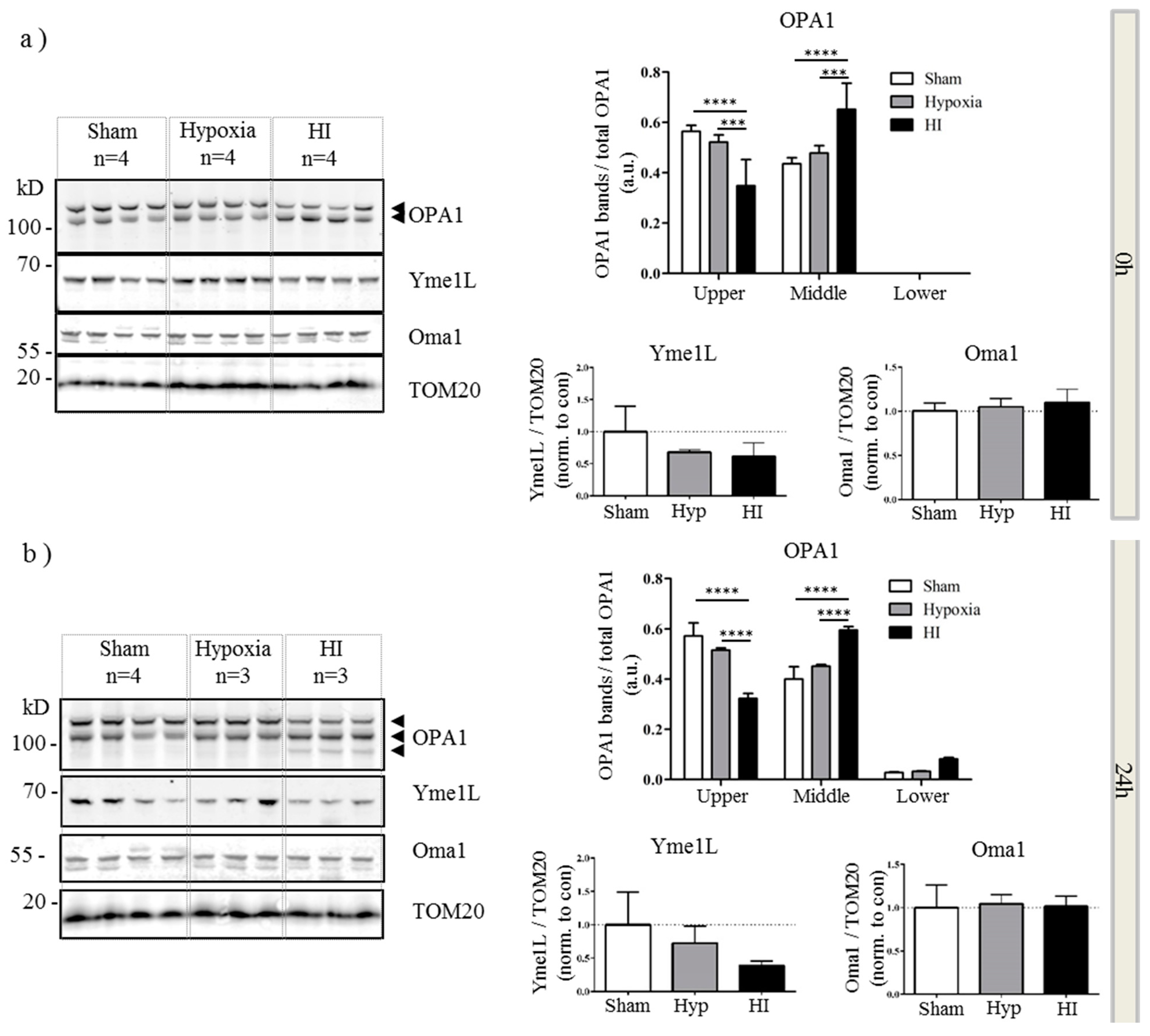
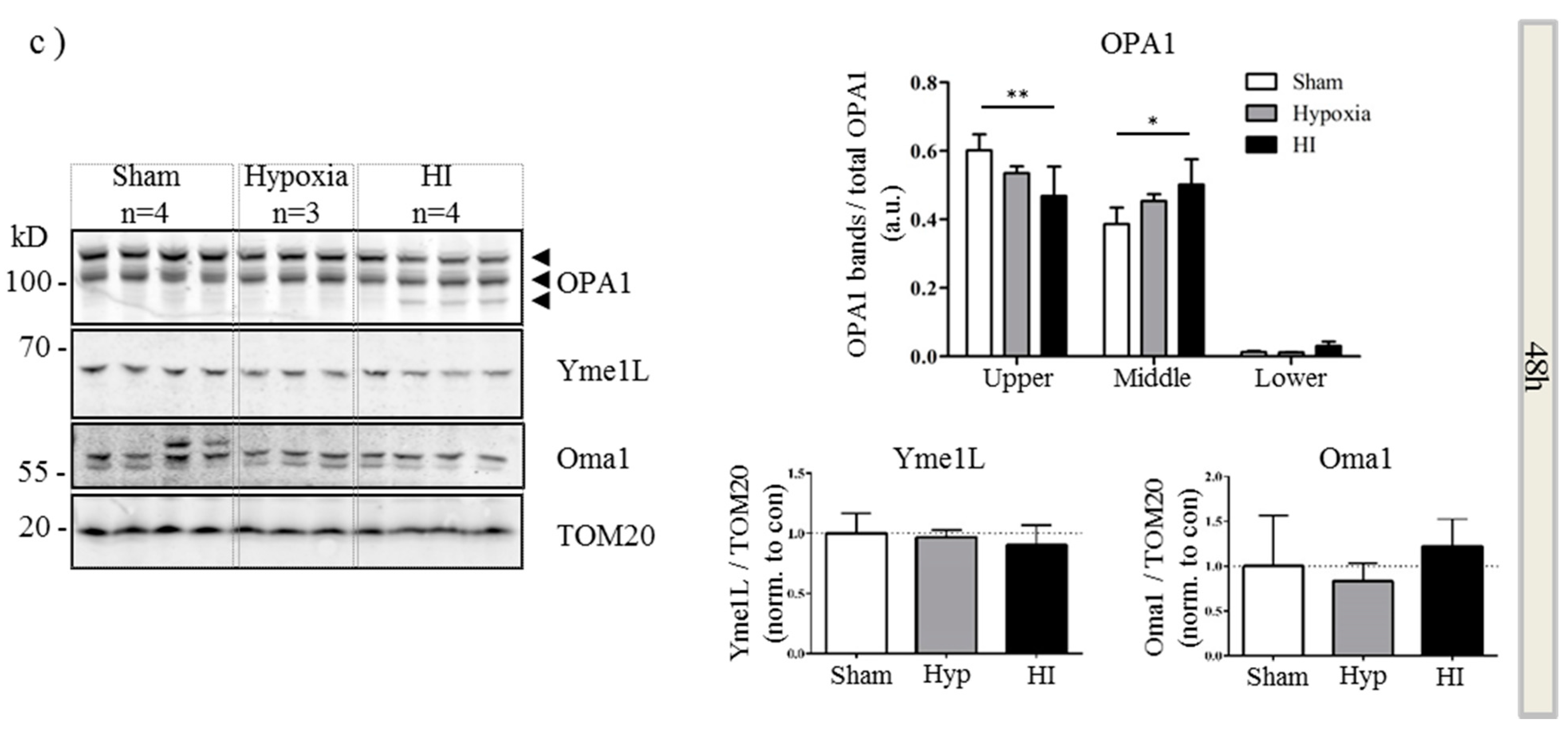
2.4. Alterations in OPA1 Processing Are Apparent in Vivo after HI
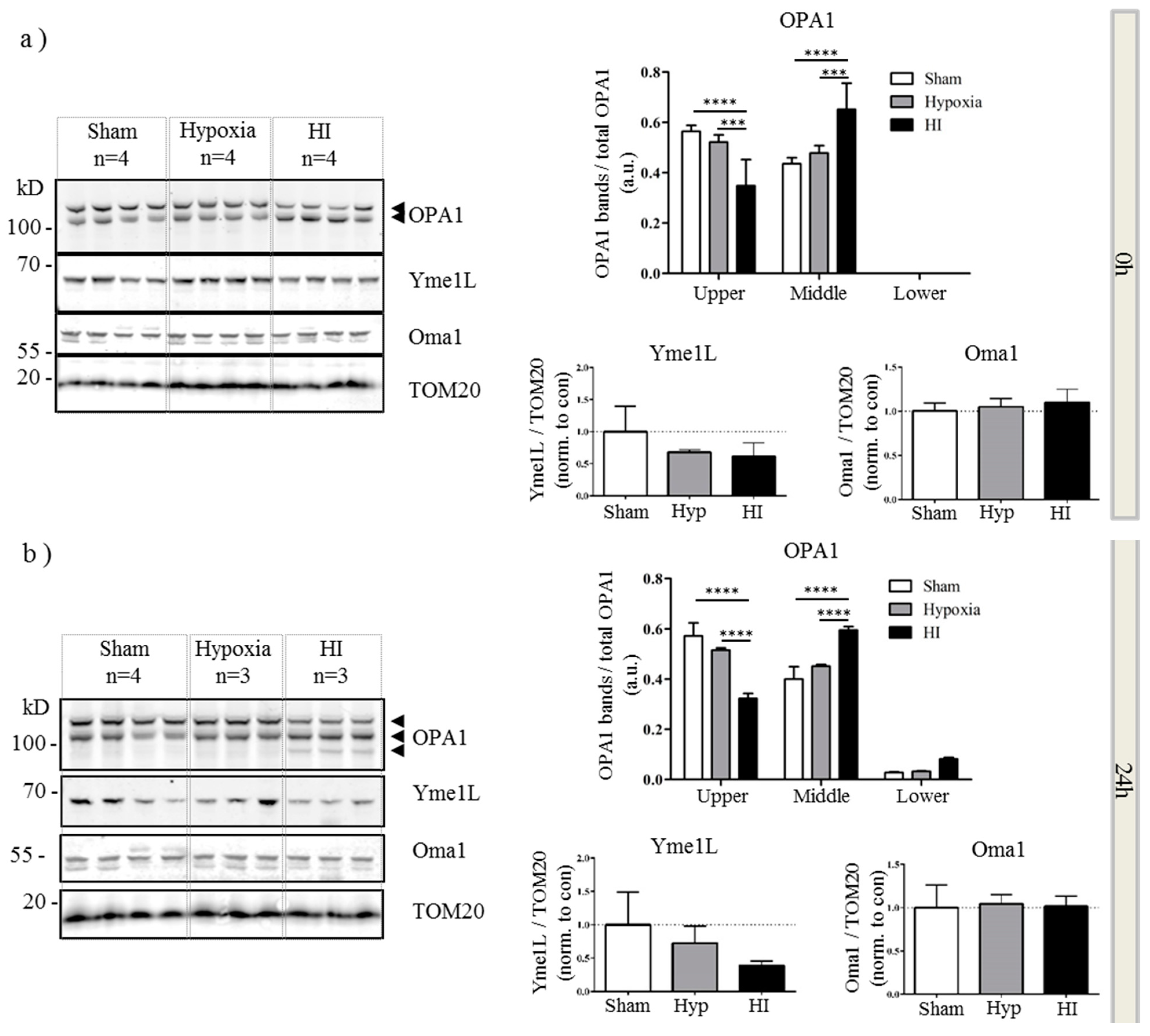
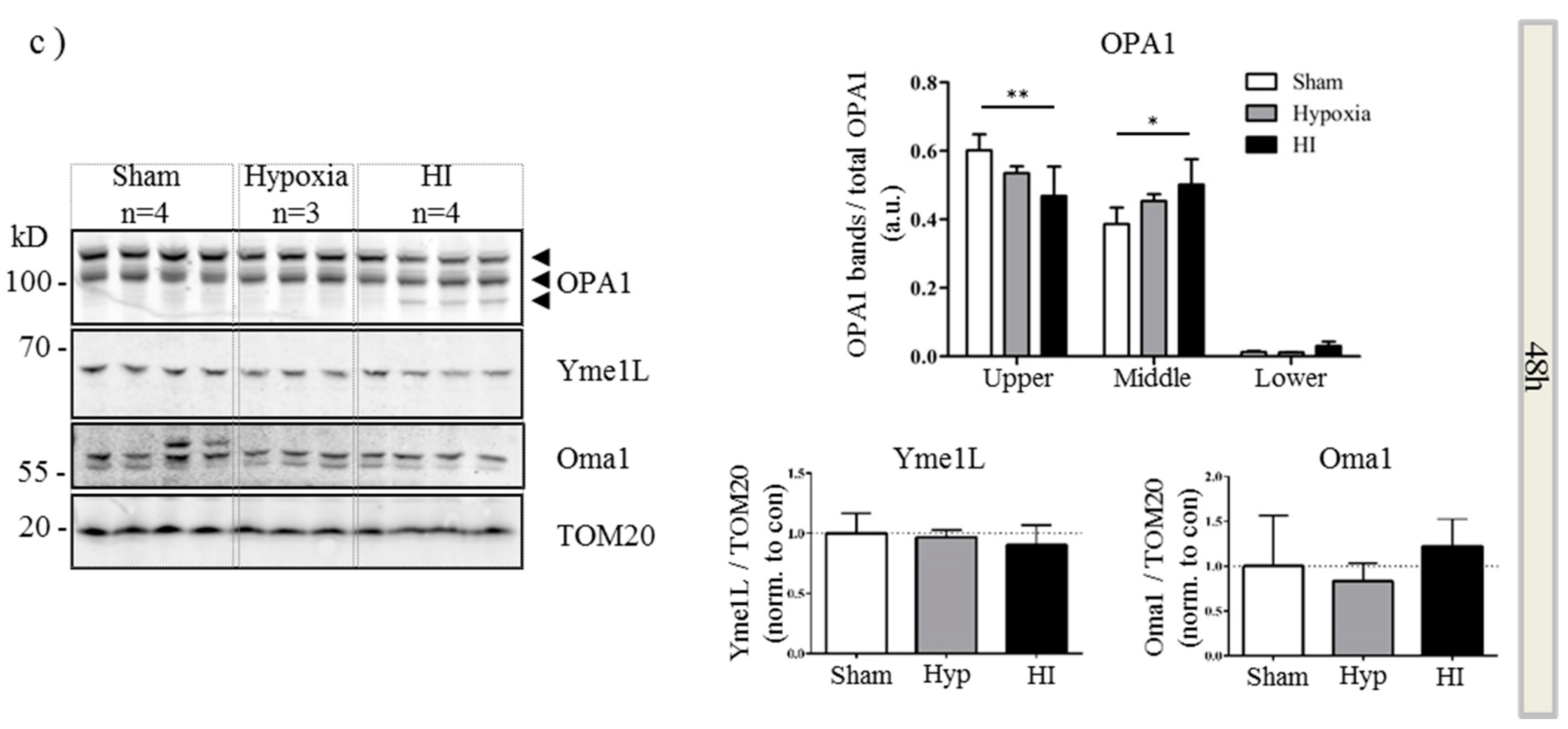
3. Discussion
4. Experimental Section
4.1. Research Ethics Statement
4.2. Primary Cortical Neuron Preparation
4.3. Neonatal Hypoxia-Ischemia
4.4. Oxygen-Glucose Deprivation (OGD) of Primary Neurons
4.5. MTT Assay
4.6. Citrate Synthase Assay
4.7. qRT-PCR
4.8. Subcellular Fractionation
4.9. Western Blot
4.10. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Evans, K.; Rigby, A.S.; Hamilton, P.; Titchiner, N.; Hall, D.M. The relationships between neonatal encephalopathy and cerebral palsy: A cohort study. J. Obstet. Gynaecol. 2001, 21, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Wells, L.; Dodd, K. The continuing fall in incidence of hypoxic-ischaemic encephalopathy in term infants. BJOG 2000, 107, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Lawn, J.E.; Bahl, R.; Bergstrom, S.; Bhutta, Z.A.; Darmstadt, G.L.; Ellis, M.; English, M.; Kurinczuk, J.J.; Lee, A.C.; Merialdi, M.; et al. Setting research priorities to reduce almost one million deaths from birth asphyxia by 2015. PLoS Med. 2011, 8, e1000389. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Perlman, J.M. Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics 1997, 100, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Wyatt, J.S.; Cady, E.B.; Delpy, D.T.; Baudin, J.; Stewart, A.L.; Hope, P.L.; Hamilton, P.A.; Reynolds, E.O. Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1989, 25, 445–451. [Google Scholar] [CrossRef]
- Blumberg, R.M.; Cady, E.B.; Wigglesworth, J.S.; McKenzie, J.E.; Edwards, A.D. Relation between delayed impairment of cerebral energy metabolism and infarction following transient focal hypoxia-ischaemia in the developing brain. Exp. Brain Res. 1997, 113, 130–137. [Google Scholar] [CrossRef]
- Gilland, E.; Bona, E.; Hagberg, H. Temporal changes of regional glucose use, blood flow, and microtubule-associated protein 2 immunostaining after hypoxia-ischemia in the immature rat brain. J. Cereb. Blood Flow Metab. 1998, 18, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Lorek, A.; Takei, Y.; Cady, E.B.; Wyatt, J.S.; Penrice, J.; Edwards, A.D.; Peebles, D.; Wylezinska, M.; Owen-Reece, H.; Kirkbride, V.; et al. Delayed (“secondary”) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: Continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1994, 36, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Thornberg, E.; Blennow, M.; Kjellmer, I.; Lagercrantz, H.; Thiringer, K.; Hamberger, A.; Sandberg, M. Excitatory amino acids in the cerebrospinal fluid of asphyxiated infants: Relationship to hypoxic-ischemic encephalopathy. Acta Paediatr. 1993, 82, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Puka-Sundvall, M.; Gajkowska, B.; Cholewinski, M.; Blomgren, K.; Lazarewicz, J.W.; Hagberg, H. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain Res. Dev. Brain Res. 2000, 125, 31–41. [Google Scholar] [CrossRef]
- Van den Tweel, E.R.; Nijboer, C.; Kavelaars, A.; Heijnen, C.J.; Groenendaal, F.; van Bel, F. Expression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia-ischemia in the neonatal rat brain. J. Neuroimmunol. 2005, 167, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Wallin, C.; Puka-Sundvall, M.; Hagberg, H.; Weber, S.G.; Sandberg, M. Alterations in glutathione and amino acid concentrations after hypoxia-ischemia in the immature rat brain. Brain Res. Dev. Brain Res. 2000, 125, 51–60. [Google Scholar] [CrossRef]
- Northington, F.J.; Zelaya, M.E.; O’Riordan, D.P.; Blomgren, K.; Flock, D.L.; Hagberg, H.; Ferriero, D.M.; Martin, L.J. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 2007, 149, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Portera-Cailliau, C.; Price, D.L.; Martin, L.J. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J. Comp. Neurol. 1997, 378, 70–87. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Thornton, C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014, 13, 217–232. [Google Scholar] [CrossRef]
- Thornton, C.; Rousset, C.I.; Kichev, A.; Miyakuni, Y.; Vontell, R.; Baburamani, A.A.; Fleiss, B.; Gressens, P.; Hagberg, H. Molecular mechanisms of neonatal brain injury. Neurol. Res. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Gilland, E.; Puka-Sundvall, M.; Hillered, L.; Hagberg, H. Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature rat brain: Involvement of NMDA-receptors. J. Cereb. Blood Flow Metab. 1998, 18, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Puka-Sundvall, M.; Wallin, C.; Gilland, E.; Hallin, U.; Wang, X.; Sandberg, M.; Karlsson, J.; Blomgren, K.; Hagberg, H. Impairment of mitochondrial respiration after cerebral hypoxia-ischemia in immature rats: Relationship to activation of caspase-3 and neuronal injury. Brain Res. Dev. Brain Res. 2000, 125, 43–50. [Google Scholar] [CrossRef]
- Rosenberg, A.A.; Parks, J.K.; Murdaugh, E.; Parker, W.D., Jr. Mitochondrial function after asphyxia in newborn lambs. Stroke 1989, 20, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Huang, Z.; Qiu, L.; Xu, F.; Vahsen, N.; Nilsson, M.; Eriksson, P.S.; Hagberg, H.; Culmsee, C.; et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007, 14, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.E.; Han, B.H.; Choi, J.; Knudson, C.M.; Korsmeyer, S.J.; Parsadanian, M.; Holtzman, D.M. BAX contributes to apoptotic-like death following neonatal hypoxia-ischemia: Evidence for distinct apoptosis pathways. Mol. Med. 2001, 7, 644–655. [Google Scholar] [PubMed]
- Wang, X.; Han, W.; Du, X.; Zhu, C.; Carlsson, Y.; Mallard, C.; Jacotot, E.; Hagberg, H. Neuroprotective effect of Bax-inhibiting peptide on neonatal brain injury. Stroke 2010, 41, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed]
- Wappler, E.A.; Institoris, A.; Dutta, S.; Katakam, P.V.; Busija, D.W. Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLoS ONE 2013, 8, e63206. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Griffoin, J.M.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.E., 3rd; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Vannucci, S.J. A model of perinatal hypoxic-ischemic brain damage. Ann. N. Y. Acad. Sci. 1997, 835, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Biala, A.K.; Dhingra, R.; Kirshenbaum, L.A. Mitochondrial dynamics: Orchestrating the journey to advanced age. J. Mol. Cell. Cardiol. 2015, 83, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Supale, S.; Li, N.; Brun, T.; Maechler, P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol. Metab. 2012, 23, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Iijima, T. Mitochondrial membrane potential and ischemic neuronal death. Neurosci. Res. 2006, 55, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Wyatt, J.S.; Hamilton, P.A.; Cady, E.B.; Delpy, D.T.; Hope, P.L.; Reynolds, E.O. Phosphorus metabolites and intracellular pH in the brains of normal and small for gestational age infants investigated by magnetic resonance spectroscopy. Pediatr. Res. 1989, 25, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. 2015, 16, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Stiburek, L.; Cesnekova, J.; Kostkova, O.; Fornuskova, D.; Vinsova, K.; Wenchich, L.; Houstek, J.; Zeman, J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 2012, 23, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; Heijnen, C.J.; van der Kooij, M.A.; Zijlstra, J.; van Velthoven, C.T.; Culmsee, C.; van Bel, F.; Hagberg, H.; Kavelaars, A. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann. Neurol. 2011, 70, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Qiu, L.; Wang, X.; Hallin, U.; Cande, C.; Kroemer, G.; Hagberg, H.; Blomgren, K. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J. Neurochem. 2003, 86, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Xu, F.; Fukuda, A.; Wang, X.; Fukuda, H.; Korhonen, L.; Hagberg, H.; Lannering, B.; Nilsson, M.; Eriksson, P.S.; et al. X chromosome-linked inhibitor of apoptosis protein reduces oxidative stress after cerebral irradiation or hypoxia-ischemia through up-regulation of mitochondrial antioxidants. Eur. J. Neurosci. 2007, 26, 3402–3410. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Neuronal hypoxia disrupts mitochondrial fusion. Neuroscience 2015, 301, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.; Bright, N.J.; Sastre, M.; Muckett, P.J.; Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem. J. 2011, 434, 503–512. [Google Scholar] [CrossRef]
- Fleiss, B.; Chhor, V.; Rajudin, N.; Lebon, S.; Hagberg, H.; Gressens, P.; Thornton, C. The anti-inflammatory effects of the small molecule Pifithrin-μ on BV2 microglia. Dev. Neurosci. 2015, 37, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Rizk, A.; Paul, G.; Incardona, P.; Bugarski, M.; Mansouri, M.; Niemann, A.; Ziegler, U.; Berger, P.; Sbalzarini, I.F. Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh. Nat. Protoc. 2014, 9, 586–596. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baburamani, A.A.; Hurling, C.; Stolp, H.; Sobotka, K.; Gressens, P.; Hagberg, H.; Thornton, C. Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2015, 16, 22509-22526. https://doi.org/10.3390/ijms160922509
Baburamani AA, Hurling C, Stolp H, Sobotka K, Gressens P, Hagberg H, Thornton C. Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury. International Journal of Molecular Sciences. 2015; 16(9):22509-22526. https://doi.org/10.3390/ijms160922509
Chicago/Turabian StyleBaburamani, Ana A., Chloe Hurling, Helen Stolp, Kristina Sobotka, Pierre Gressens, Henrik Hagberg, and Claire Thornton. 2015. "Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury" International Journal of Molecular Sciences 16, no. 9: 22509-22526. https://doi.org/10.3390/ijms160922509