1. Introduction
A bacteriophage, living within a host bacterium, is a virus that consists of DNA or RNA, structural proteins and a few other packaged proteins [
1]. It replicates itself by making use of the host biosynthetic machinery, infects host bacterium and eventually destroys or hijacks the bacterial cell [
2]. Bacteriophages play some positive, as well as negative roles in biology, ecology and health. In the positive field, bacteriophages are extensively applied to bacterial identification, bacterial pathogenesis, organismal ecology and evolutionary biology [
3]. In particular, clinical phage therapy aiming at different bacterial diseases may provide a novel perspective to deal with the growing threat of antibiotic resistance in pathogenic bacteria [
4]. On the other hand, the pollution of bacteriophages could cause a great loss to the fermentation industry.
Bacteriophage proteins are of vital importance to determine the functions and mechanisms of bacteriophages. Bacteriophage proteins include structural (virion) proteins and non-structural (non-virion) proteins. Bacteriophage virion proteins, coded by the bacteriophage genome, are components of the mature assembled bacteriophage particles. They may include capsid proteins, envelope proteins and virus particle enzymes [
5]. These virion proteins determine the specificity for recognizing host bacteria and perform great functions in bacteriophage virus recombination, receptor recognition, bacteria attachment and penetration [
6]. Bacteriophage non-virion proteins, also encoded by the bacteriophage genome and produced in infected cells, are not packaged in the mature bacteriophage particles [
5]. These non-virion proteins, mostly enzymes and regulatory proteins, play important roles in bacteriophage replication, transcription and polyprotein processing [
7].
Due to the major contributions of virion proteins in deciding the functions and applications of bacteriophages, identifying bacteriophage virion proteins is crucial to gain insight into the relationship between bacteriophage and host bacteria, as well as the influence of bacteriophages on the development of new pathogens and antibacterial drugs. The prediction of virion components of the bacteriophages based on the homology of genomic data leads to bad results [
8]. With the avalanche of genome sequences generated in the post-genomic age, more than 70% of bacteriophage sequences in the viral reference sequence database that encode proteins do not have annotated functions [
1]. Therefore, it would be urgent to develop computational methods for rapidly and effectively identifying bacteriophage virion proteins from bacteriophage sequences.
In recent years, several computational methods have been reported for bacteriophage gene function prediction and virion protein prediction. Li
et al. [
9] developed the novel support vector machine-based system called SynFPS to perform gene function prediction, which utilized gene-to-gene distances based on the K-means clustering technique to identify closely-related genomes. Feng
et al. [
10] developed a naive Bayes-based predictor to identify bacteriophage virion proteins, using primary amino acid and dipeptide composition as the encoding scheme. Later, Ding
et al. [
11] utilized support vector machine to establish the prediction model for bacteriophage virion protein prediction. In this model, ANOVA (analysis of variance) was applied to select the high discriminative features derived from g-gap dipeptide compositions. The aforementioned methods have their own merits and achieve acceptable results, but the following shortcomings still need to be taken into consideration. (1) Previous studies relied on composition-based features, which were extracted merely based on the alphabetic sequence of the amino acids, but failed to capture some useful features based on physicochemical properties, evolutionary information,
etc. In general, a single feature cannot preserve enough discriminative information for protein attribute predictions, and multiple features can complement each other to enhance the performance and robustness of a predictor [
12,
13]. In view of the above-mentioned fact, hybrid features have been increasingly used in recent studies instead of single features for constructing classifiers [
14,
15]; (2) The existing methods were all based on individual classifiers, which could have their own inherent defects, leading to unsatisfactory predictions [
16]. It is generally accepted that the ensemble predictor, which integrates multiple basic classifiers of diverse learning policies (or diversely trained), can achieve better prediction performance than its component classifiers for protein attribute predictions [
17]. Thus, the ensemble predictor has been considered as the future direction for protein classification problems [
18].
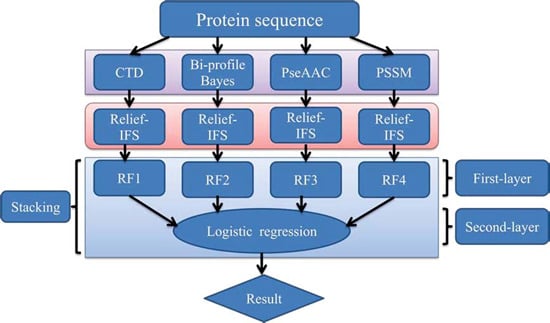
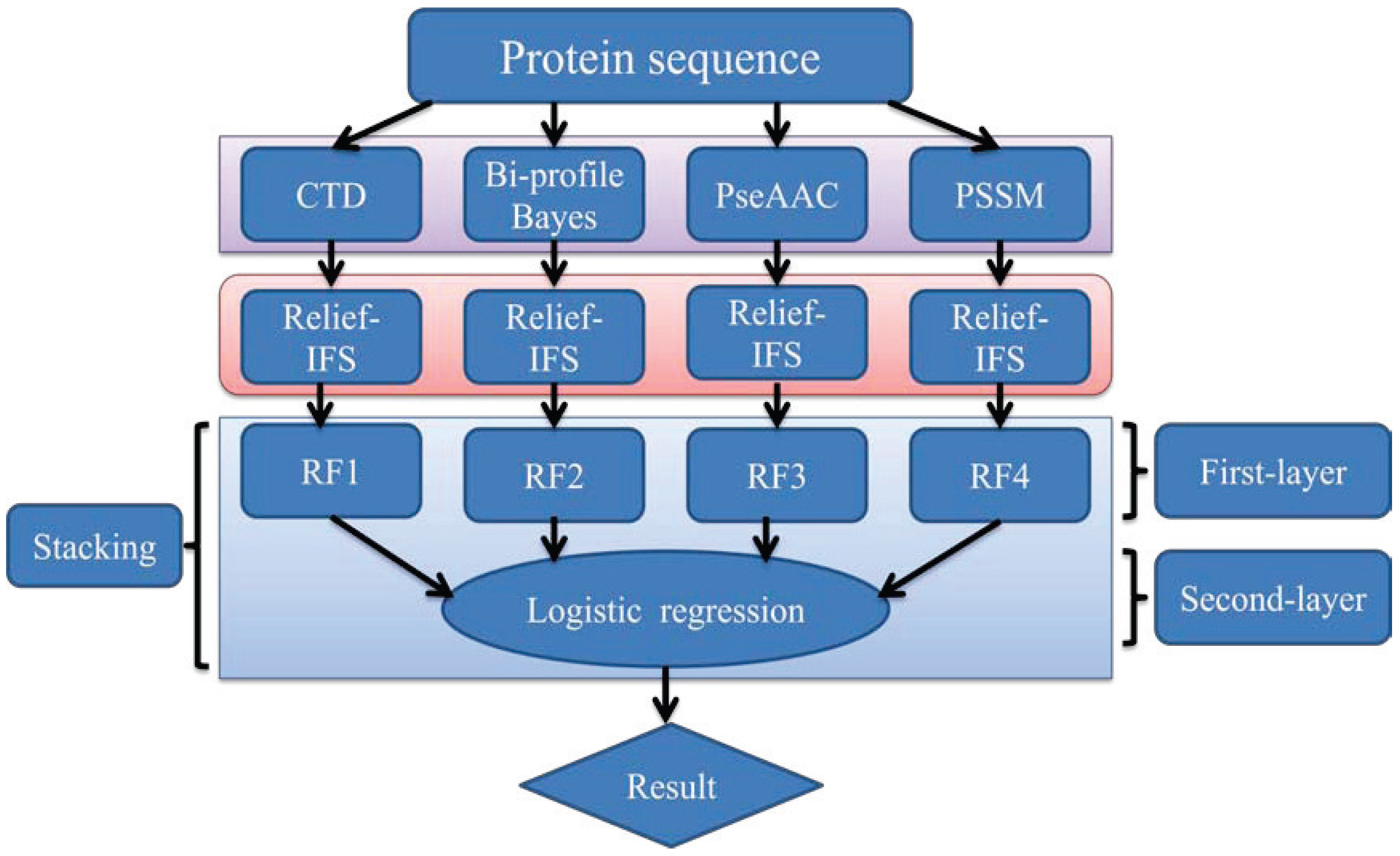
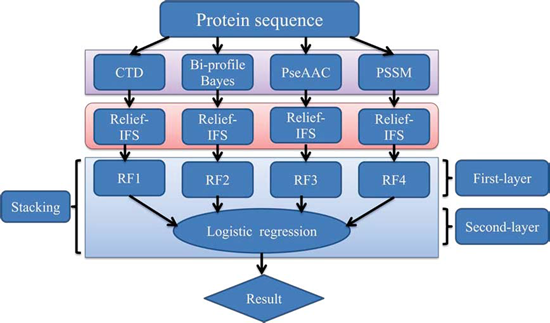
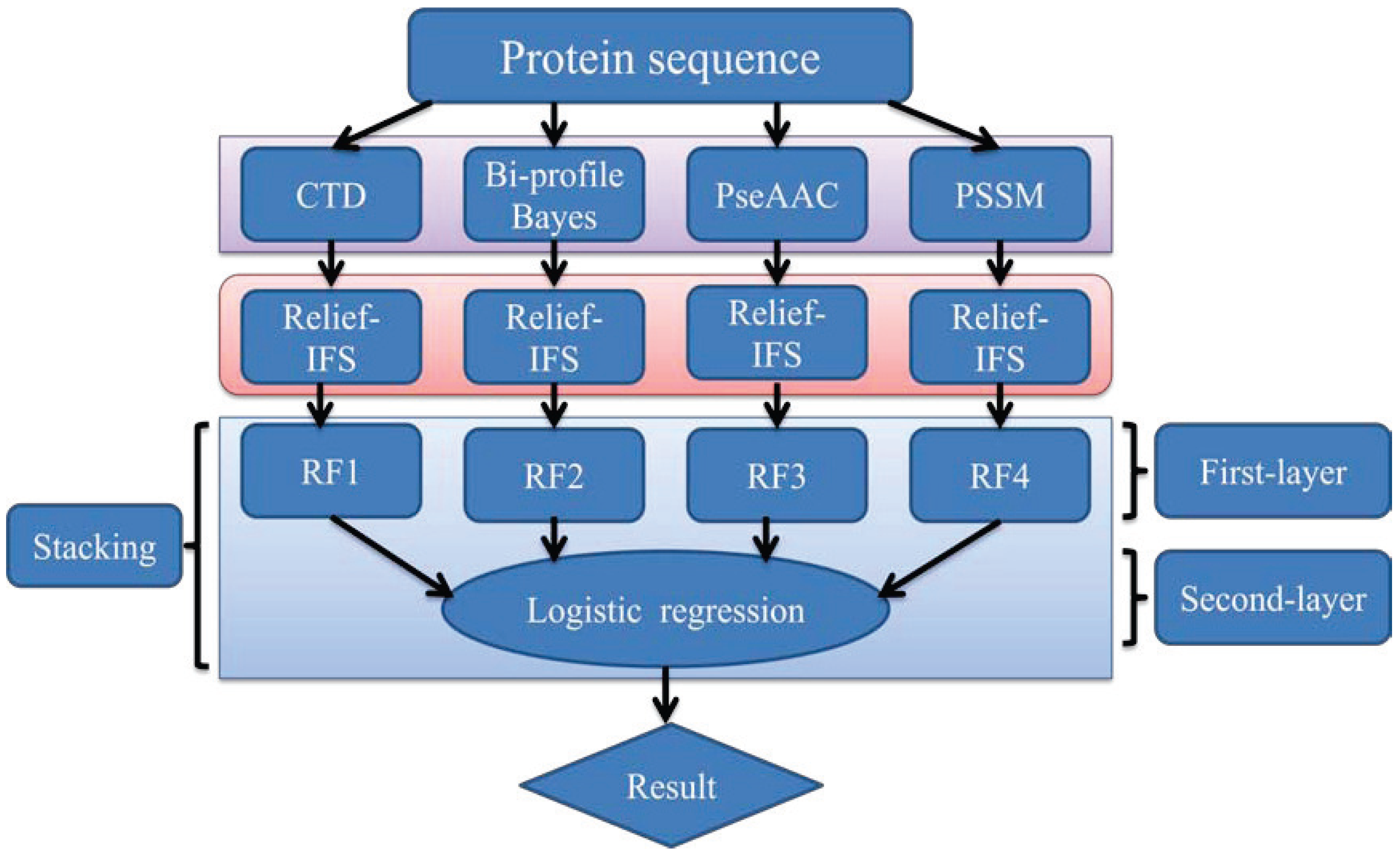
In order to address the above-mentioned limitations and to improve the performance for bacteriophage virion protein prediction, this study puts forward a stacking-based ensemble method with hybrid features incorporating CTD (composition, transition and distribution), bi-profile Bayes, PseAAC (pseudo-amino acid composition) and PSSM (position-specific scoring matrix). The proposed method is implemented in the following steps. (1) The benchmark dataset is obtained from Universal Protein Resource (UniProt) and divided into a training dataset and an independent testing dataset; (2) The protein sequences are converted into four different numerical feature vectors based on CTD, bi-profile Bayes, PseAAC and PSSM, respectively; (3) The relief algorithm combined with incremental feature selection (IFS) method is adopted to obtain optimal feature subsets for individual feature spaces; (4) The ensemble method is then developed by stacking the predictions of individual classifiers trained by the above-mentioned distinct feature spaces. A summary of the computational framework of our method is illustrated in
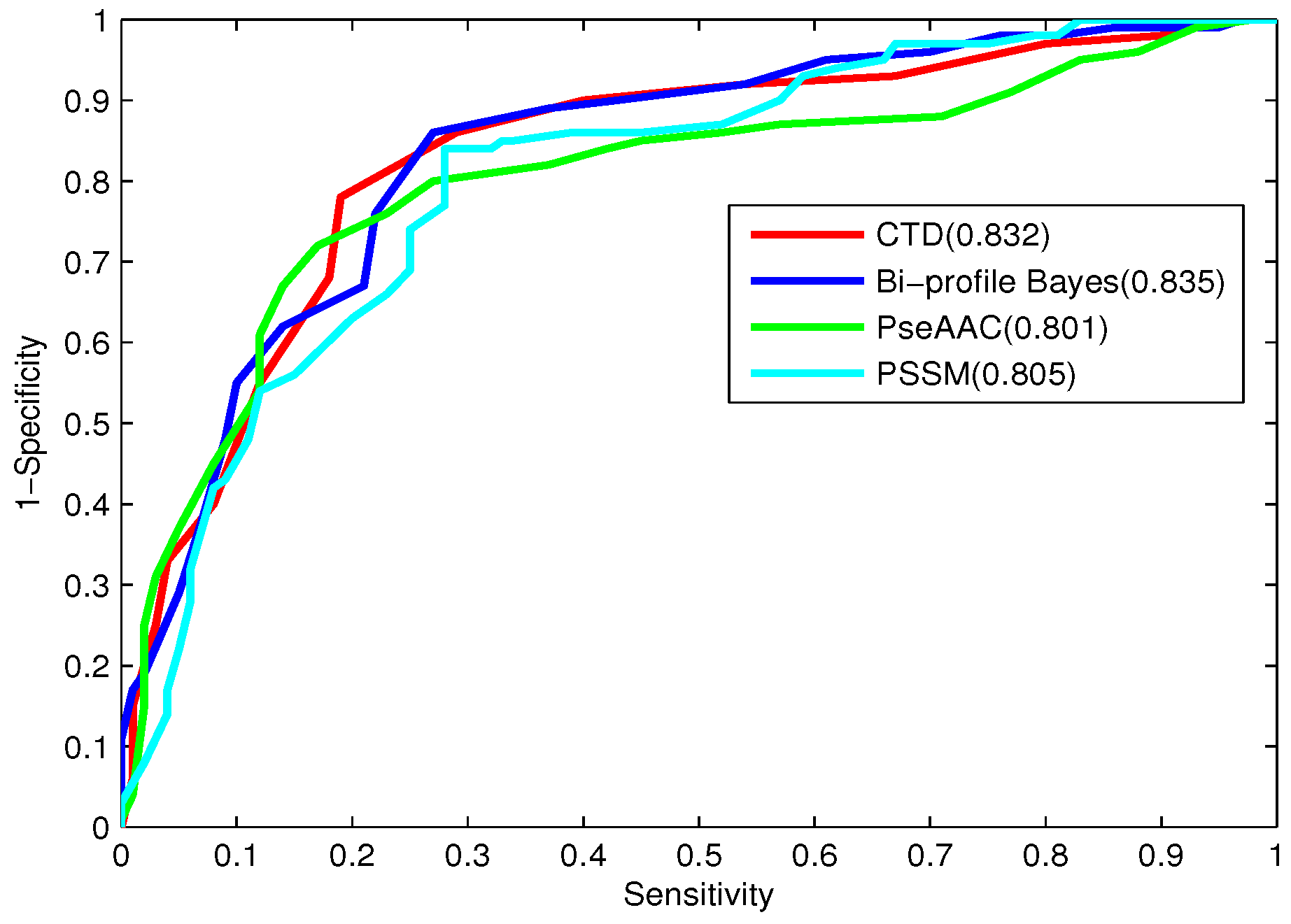
Figure 1. To evaluate the performance of our ensemble predictor objectively, the predictive capability of the present model is tested on the independent testing dataset and compared with that of [
10,
11].
Figure 1.
The computational framework of the proposed method. CTD: composition, transition and distribution; PseAAC: pseudo-amino acid composition; PSSM: position-specific scoring matrix; IFS: incremental feature selection; RF: random forest.
Figure 1.
The computational framework of the proposed method. CTD: composition, transition and distribution; PseAAC: pseudo-amino acid composition; PSSM: position-specific scoring matrix; IFS: incremental feature selection; RF: random forest.
3. Materials and Methods
3.1. Data Collection
Bacteriophage virion protein sequences applied in this study are collected from the UniProt database [
20] by searching “organism: phage AND subcellular location: virion AND reviewed: yes”. Due to the number of bacteriophage non-virion protein sequences being extremely large, protein sequences that were created after 2000 in the database and that match the search criterion “organism: phage NOT subcellular location: virion AND reviewed: yes” are selected as the bacteriophage non-virion proteins. Following the above procedures, 253 bacteriophage virion protein sequences and 248 bacteriophage non-virion protein sequences are obtained.
In order to obtain a reliable and high quality dataset, the following criteria are further performed. (1) The sequences that are fragments of other proteins are excluded because their information is redundant and lack integrity; (2) The sequences containing nonstandard letters, such as “B”, “X” or “Z”, are excluded, because their meanings are ambiguous.
It is well known that the benchmark dataset containing highly similar sequences can overestimate the performance and reduce the robustness of a predictor. Thus, a cutoff threshold is introduced to avoid homology bias and redundancy. The smaller the cutoff threshold is, the more stringent the benchmark dataset will be [
21]. In order to avoid the smaller cutoff threshold value causing the benchmark dataset to be too small to have statistical significance, the sequence identity cutoff threshold is set as 80% to dislodge the redundant sequences using the CD-HIT program [
22]. After the above screening procedures, the final benchmark dataset consists of 168 bacteriophage virion protein sequences and 192 bacteriophage non-virion protein sequences.
In order to validate the proposed ensemble model objectively, 100 bacteriophage virion proteins and 100 bacteriophage non-virion proteins are respectively selected from the final benchmark dataset as the training dataset for constructing the proposed model, while the rest of the 68 bacteriophage virion proteins and 92 bacteriophage non-virion proteins are the independent testing dataset. The training dataset and independent testing dataset are available in
Supplementary Table S1.
3.2. Feature Extraction
To develop a powerful predictor for pattern recognition problems in bioinformatics, one of the key steps is to represent the protein sequences with appropriate descriptors that can truly reflect their intrinsic correlation with the target sequences to be predicted [
21]. A single-feature descriptor cannot preserve enough discriminative information for protein attribute predictions, which may lead to unsatisfactory predictive performance. In such circumstances, the idea of hybrid features is adopted as a common way for enhancing the discrimination power of a predictor. In this study, hybrid features extracted from CTD, bi-profile Bayes, PseAAC and PSSM are utilized for encoding protein sequences.
3.2.1. Composition, Transition and Distribution
Amino acid composition is one of the most basic characteristics of proteins. To avoid the notorious dimensional disaster, the 20 native amino acids can be clustered into a smaller number of representative residues, called the reduced amino acid alphabet (RAAA) [
23], as shown in
Table 7.
Compared to the traditional amino acid composition, RAAA not only simplifies the complexity of the protein system, but also improves the ability to find structurally-conserved regions and the structural similarity of entire proteins [
24].
The CTD method, introduced by Dubchak
et al. [
25], can effectively extract the global information of protein sequences. Recently, the CTD method has been widely applied to protein function predictions and structure studies [
26,
27]. Based on the cluster profiles (CP) in
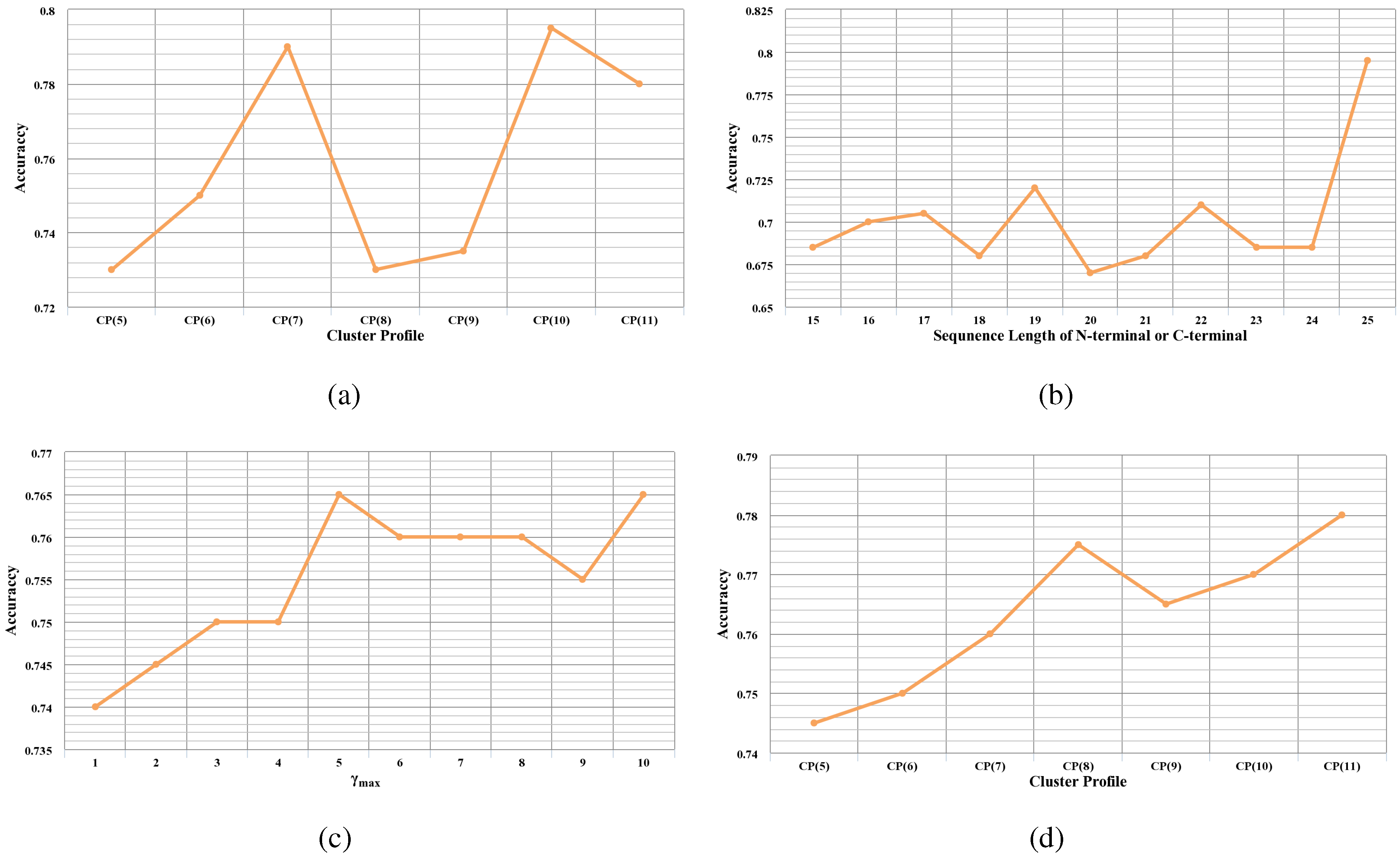
Table 7, three global descriptors, composition, transition and distribution, are employed in this study to describe the properties of protein sequences.
Table 7.
Scheme for the reduced amino acid alphabets.
Table 7.
Scheme for the reduced amino acid alphabets.
| Reference | Cluster Profiles | Reduced Amino Acid Alphabets |
|---|
| [23] | CP(5) | G-IVFYW-ALMEQRK-P-NDHSTC |
| [28] | CP(6) | RDENQKH-LIVAMF-STYW-P-G-C |
| [29] | CP(7) | AILVGP-DE-HKR-FWY-NQ-ST-CM |
| [23] | CP(8) | G-IV-FYW-ALM-EQRK-P-ND-HSTC |
| [23] | CP(9) | G-IV-FYW-ALM-EQRK-P-ND-HS-TC |
| [30] | CP(10) | FWY-DE-H-K-R-C-M-QN-ST-AGILVP |
| [23] | CP(11) | G-IV-FYW-A-LM-EQRK-P-ND-HS-T-C |
For a given
, the composition is defined as:
where
denotes the number of each group in
and
L is the length of the whole sequence.
For a given
, the transition descriptor is defined as:
where
and
.
is one of the amino acid groups in
.
is the number of the dipeptide encoded as “
”.
Distribution measures the respective locations of the first, 25%, 50%, 75% and 100% of each amino acid group in
, which can be calculated by:
where
is the chain length within which the first of the amino acids of group
i is located.
measure the chain lengths within which the 25%, 50%, 75% and 100% of the amino acids of group
i are located, respectively.
Based on the 7 different cluster profiles in
Table 7, the prediction results obtained by the proposed CTD features are investigated and compared.
3.2.2. Bi-Profile Bayes
Statistical differences usually exist in the position-specific amino acid composition of protein sequences between positive and negative datasets. In this study, the bi-profile Bayes method is utilized to extract the statistical information about the position-specific amino acid composition between positive and negative datasets.
A previous study has shown that signal peptides play a vital role in the structures and functions of proteins [
31]. Generally, important targeting signal peptides often occur at the N-terminal and retention signal peptides for protein sorting at the C-terminal of proteins [
32]. In order to extract significant signal peptide information of a protein, the sequence segments at N-terminus and C-terminus are considered.
For the two-class classification problem, suppose
and
correspond to the negative class and the positive class, respectively; the amino acid composition at each position of the N-terminus and C-terminus with the length of
m is calculated by the following formula.
where
represents the amino acid information extracted from the negative dataset;
represents the amino acid information extracted from the positive dataset;
are the posterior probabilities of 20 native amino acids at the
i-th position of the N-terminus, represented by
; and
are the posterior probabilities of 20 native amino acids at the
i-th position of the C-terminus, formulated as
.
Based on the bi-profile Bayes method, a test protein sample can be formulated as:
where
denote the posterior probabilities of each amino acid at each position of the N-terminus of the test protein sample in the positive dataset, which are extracted from
;
and denote the posterior probabilities of each amino acid at each position of the C-terminus of the test protein sample in the positive dataset, which are extracted from
.
Similarly, , extracted from , represent the posterior probabilities of each amino acid at each position of the N-terminus in the negative dataset; and , extracted from , represent the posterior probabilities of each amino acid at each position of the C-terminus in the negative dataset.
The length of the N- or C-terminus can affect the predictive performance of a classifier. Therefore, different lengths of the N- or C-terminus are investigated variably from 15 to 25 residues to find out the discriminative one.
3.2.3. Pseudo-Amino Acid Composition
Physicochemical properties of amino acids have a crucial influence on protein structures and functions [
33]. Therefore, extracting features from protein sequences based on appropriate physicochemical properties may prospectively improve the performance of a predictor.
Previous studies have shown that certain physicochemical properties are related to the functions and structures of bacteriophage virion proteins. Coia
et al. [
34] have revealed that amino acids with small side-chain masses are prone to occur in bacteriophage virion proteins. Marvin
et al. [
35] have demonstrated that certain physicochemical properties, such as amphipathicity and charge, play vital roles in the functions of bacteriophage virion proteins. Kuzmicheva
et al. [
36] have reported that bacteriophage virion proteins should preserve small amino acids sharing diminutive positively-charged surfaces and higher occupied molecular orbitals.
Sequence order information represents important properties of protein sequences for prediction purposes, which may affect the predictive performance of a predictor [
37]. The PseAAC approach, proposed by Chou [
38], takes into consideration physicochemical properties and sequence order information of protein sequences.
In order to extract discriminative physicochemical features of protein sequences, 10 widely-used physicochemical properties of amino acids are adopted from the AAIndex (amino acid index) database [
39] to calculate the correlations between residues along a protein sequence using the PseAAC method. They are the number of atoms, the number of potential hydrogen bonds, the number of electrostatic charges, the isoelectric point, mass, flexibility, hydrophilicity, hydrophobicity, propensity and the electron-ion interaction potential.
In terms of the 10 physicochemical properties mentioned above, based on PseAAC, a given protein sequence with
L amino acids can be formulated as:
where
T denotes a transpose operator;
is the γ-th tier correlation factor based on the
j-th physicochemical property; and
is the
j-th physicochemical property value of the
i-th residue along the protein sequence. γ is closely related to sequence order information and plays an important role in the performance of a predictor. Therefore, we evaluate the discrimination power of the features based on different γ varying from 1 to 10.
3.2.4. Position-Specific Scoring Matrix
Protein sequences have stemmed from a very finite number of ancestral species protein sequences, involving changes, insertions and deletions of single or several residues [
40]. As a result, the original and resultant protein sequences may have few sequence similarities, but they may still share some structure similarities and the same functions [
41]. Therefore, evolutionary conservations can determine important biological functions and contribute to biological analysis [
42].
The position-specific scoring matrix (PSSM) is adopted in this study to extract evolution information, which has been widely employed in protein attribute prediction problems [
43,
44]. To obtain the PSSM profiles, the Position-Specific Iterative BLAST (PSI-BLAST) [
45], a powerful sequence searching program, is applied to search the non-redundant (NR) database for multiple sequence alignment through 3 iterations using the E-value cutoff of 0.0001. For a given protein sequence with
L amino acids, the corresponding PSSM has
elements and is defined as:
where the values in the
i-th row are the probabilities of the
i-th residue in a given protein sequence mutating to 20 native amino acids. Then, the PSSM is normalized with the following sigmoid function to scale each element to a range of 0 to 1:
where
x is the original PSSM value.
To obtain a uniform number of features, we sum up all of the columns and rows in the PSSM corresponding to the amino acids in the same group of cluster profiles as indicated in
Section 3.2.1. For the cluster profile with
N groups, the PSSM profile is transformed to a
matrix following the above procedures. The elements of the
matrix are employed to encode each protein sequence. The discrimination power of the proposed PSSM conservation scores based on 7 cluster profiles is evaluated, respectively. Then, the cluster profile that achieves the best performance is chosen to extract features from PSSM.
3.3. Feature Selection
Through the feature extraction methods, protein sequences are encoded into various numerical feature vectors. However, not all of the extracted features contribute to the prediction. Much information redundancy or noise may exist in the extracted features, which will deteriorate the performance of a predictor and result in dimensional disaster [
46].
Feature selection is essential to overcome the above troubles. It is a critical step in the classification problems to select the discriminative features and decrease model complexity, which can not only lower computation complexity, but also build a high-quality and robust prediction model [
47]. In this study, the Relief algorithm combined with IFS is employed to acquire the optimal feature subsets of individual feature spaces.
Relief: The Relief algorithm, originally proposed by Kira [
48], is used to depict the relevance between the features and class labels. Based on the ability of the feature to distinguish the near samples, the Relief algorithm is a feature-weighting algorithm that can be used to estimate the quality of each feature. During the process, the Relief algorithm endows each feature with a weighting iteratively as formulated by:
where
and
are separately the current and next weighting values.
p stands for a given feature, and
denotes the
i-th sample sequence.
, referred to as a near hit, represents the nearest neighbor samples from the same class label against
.
, referred to as a near miss, represents the nearest neighbor samples from the different class labels against
.
Y and
S are the sample sets with the same and different class labels against
, respectively.
m is the number of random samples, and the function of
is used for calculating the distance between the random samples.
The ranked feature list can be obtained based on
, represented as:
where
represents the feature with the highest value of
,
with the second highest value of
,⋯, and
with the lowest value of
.
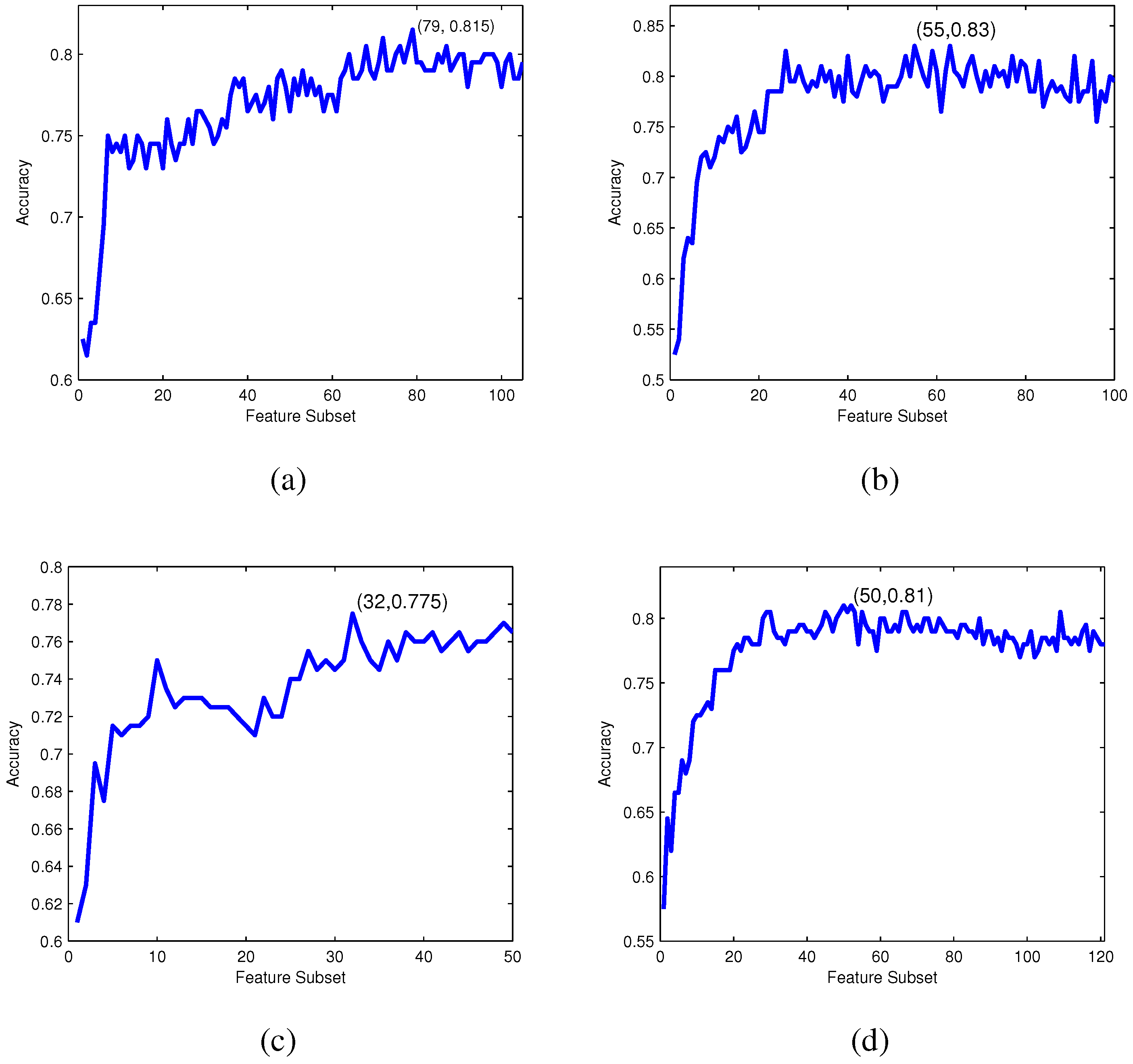
Incremental feature selection: Based on the ranked feature list according to their relevance to the classes evaluated by Relief, IFS, one of the well-known searching strategies of feature selection, is employed to determine the optimal feature subset.
The IFS procedure starts with an empty subset and adds features in the ranked feature list one by one from higher to lower rank into the feature subset [
49]. A new subset is generated when a new feature in the ranked list is added, and the
i-th feature subset can be formulated as:
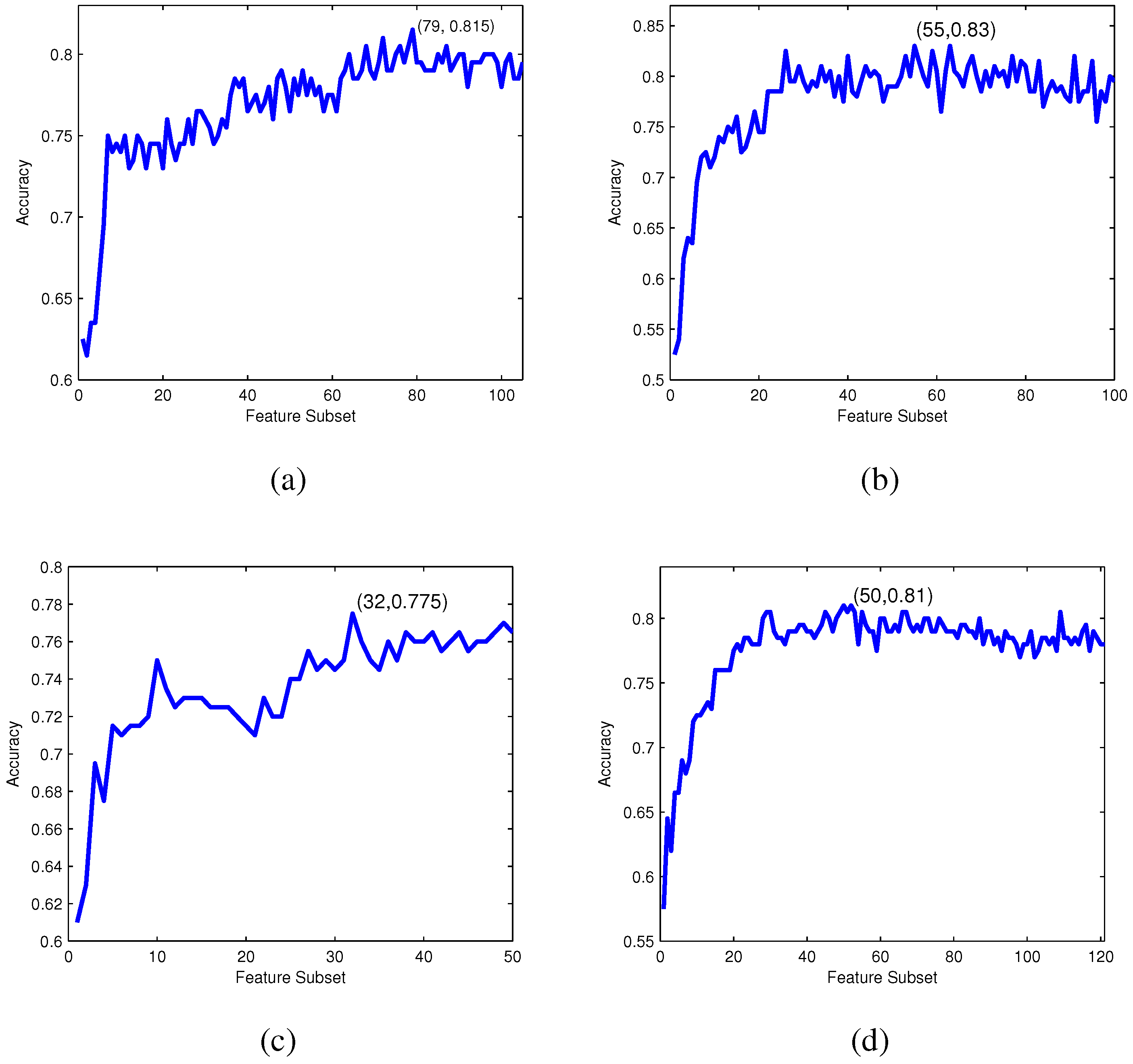
For each feature subset , an RF-based predictor is constructed and evaluated by 10-fold cross-validation. The IFS curve can be drawn with accuracy values as the y-axis and index i of as the x-axis. The optimal feature subset is obtained when the IFS curve reaches its peak.
3.4. Machine Learning Method
Random Forest: The RF method, developed by L. Breiman [
50], is a popular machine learning method that has been successfully employed in various biological prediction problems [
51,
52]. It is an ensemble classifier generating multiple decision trees, where each decision tree is constructed based on the training dataset and produces a classification label. To predict a test sample, its feature vector is put into each of the decision trees in the forest, and each tree gives a vote suggesting one class. Then, the RF method will choose the final classification label with the most votes over all of the decision trees as the output class [
53]. Refer to [
50] for a detailed description of the RF method. In this study, 4 random forest models are developed, and each one is trained by the individual feature space described in
Section 3.2.
Logistic regression: Logistic regression (LR), a probabilistic statistical classification model, is a machine learning framework suitable to establish the logistic relationship between the two class labels and the numerical feature vectors [
54]. It has been widely applied to various pattern recognition problems, such as the prediction of flexible regions in proteins [
55], the determination of protein folding kinetic types [
56] and the prediction of lysine acetylation site [
33].
Given a protein sequence and its feature vector, the likelihood can be calculated by the logistic regression model. The feature vector
combined with the class label is used as the input:
where the optimized regression coefficients
are obtained based on the training dataset.
In this study, the logistic regression approach integrates the outputs of the 4 RF models to provide the final prediction score
P, which is formulated as:
where the constant term
and the coefficient
of each RF output
are deduced through the regression process based on the training dataset.
For protein sequences in the test dataset, P has values between 0 and 1, which will be optimized to maximize the accuracy for bacteriophage virion protein prediction.
The WEKA (Waikato Environment for Knowledge Analysis) software package [
57] is used for the above-mentioned machine learning algorithms, where default parameters are employed for 4 RF models.
3.5. Ensemble Learning Method
Every single classifier has its own shortcomings, and it will not always perform well on all datasets [
14]. Ensemble learning emerges as a promising measure to overcome this problem. Unlike the single-prediction algorithm, ensemble learning consists of multiple basic individual classifiers and then aggregates the outputs of all independent classifiers to tackle the same classification task [
58]. Due to the procedure of consulting several experts for suggestions before giving the final results, the ensemble learning is supposed to significantly improve the performance of a prediction method [
59].
In this study, 4 different random forest models as the first layer are constructed. Then, two popular methods, majority voting and stacking, are employed to combine the 4 random forest models to give the final prediction results.
Majority voting: The majority voting strategy, the simplest way to combine the predicted class labels of multiple classifiers [
60], is used to aggregate the independent classifiers. The target protein sequence is assigned to the predicted class label whose score function has the maximum value, that is the corresponding class label achieves the highest number of votes [
60].
For a query protein, denote the output of the
i-th RF model by:
where
j denotes the predicted class label.
separately represent the sample predicted as the positive class
and the sample predicted as the negative class
.
The output of the 4 RF models is given as a 4-dimensional vector
. The query protein is assigned to the positive class when
according to the following formula:
Otherwise, it is predicted as the negative class.
Stacking: Stacking is an approach for combining multiple classifiers and achieves the highest generalization accuracy [
61]. Compared to the majority voting method, stacking will learn twice in two layers. In the first layer, a number of individual classifiers are generated using the original feature vectors as the input. In the second layer, a specific classifier is employed to combine the predicted probabilities for every class label from the classifiers in the first layer [
62].
In this study, 4 random forest models are generated as the first layer, each of which is trained by 4 different feature spaces, respectively. Then, the logistic regression approach integrates the 4 independent prediction scores from the first layer to give the final results.
Majority voting and stacking both have their own particular merits. The former is the simplest combination strategy, without cross-validation and the training of the second-layer classifier, which is computationally inexpensive. The latter provides the function of being highly extensible to more classifier algorithms at both the first and second layers, regardless of their internal structures [
62].
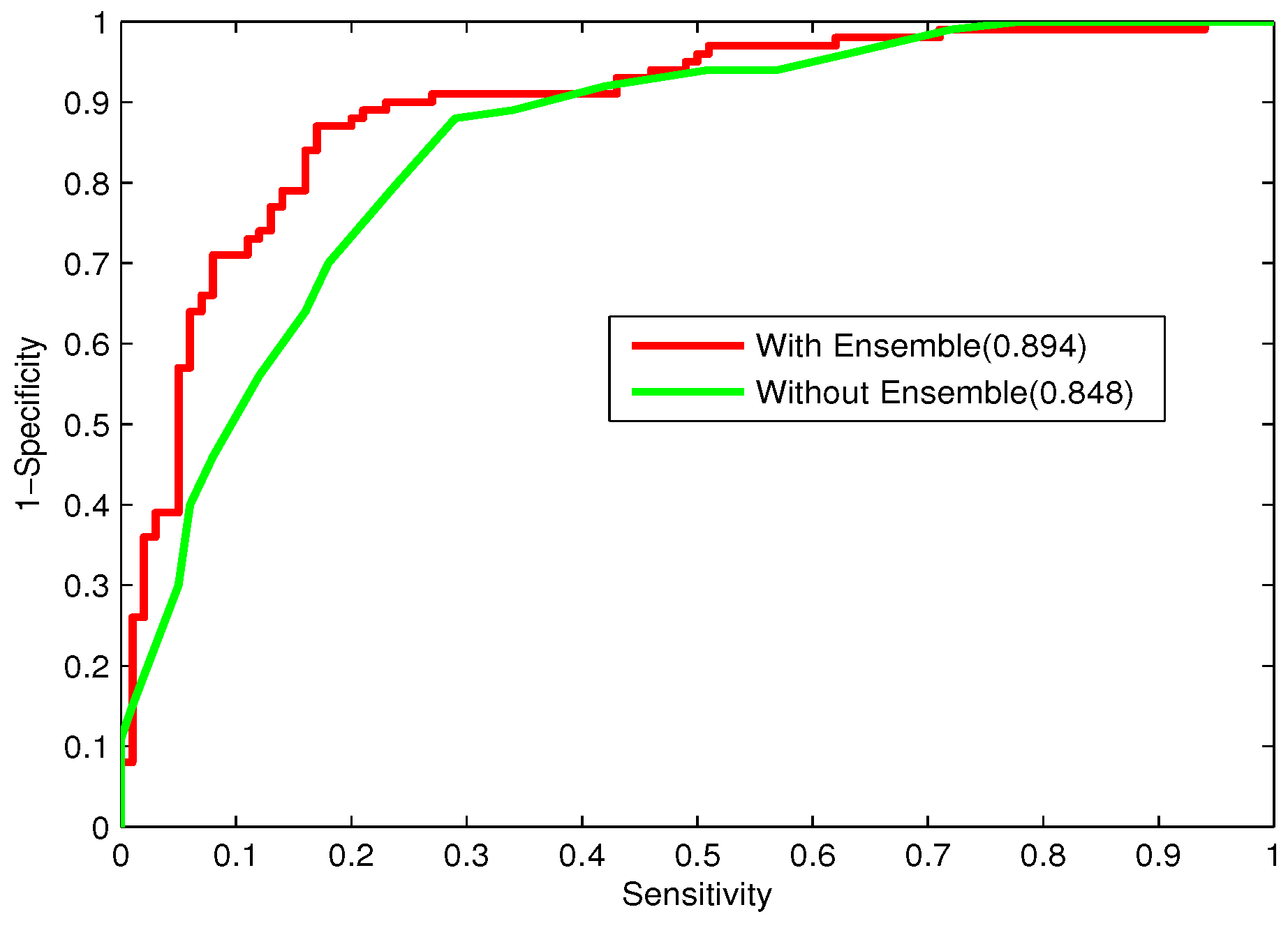
In the study, the performance of majority voting and stacking will be compared.
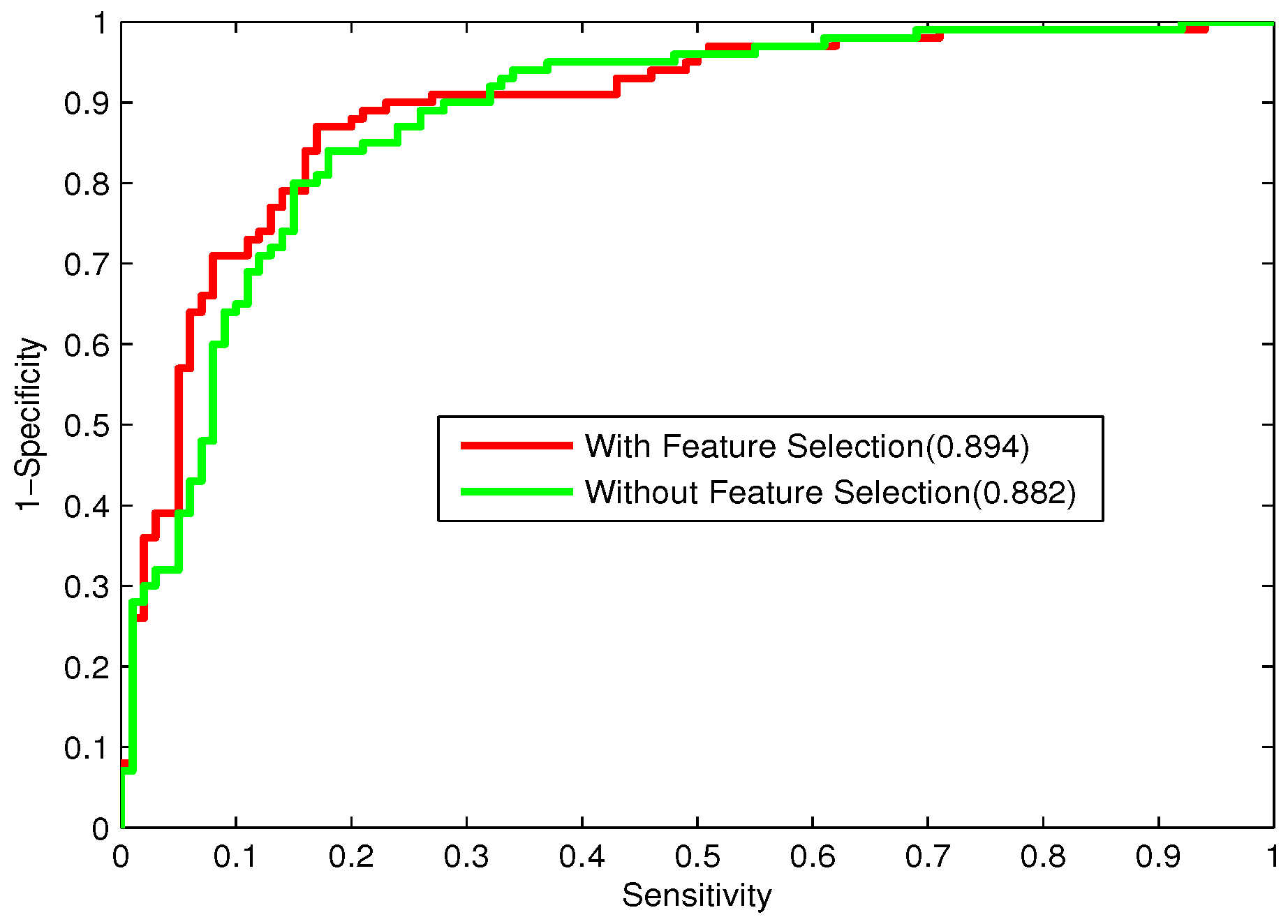
3.6. Performance Measures
The independent dataset test, the sub-sampling test (e.g., 5-fold or 10-fold cross-validation) and the jackknife test are widely applied to examine the performance of a predictor [
63]. The jackknife test is deemed as the most objective method in statistical prediction, which can always obtain a unique outcome for a given benchmark dataset [
59]. To reduce the computational complexity, 10-fold cross-validation is adopted in this study. During 10-fold cross-validation, the training dataset is randomly divided into 10 equally-sized folds. Each fold is used as the testing set, and the remaining 9 folds as the training set. This process is repeated 10 times with different combinations of training and testing datasets. Then, the ultimate result is obtained by averaging the 10 prediction results.
To illuminate the performance of the predictor more intuitively, 4 common measurements: sensitivity (
, specificity (
), accuracy (
) and Matthew’s correlation coefficient
are adopted, which are formulated as:
where
,
,
and
stand for the numbers of true positive, true negative, false positive and false negative, respectively.
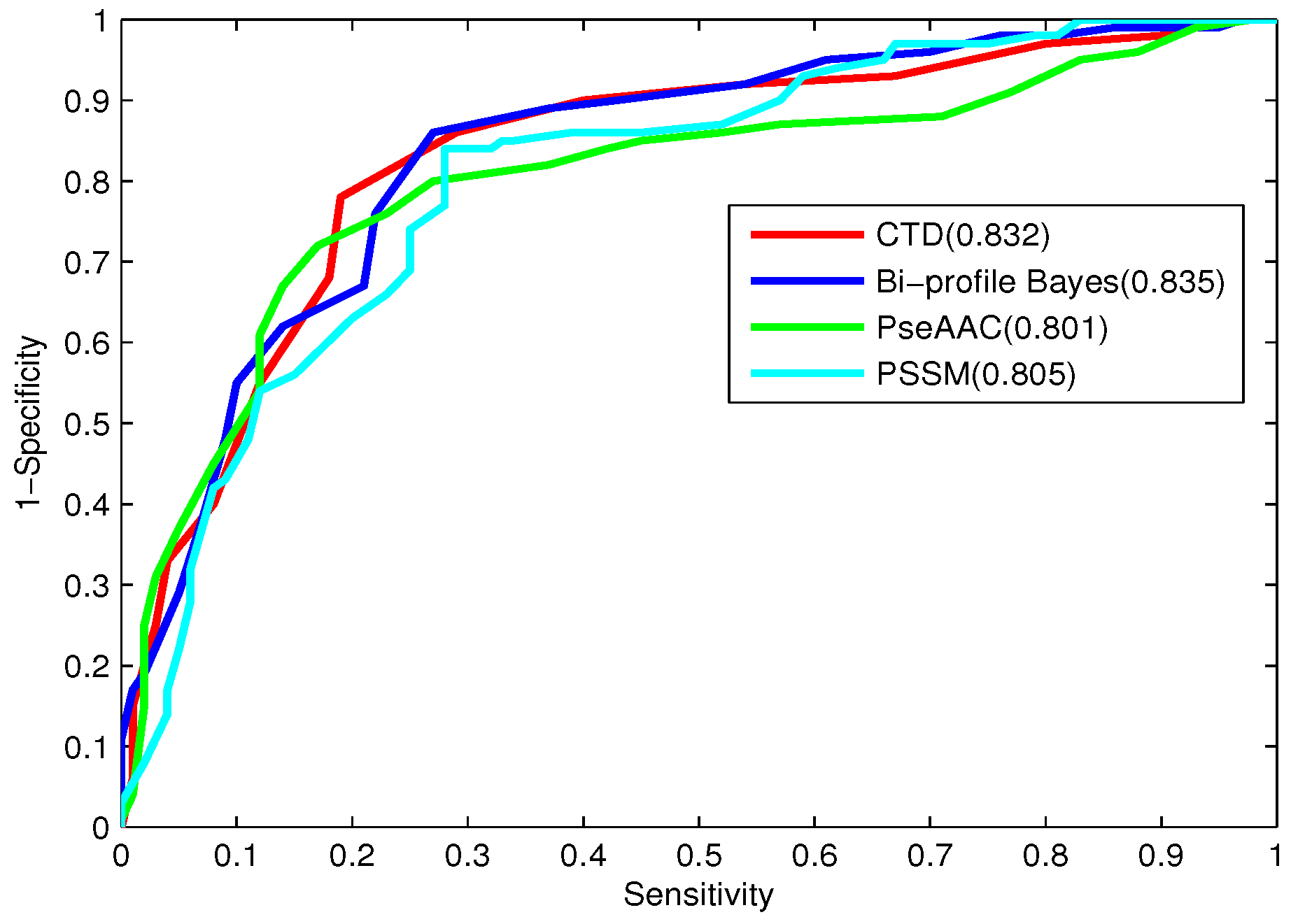
To further evaluate the performance of the classifier, the receiver operating characteristic (ROC) curve, one of the most valid measures in pattern recognition, is also employed [
64]. The ROC curve is plotted with the
as the
y-axis and
as the
x-axis by varying the thresholds. The area under the ROC curve (
) is the index for model evaluation obtained from the ROC curve. The higher
value corresponds to the better performance of the classifier.
4. Conclusions
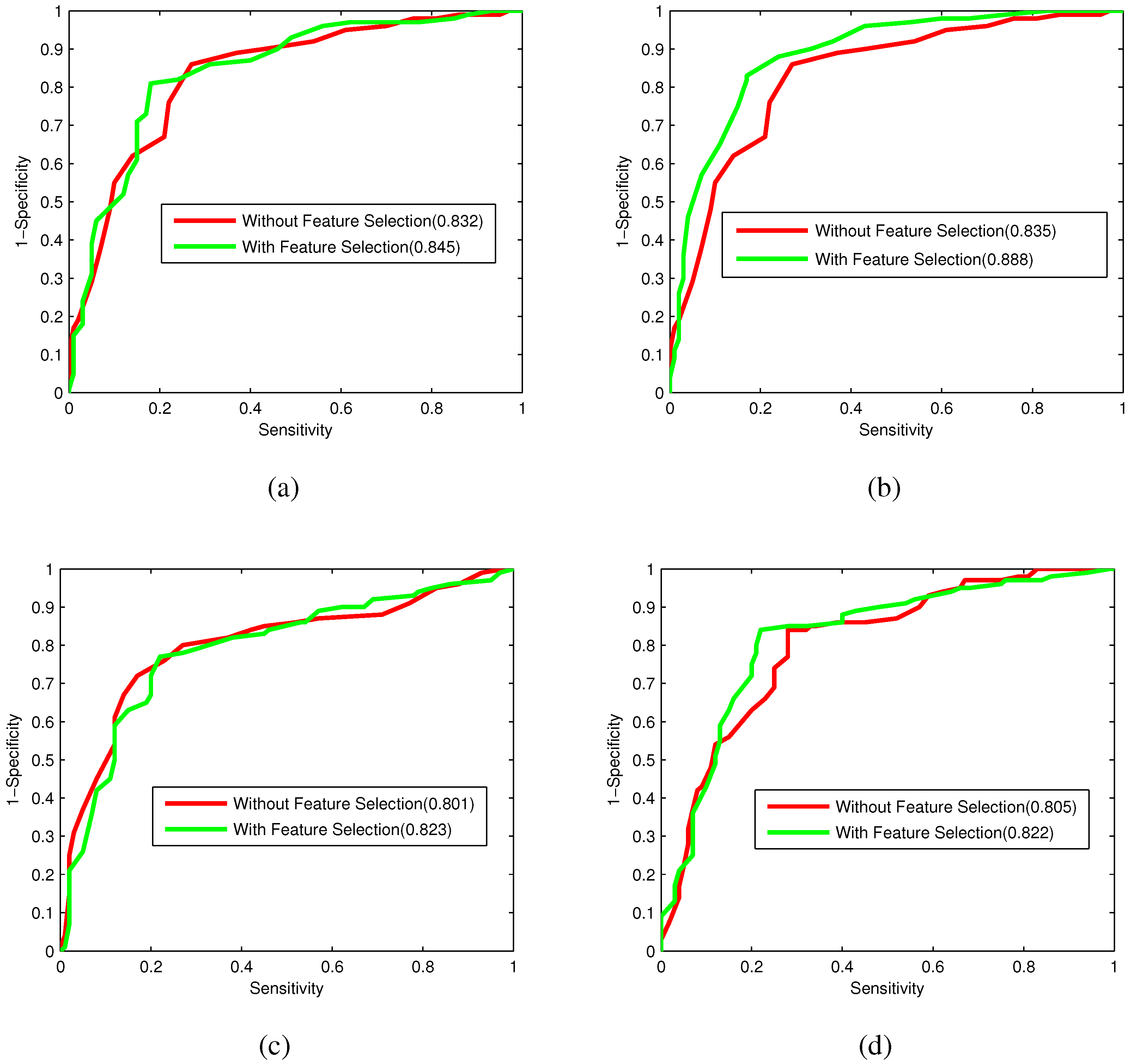
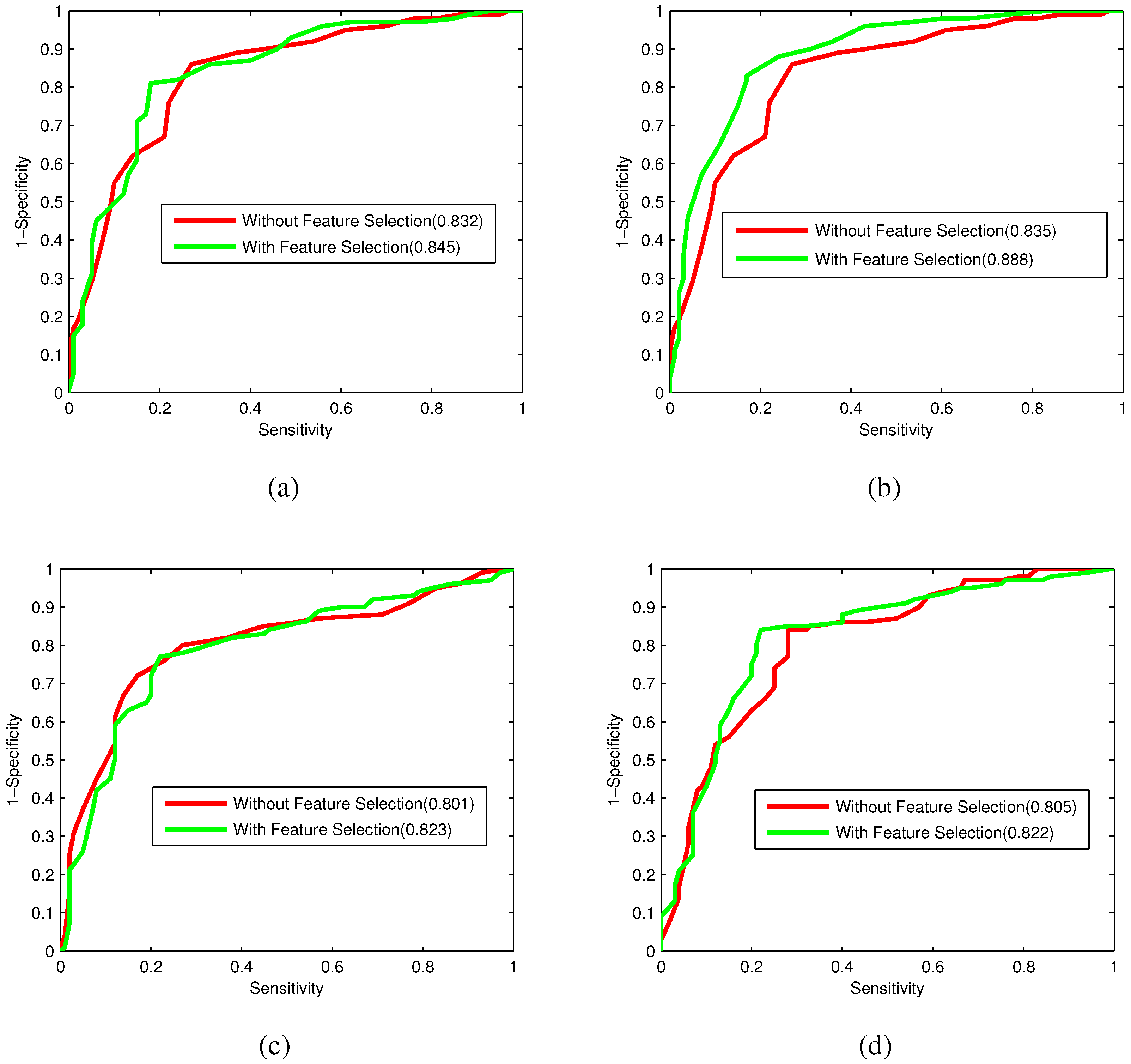
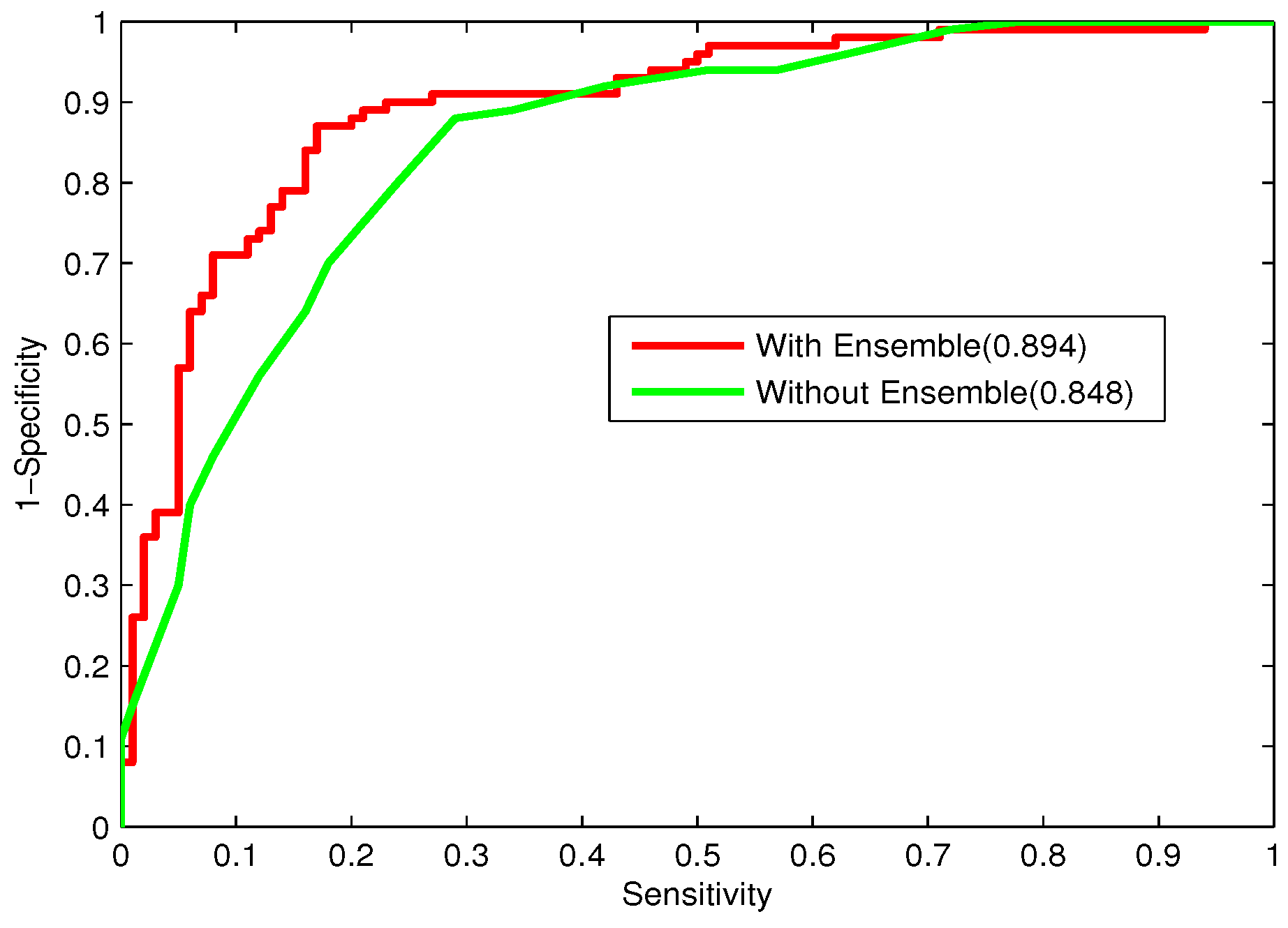
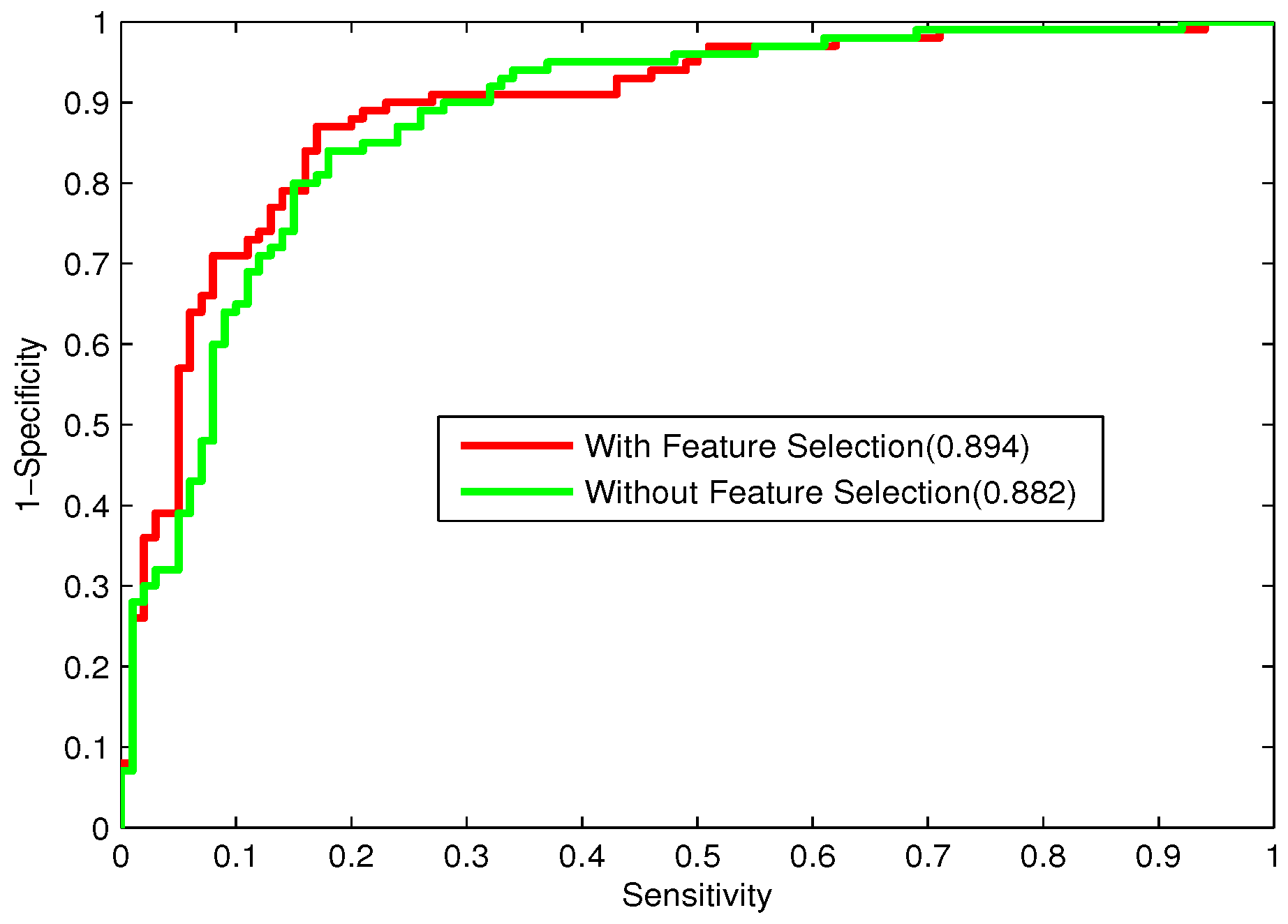
Bacteriophage virion proteins have the functions of specificity determination for the host bacteria, antigenicity of bacteriophages and clinical phage therapy. Due to the major functions of bacteriophage virion proteins in deciding applications of bacteriophages, identifying bacteriophage virion proteins is crucial for understanding the relationship between bacteriophage and host bacteria and the influence of bacteriophages on the development of new pathogens and antibacterial drugs. It is urgent to develop computational methods for rapidly and effectively identifying bacteriophages virion proteins. An ensemble method for bacteriophage virion protein prediction has been presented in this study with hybrid features extracted from CTD, bi-profile Bayes, PseAAC and PSSM. To solve the dimensional disaster and to improve the performance, Relief combined with the IFS method is adopted to obtain the optimal feature subsets. The s of individual RF modules based on four individual optimal features are 0.845, 0.888, 0.823 and 0.822, respectively, indicating the discrimination power of the selected features. The four RF models are combined by a stacking strategy with an accuracy of 0.85. To evaluate the prediction performance objectively, the proposed ensemble method is tested on the independent testing dataset and compared to previous studies. The prediction results of our proposed method are superior to those of the earlier studies. Thus, the proposed method may provide a useful tool for understanding the relationship between bacteriophage and host bacteria. Furthermore, it may provide crucial clues to develop new pathogens and antibacterial drugs in the future.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}