
BMP9-Induced Survival Effect in Liver Tumor Cells Requires p38MAPK Activation

Abstract
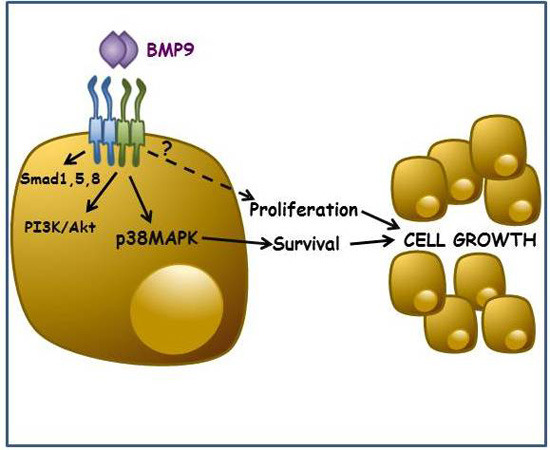
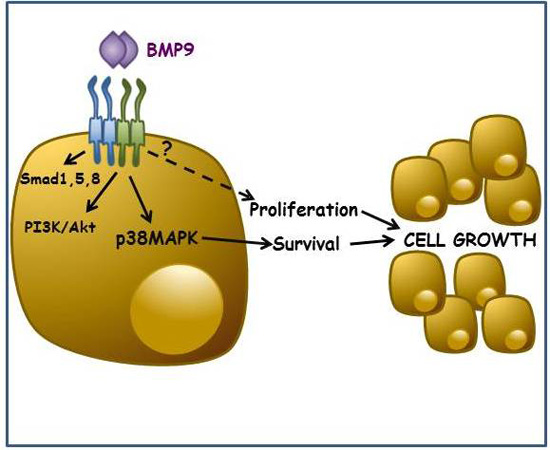
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
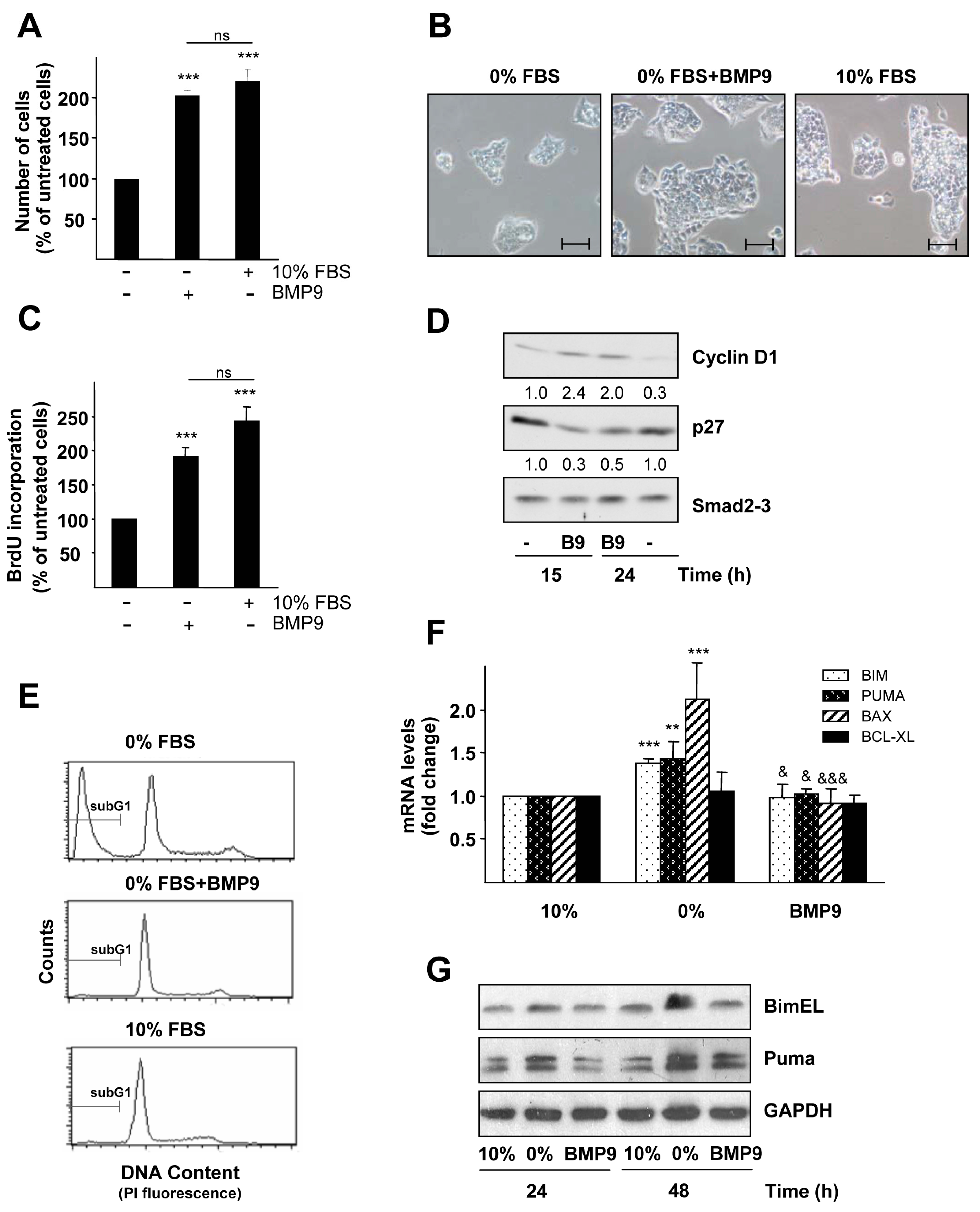
2.1. BMP9 Promotes HepG2 Cell Growth through Cell Cycle Regulation and Survival
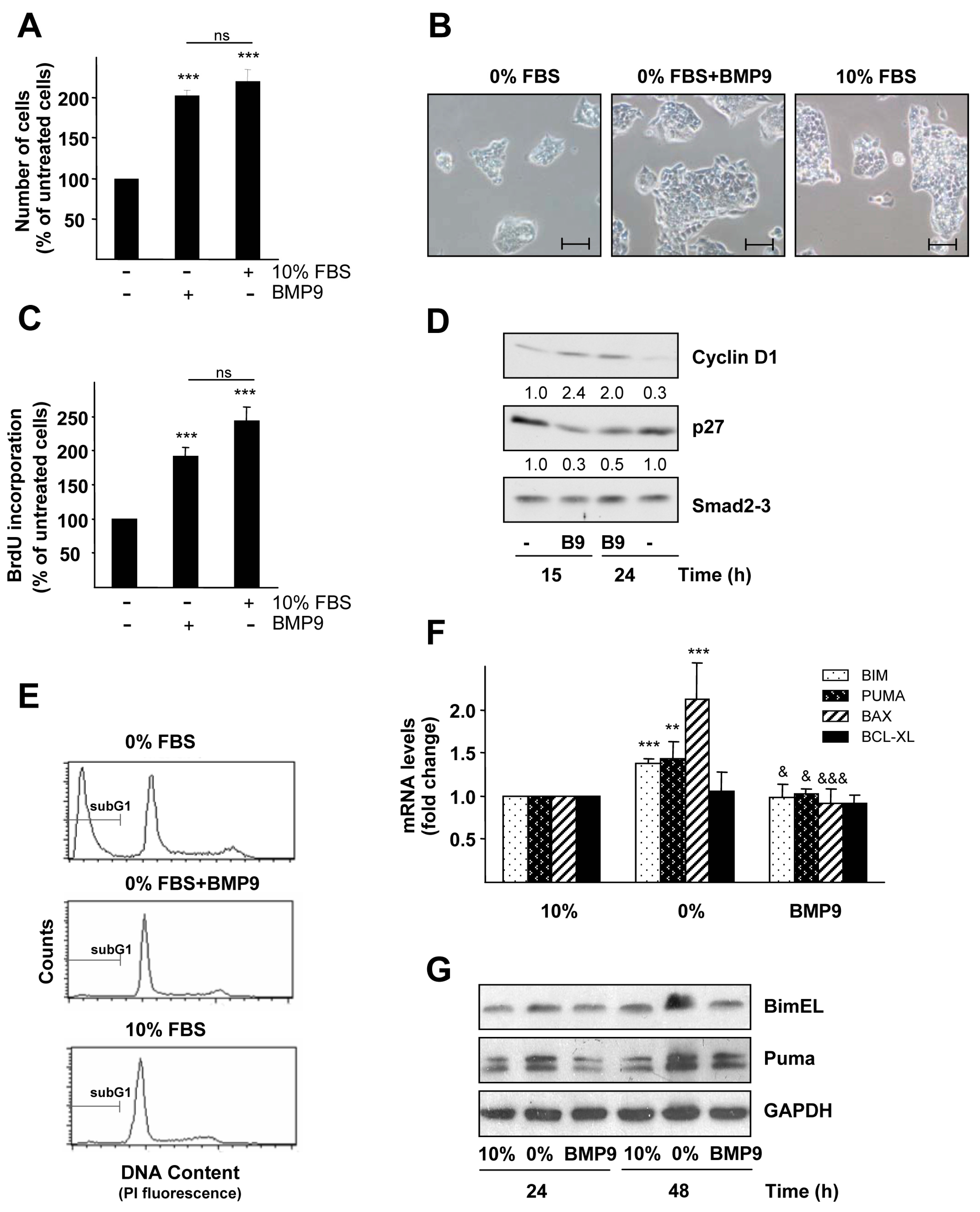
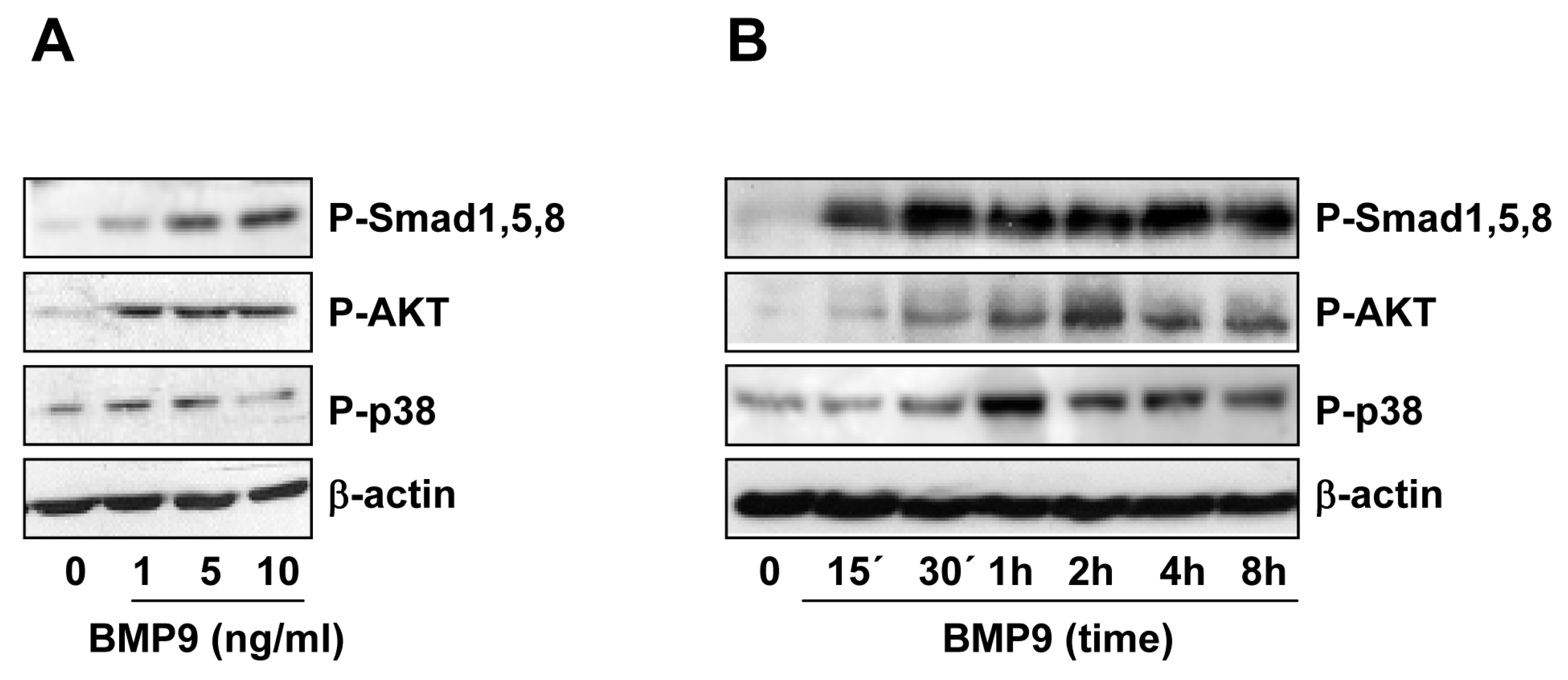
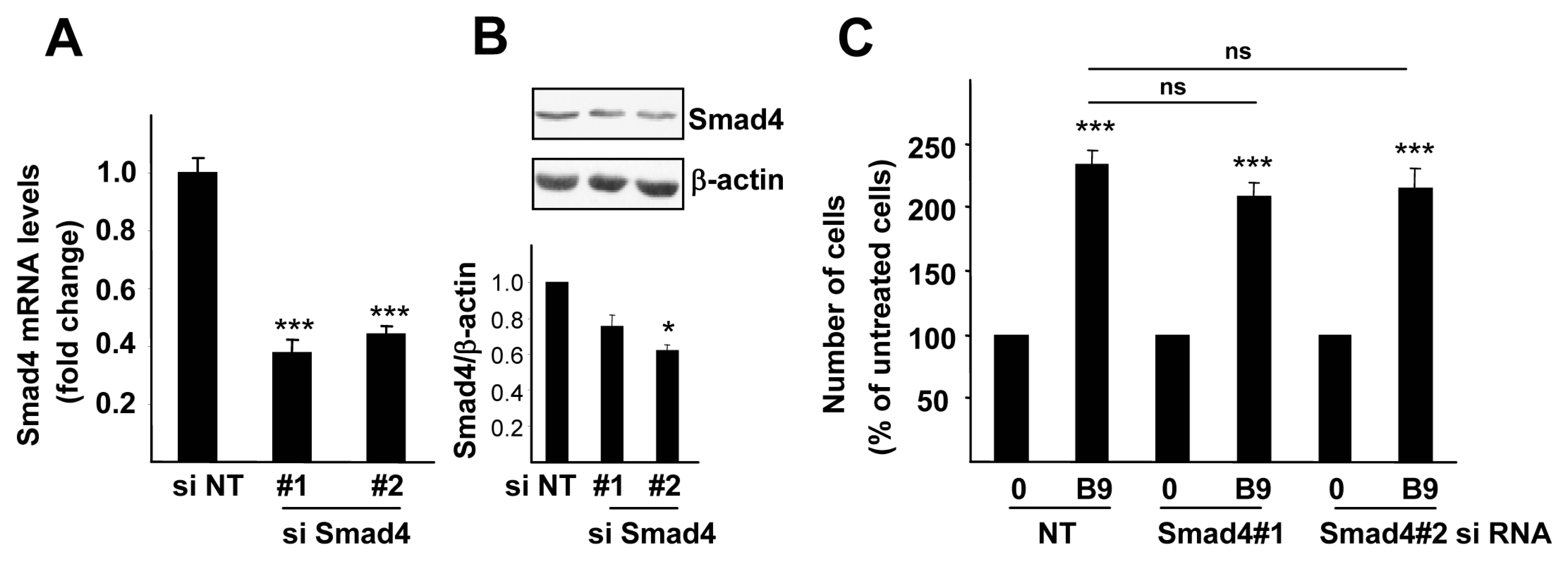
2.2. BMP9 Activates Non-Canonical Pathways in Hepg2 Cells

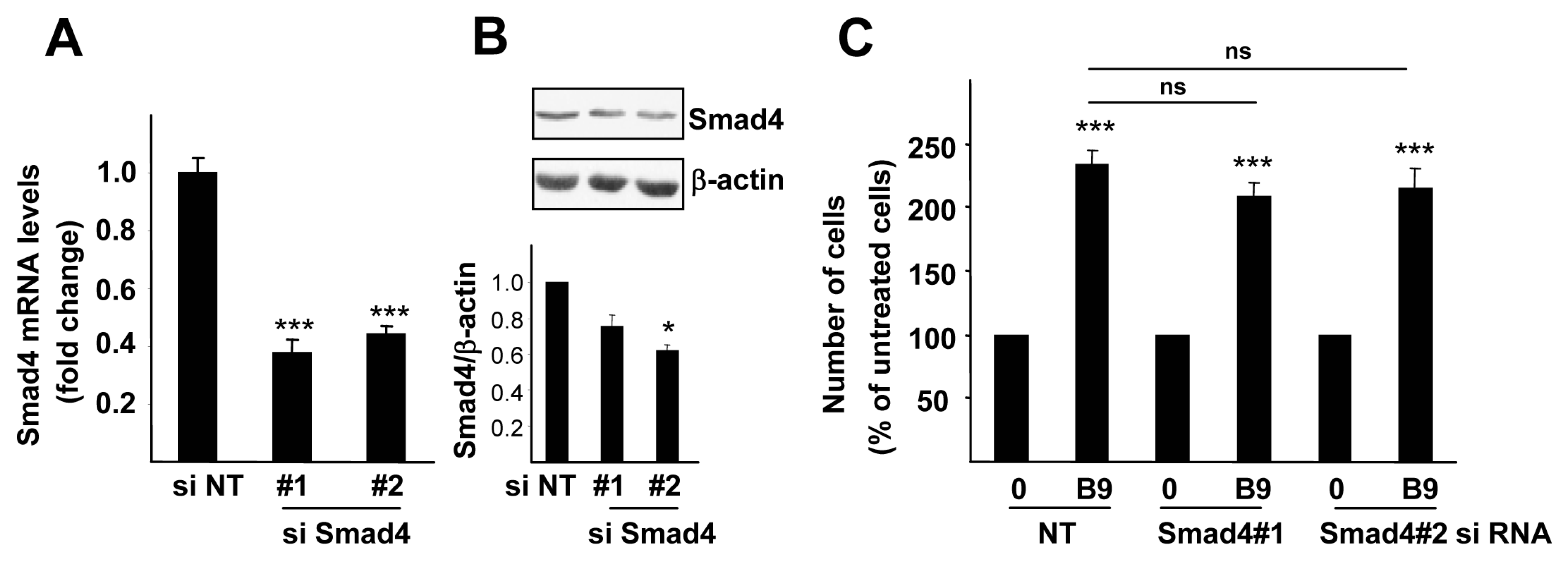
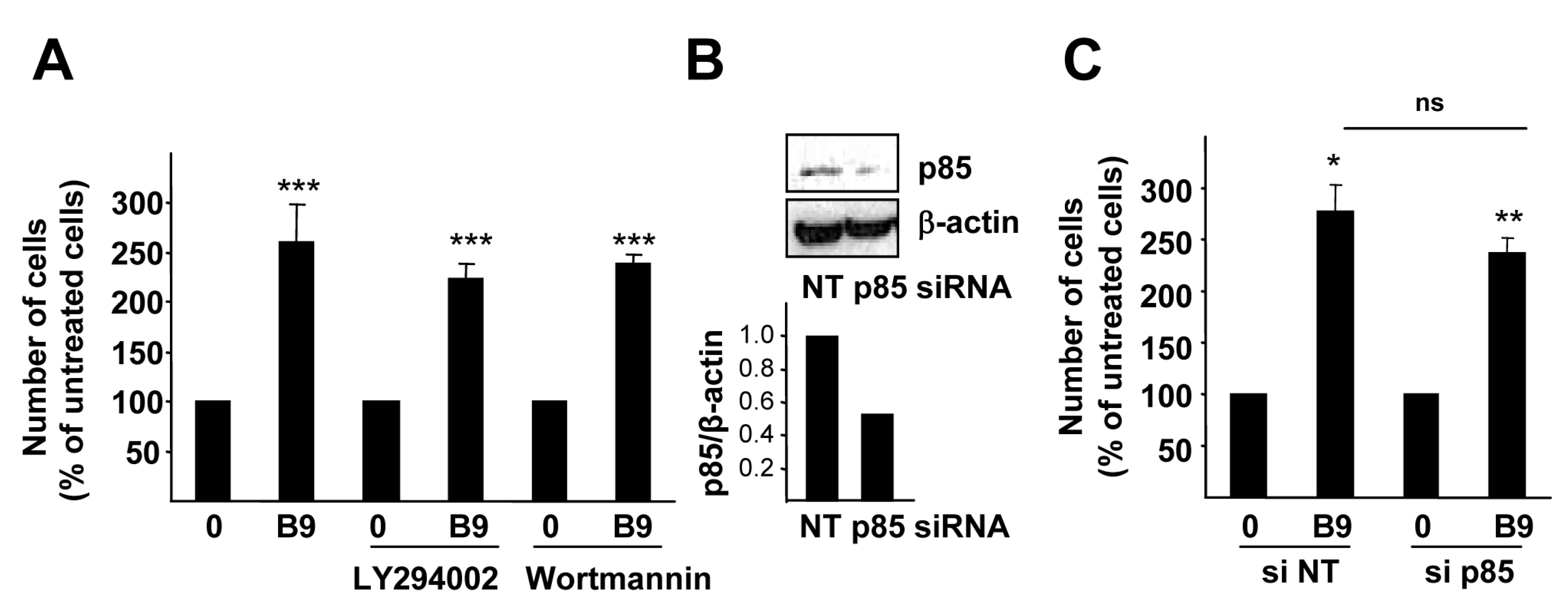
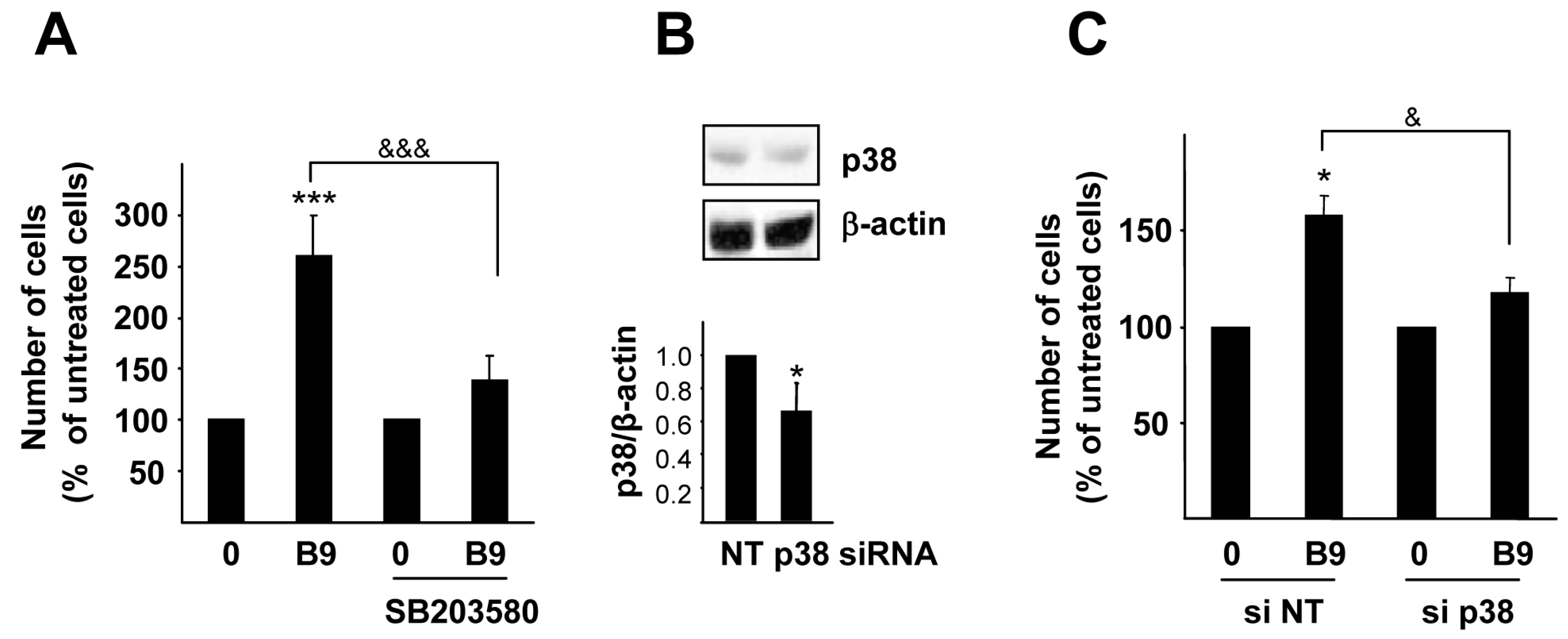
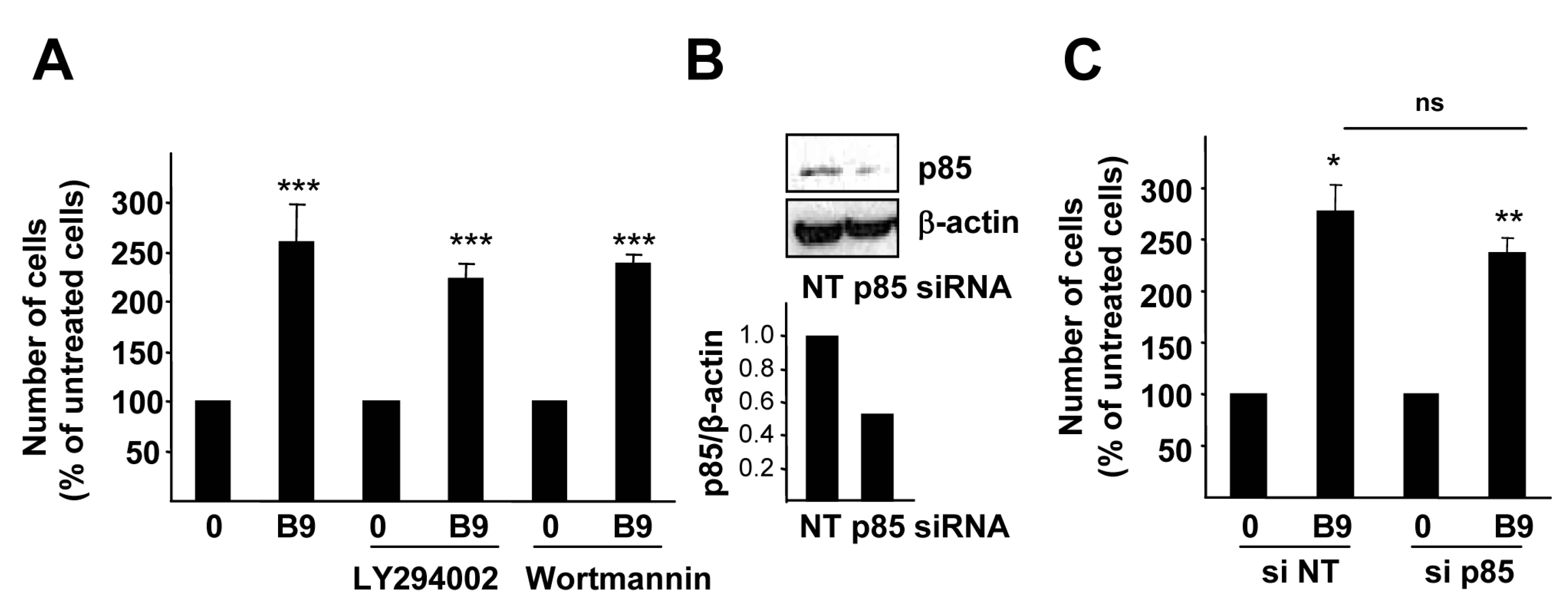
2.3. p38MAPK, but Not PI3K Activation, Is Required for the BMP9 Growth Promoting Effect in HepG2 Cells

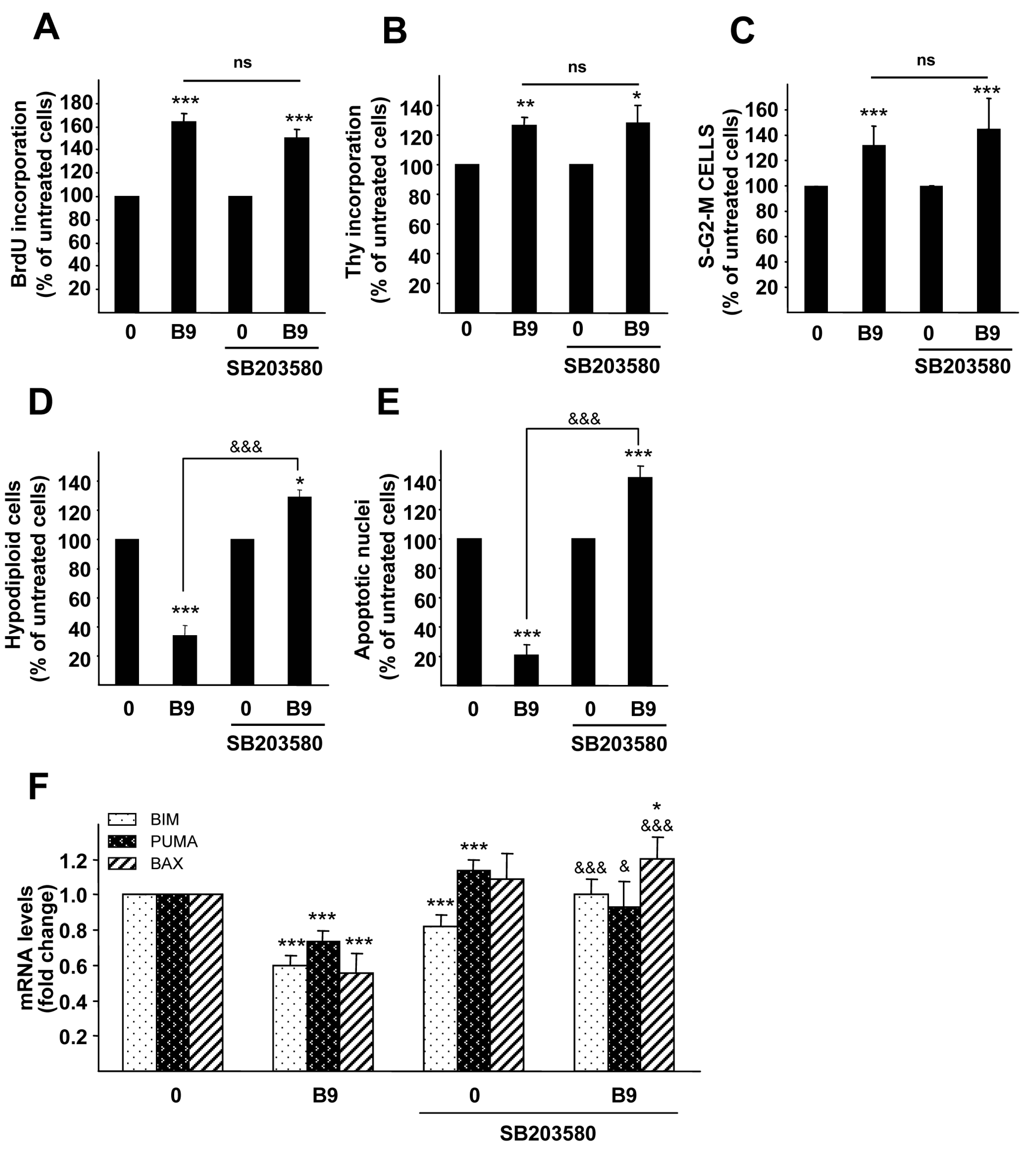
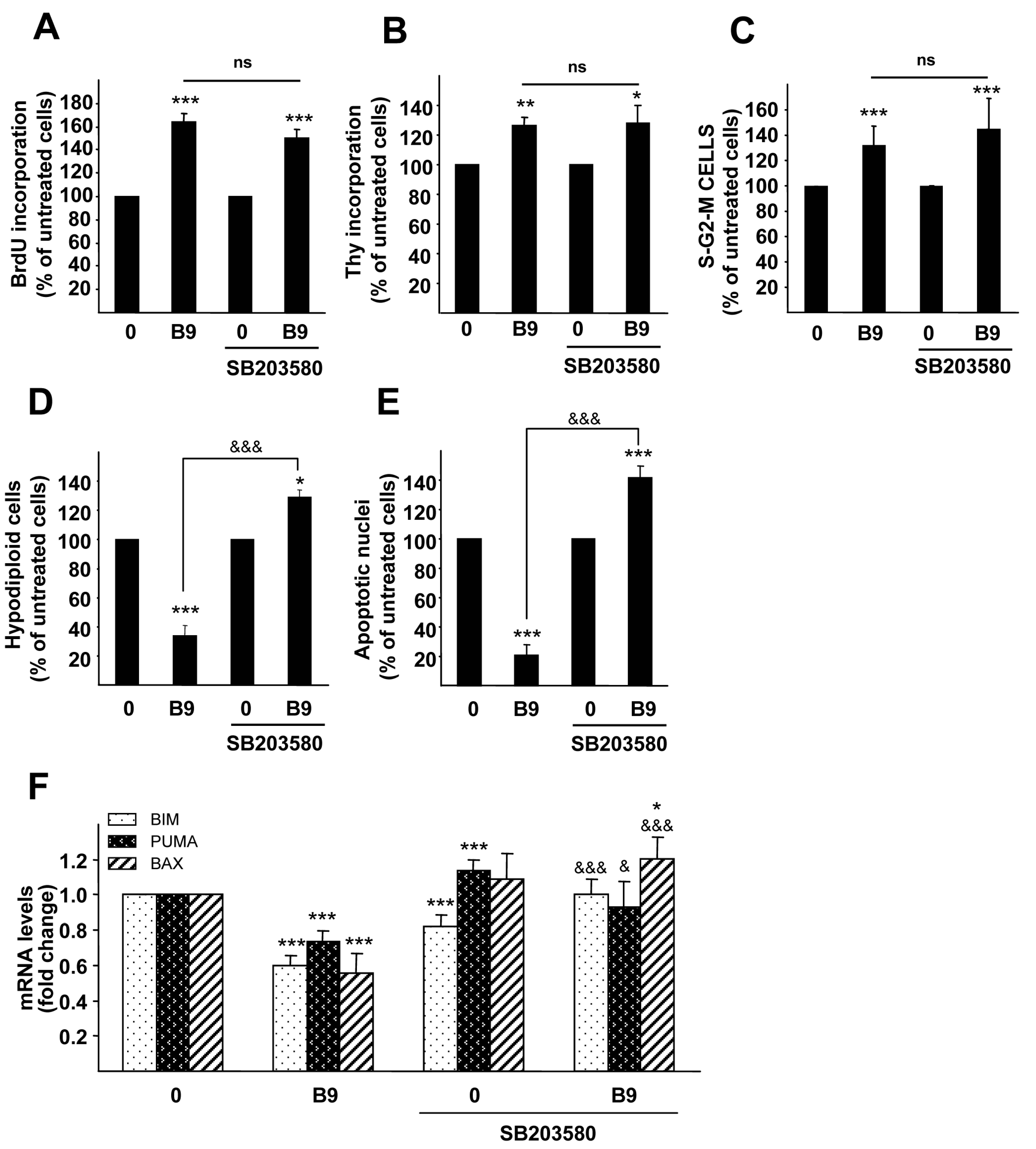
2.4. p38MAPK Is Required for the BMP9 Survival Effect on Serum Starvation-Triggered Apoptosis in HepG2 Cells
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Culture
4.3. Analysis of Cell Number
4.4. Cell Viability Analysis
4.5. BrdU Incorporation Assay
4.6. Thymidine Incorporation Assay
4.7. Analysis of Cell DNA Content by Flow Cytometry
4.8. Western Blotting
4.9. siRNA Knockdown Assays
4.10. Measurement of Apoptotic Index
4.11. Quantitative Reverse Transcriptase-Polymerase Chain Reaction Analysis
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Villanueva, A.; Llovet, J.M. Targeted therapies for hepatocellular carcinoma. Gastroenterology 2011, 140, 1410–1426. [Google Scholar] [CrossRef] [PubMed]
- Zender, L.; Villanueva, A.; Tovar, V.; Sia, D.; Chiang, D.Y.; Llovet, J.M. Cancer gene discovery in hepatocellular carcinoma. J. Hepatol. 2010, 52, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Wozney, J.M.; Rosen, V.; Celeste, A.J.; Mitsock, L.M.; Whitters, M.J.; Kriz, R.W.; Hewick, R.M.; Wang, E.A. Novel regulators of bone formation: Molecular clones and activities. Science 1988, 242, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.M.; Dunn, N.R.; Hogan, B.L.; Zaret, K.S. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001, 15, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Grompe, M. Generation and regeneration of cells of the liver and pancreas. Science 2008, 322, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Sanchez, A.; Fabregat, I. BMPs and liver: more questions than answers. Curr. Pharm. Des. 2012, 18, 4114–4125. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Kuo, K.K.; Kuo, T.L.; Lee, K.T.; Cheng, K.H. The activation of MEK/ERK signaling pathway by bone morphogenetic protein 4 to increase hepatocellular carcinoma cell proliferation and migration. Mol. Cancer Res. 2012, 10, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Maegdefrau, U.; Amann, T.; Winklmeier, A.; Braig, S.; Schubert, T.; Weiss, T.S.; Schardt, K.; Warnecke, C.; Hellerbrand, C.; Bosserhoff, A.K. Bone morphogenetic protein 4 is induced in hepatocellular carcinoma by hypoxia and promotes tumour progression. J. Pathol. 2009, 218, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Maegdefrau, U.; Bosserhoff, A.K. BMP activated Smad signaling strongly promotes migration and invasion of hepatocellular carcinoma cells. Exp. Mol. Pathol. 2012, 92, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Hsia, Y.; Yang, W.Y.; Lin, Y.I.; Li, C.C.; Tsai, T.F.; Chang, K.W.; Shieh, G.S.; Tsai, S.F.; Wang, H.D.; et al. Identification of the common regulators for hepatocellular carcinoma induced by hepatitis B virus X antigen in a mouse model. Carcinogenesis 2012, 33, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xiong, L.; Zou, L.; Zhao, J. Upregulation of Bone Morphogenetic Protein 4 is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. Pathol. Oncol. Res. 2012, 18, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Bidart, M.; Ricard, N.; Levet, S.; Samson, M.; Mallet, C.; David, L.; Subileau, M.; Tillet, E.; Feige, J.J.; Bailly, S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell Mol. Life Sci. 2011, 69, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.J.; Song, J.J.; Cox, K.; Rosen, V.; Wozney, J.M. Bone morphogenetic protein-9, a new member of the TGF-beta superfamily. J. Bone Miner. Res. 1994, Suppl. 1, 136. [Google Scholar]
- Herrera, B.; Garcia-Alvaro, M.; Cruz, S.; Walsh, P.; Fernandez, M.; Roncero, C.; Fabregat, I.; Sanchez, A.; Inman, G.J. BMP9 is a proliferative and survival factor for human hepatocellular carcinoma cells. PLoS ONE 2013, 8, e69535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Gu, X.; Weng, H.; Ghafoory, S.; Liu, Y.; Feng, T.; Dzieran, J.; Li, L.; Ilkavets, I.; Kruithof-de Julio, M.; et al. Bone morphogenetic protein-9 (BMP-9) induces epithelial to mesenchymal transition (EMT) in hepatocellular carcinoma cells. Cancer Sci. 2013, 104, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Sieber, C.; Kopf, J.; Hiepen, C.; Knaus, P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 2009, 20, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Jang, J.E.; Kang, C.M.; Chung, H.Y.; Kim, N.D.; Kim, K.W. Hypoxia-induced VEGF enhances tumor survivability via suppression of serum deprivation-induced apoptosis. Oncogene 2000, 19, 4621–4631. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cederbaum, A.I. Overexpression of CYP2E1 in mitochondria sensitizes HepG2 cells to the toxicity caused by depletion of glutathione. J. Biol. Chem. 2006, 281, 5128–5136. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, J.; Cederbaum, A.I. Serum deprivation-induced HepG2 cell death is potentiated by CYP2E1. Free Radic. Biol. Med. 2006, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Star, G.P.; Giovinazzo, M.; Langleben, D. Bone morphogenic protein-9 stimulates endothelin-1 release from human pulmonary microvascular endothelial cells: A potential mechanism for elevated ET-1 levels in pulmonary arterial hypertension. Microvasc. Res. 2010, 80, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Trembath, R.C.; Morrell, N.W. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009, 284, 15794–15804. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Sancho, P.; Bertran, E.; Iglesias-Serret, D.; Gil, J.; Fabregat, I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-β-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. 2009, 69, 7595–7602. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Zhou, J.P.; Liu, Y.P.; Liu, J.J.; Yang, X.N.; Jazag, A.; Zhang, Z.P.; Guleng, B.; Ren, J.L. The silencing of Pokemon attenuates the proliferation of hepatocellular carcinoma cells in vitro and in vivo by inhibiting the PI3K/Akt pathway. PLoS ONE 2012, 7, e51916. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Shi, Y.; Guo, X.H.; Ouyang, Y.B.; Wang, S.S.; Liu, D.J.; Wang, A.N.; Li, N.; Chen, D.X. Phosphorylated AKT inhibits the apoptosis induced by DRAM-mediated mitophagy in hepatocellular carcinoma by preventing the translocation of DRAM to mitochondria. Cell Death Dis. 2014, 5, e1078. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, Y.; Feng, Y.; Zhang, L.; Li, J.; Xie, Y.A.; Luo, X. Epigallocatechin-3-gallate inhibits cell growth, induces apoptosis and causes S phase arrest in hepatocellular carcinoma by suppressing the AKT pathway. Int. J. Oncol. 2014, 44, 791–796. [Google Scholar] [PubMed]
- Herrera, B.; van Dinther, M.; Ten Dijke, P.; Inman, G.J. Autocrine bone morphogenetic protein-9 signals through activin receptor-like kinase-2/Smad1/Smad4 to promote ovarian cancer cell proliferation. Cancer Res. 2009, 69, 9254–9262. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Ding, J.; Sun, W.; Fu, J.; Chen, Y.; Wu, K.; Ning, B.; Han, T.; Huang, L.; Chen, C.; et al. Cyclin G1-mediated epithelial-mesenchymal transition via phosphoinositide 3-kinase/Akt signaling facilitates liver cancer progression. Hepatology 2012, 55, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, X.; He, Y.; Ye, G.; Li, X.; Zhang, X.; Zhou, L.; Deng, F. Bone morphogenetic protein-9 induces osteogenic differentiation of rat dental follicle stem cells in P38 and ERK1/2 MAPK dependent manner. Int. J. Med. Sci. 2012, 9, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Drevelle, O.; Daviau, A.; Senta, H.; Bergeron, E.; Faucheux, N. Preventing MEK1 activation influences the responses of human osteosarcoma cells to bone morphogenetic proteins 2 and 9. Anticancer Drugs 2013, 24, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.J.; Zhao, Y.Z.; Wang, J.; He, J.W.; Weng, Y.G.; Luo, J.Y. Smads, p38 and ERK1/2 are involved in BMP9-induced osteogenic differentiation of C3H10T1/2 mesenchymal stem cells. BMB Rep. 2012, 45, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Song, T.; Wang, W.; Wang, J.; He, J.; Wu, N.; Tang, M.; He, B.; Luo, J. P38 and ERK1/2 MAPKs act in opposition to regulate BMP9-induced osteogenic differentiation of mesenchymal progenitor cells. PLoS ONE 2012, 7, e43383. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Comes, F.; Matrone, A.; Lastella, P.; Nico, B.; Susca, F.C.; Bagnulo, R.; Ingravallo, G.; Modica, S.; Lo Sasso, G.; Moschetta, A.; et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007, 14, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Heron-Milhavet, L.; LeRoith, D. Insulin-like growth factor I induces MDM2-dependent degradation of p53 via the p38 MAPK pathway in response to DNA damage. J. Biol. Chem. 2002, 277, 15600–15606. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Krainc, D.; Sherman, K.; Lipton, S.A. Antiapoptotic role of the p38 mitogen-activated protein kinase-myocyte enhancer factor 2 transcription factor pathway during neuronal differentiation. Proc. Natl. Acad. Sci. USA 2000, 97, 7561–7566. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Uzquiza, A.; Arechederra, M.; Molina, I.; Banos, R.; Maia, V.; Benito, M.; Guerrero, C.; Porras, A. C3G down-regulates p38 MAPK activity in response to stress by Rap-1 independent mechanisms: Involvement in cell death. Cell Signal. 2010, 22, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, R.G.; Sonnenberg-Hirche, C.; Smith, S.D.; Hu, C.; Barton, A.; Sadovsky, Y.; Nelson, D.M. Epidermal growth factor abrogates hypoxia-induced apoptosis in cultured human trophoblasts through phosphorylation of BAD Serine 112. Endocrinology 2008, 149, 2131–2137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shan, P.; Alam, J.; Fu, X.Y.; Lee, P.J. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J. Biol. Chem. 2005, 280, 8714–8721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shan, P.; Otterbein, L.E.; Alam, J.; Flavell, R.A.; Davis, R.J.; Choi, A.M.; Lee, P.J. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J. Biol. Chem. 2003, 278, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Alvarez, A.M.; Benito, M.; Fabregat, I. Apoptosis induced by transforming growth factor-beta in fetal hepatocyte primary cultures: Involvement of reactive oxygen intermediates. J. Biol. Chem. 1996, 271, 7416–7422. [Google Scholar] [PubMed]
- de Juan, C.; Benito, M.; Alvarez, A.; Fabregat, I. Differential proliferative response of cultured fetal and regenerating hepatocytes to growth factors and hormones. Exp. Cell Res. 1992, 202, 495–500. [Google Scholar] [CrossRef]
- Conner, E.; Wirth, P.; Kiss, A.; Santoni-Rugiu, E.; Thorgeirsson, S. Growth inhibition and induction of apoptosis by HGF in transformed rat liver epithelial cells. Biochem. Biophys. Res. Commun. 1997, 236, 396–401. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Álvaro, M.; Addante, A.; Roncero, C.; Fernández, M.; Fabregat, I.; Sánchez, A.; Herrera, B. BMP9-Induced Survival Effect in Liver Tumor Cells Requires p38MAPK Activation. Int. J. Mol. Sci. 2015, 16, 20431-20448. https://doi.org/10.3390/ijms160920431
García-Álvaro M, Addante A, Roncero C, Fernández M, Fabregat I, Sánchez A, Herrera B. BMP9-Induced Survival Effect in Liver Tumor Cells Requires p38MAPK Activation. International Journal of Molecular Sciences. 2015; 16(9):20431-20448. https://doi.org/10.3390/ijms160920431
Chicago/Turabian StyleGarcía-Álvaro, María, Annalisa Addante, Cesáreo Roncero, Margarita Fernández, Isabel Fabregat, Aránzazu Sánchez, and Blanca Herrera. 2015. "BMP9-Induced Survival Effect in Liver Tumor Cells Requires p38MAPK Activation" International Journal of Molecular Sciences 16, no. 9: 20431-20448. https://doi.org/10.3390/ijms160920431