
Structural Features of the ATP-Binding Cassette (ABC) Transporter ABCA3

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Functional Features of ABC Transporters
2.1. Mechanism of Transport
2.2. In Vitro and in Vivo Models to Study the Functional Role of ABCA3
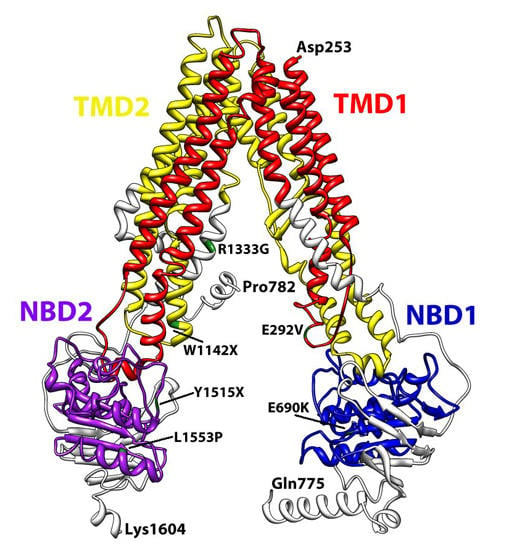
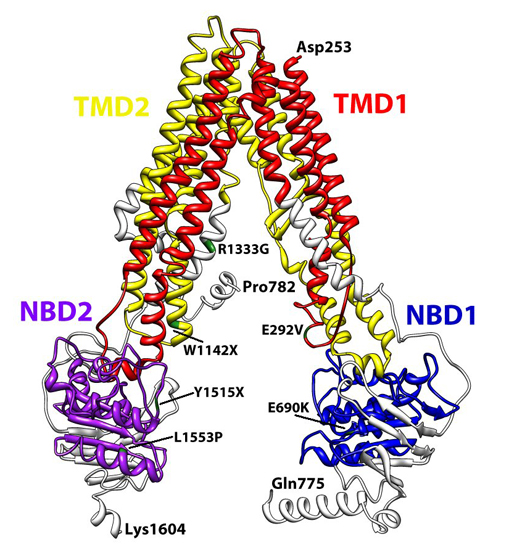
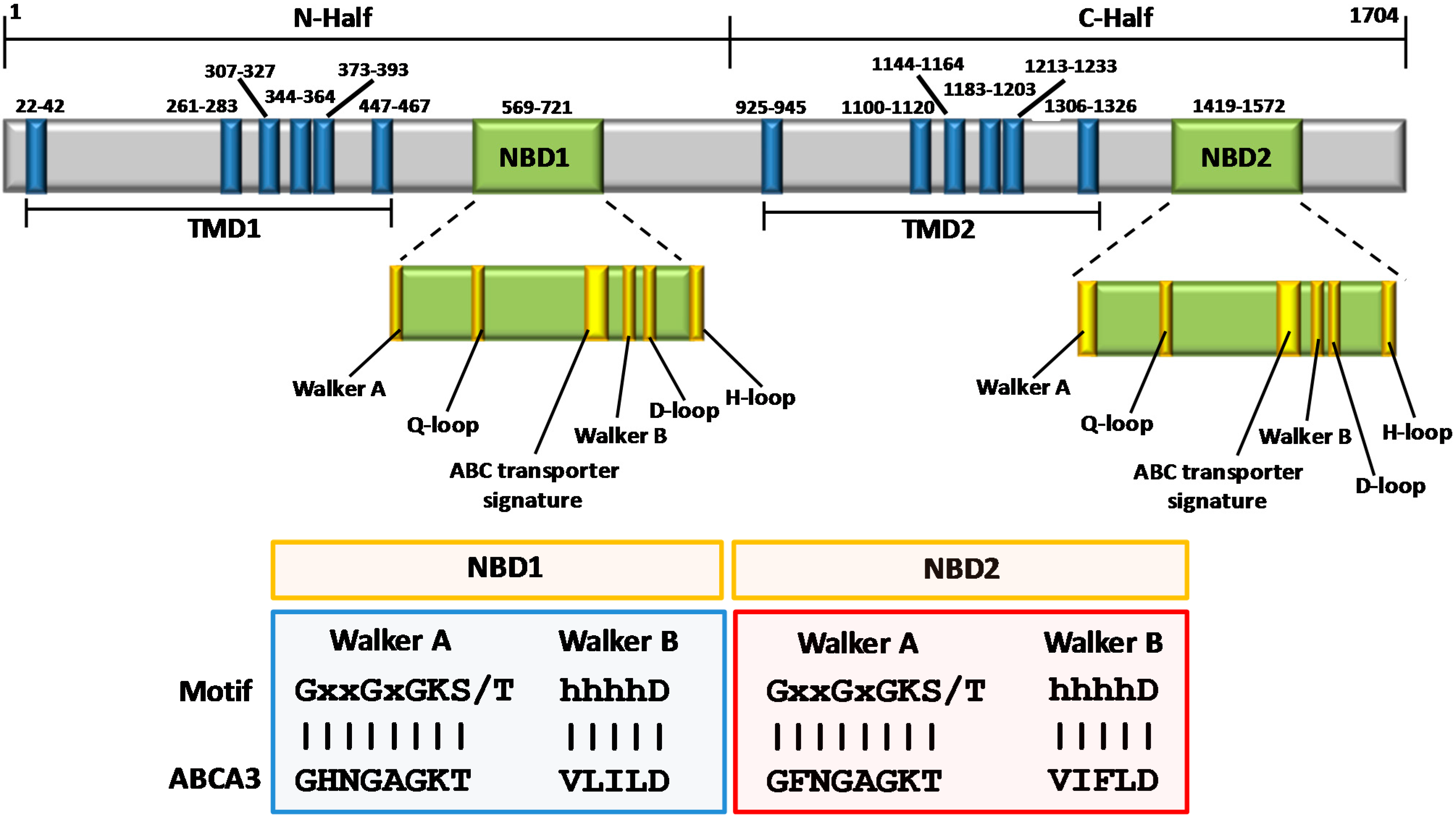
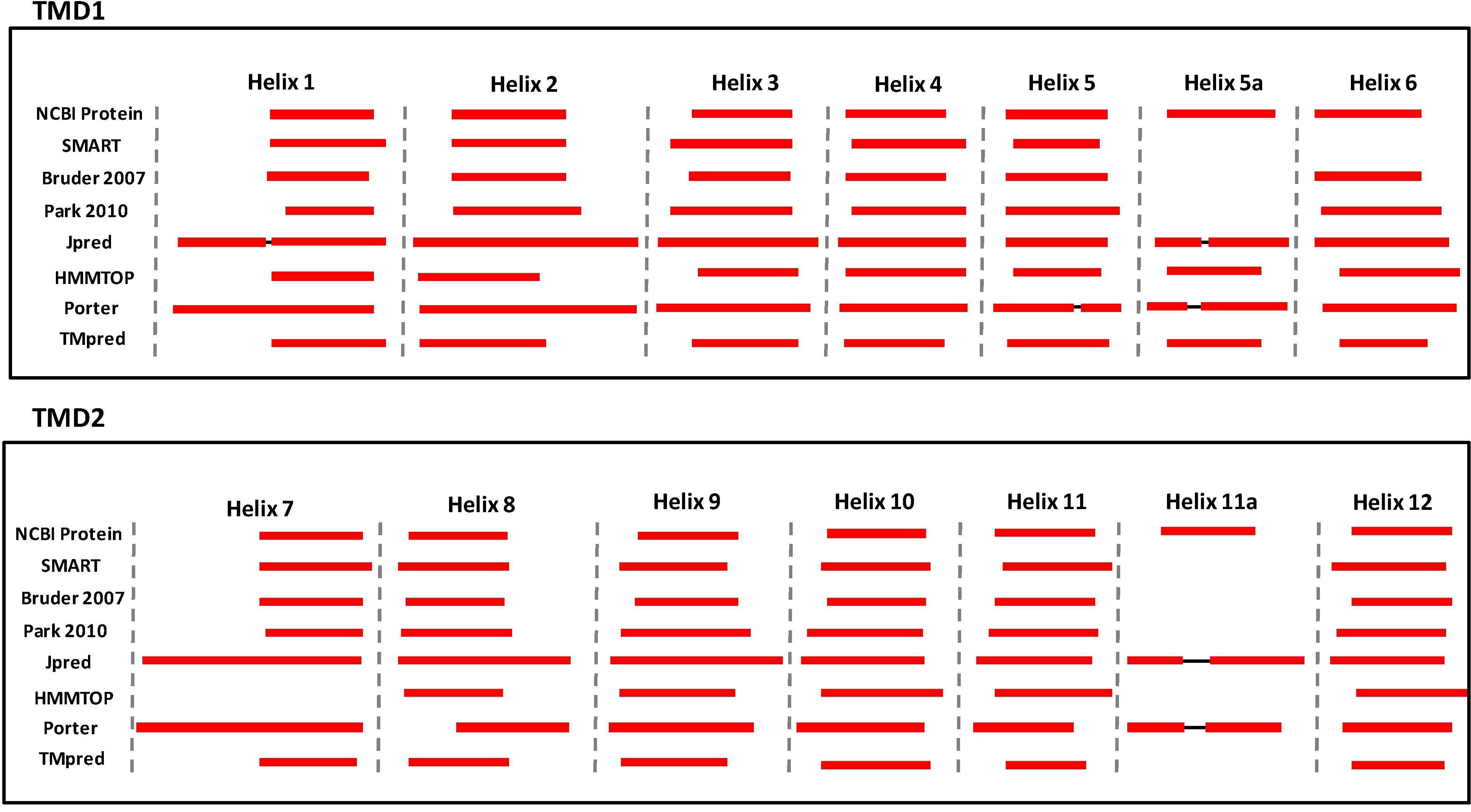
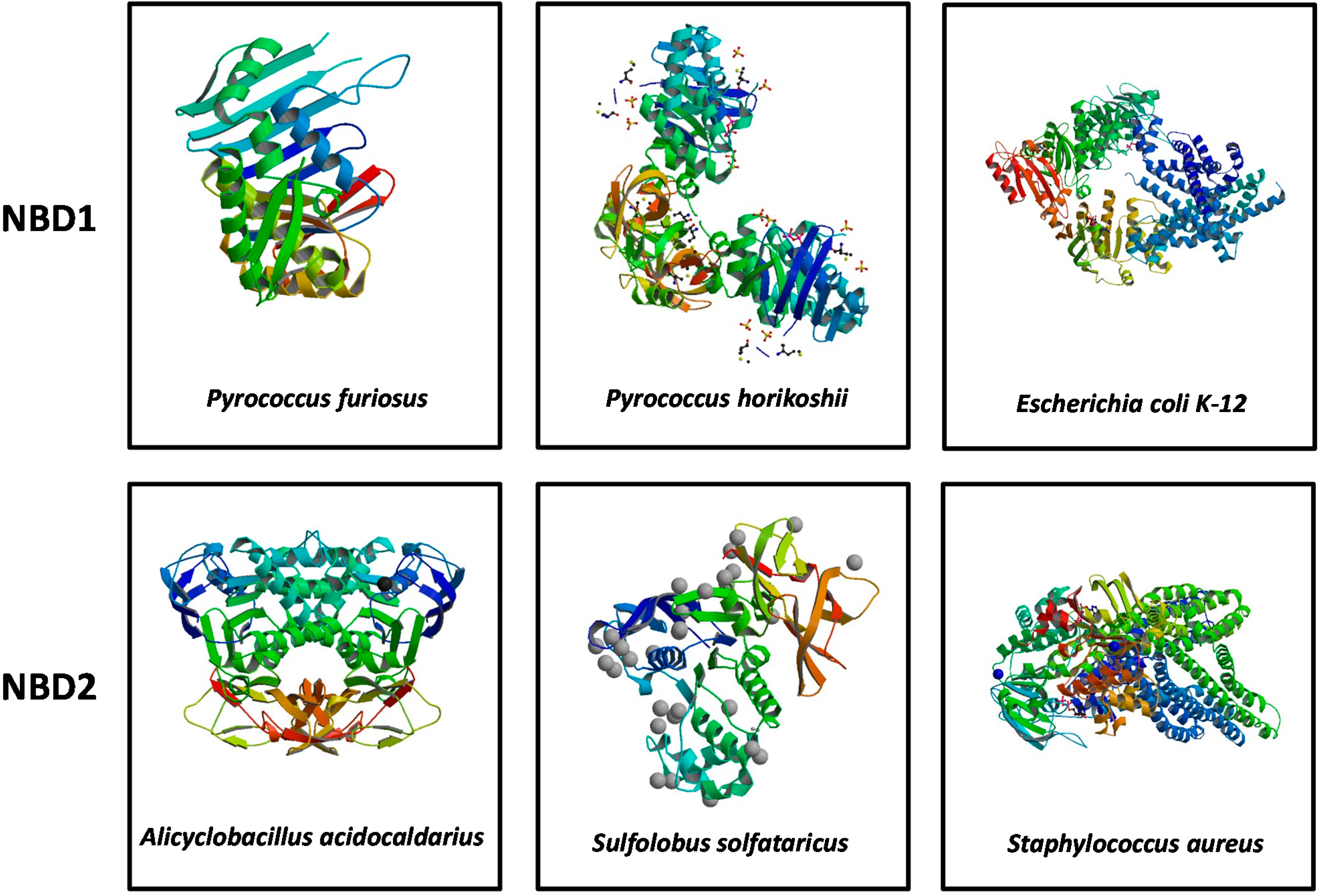
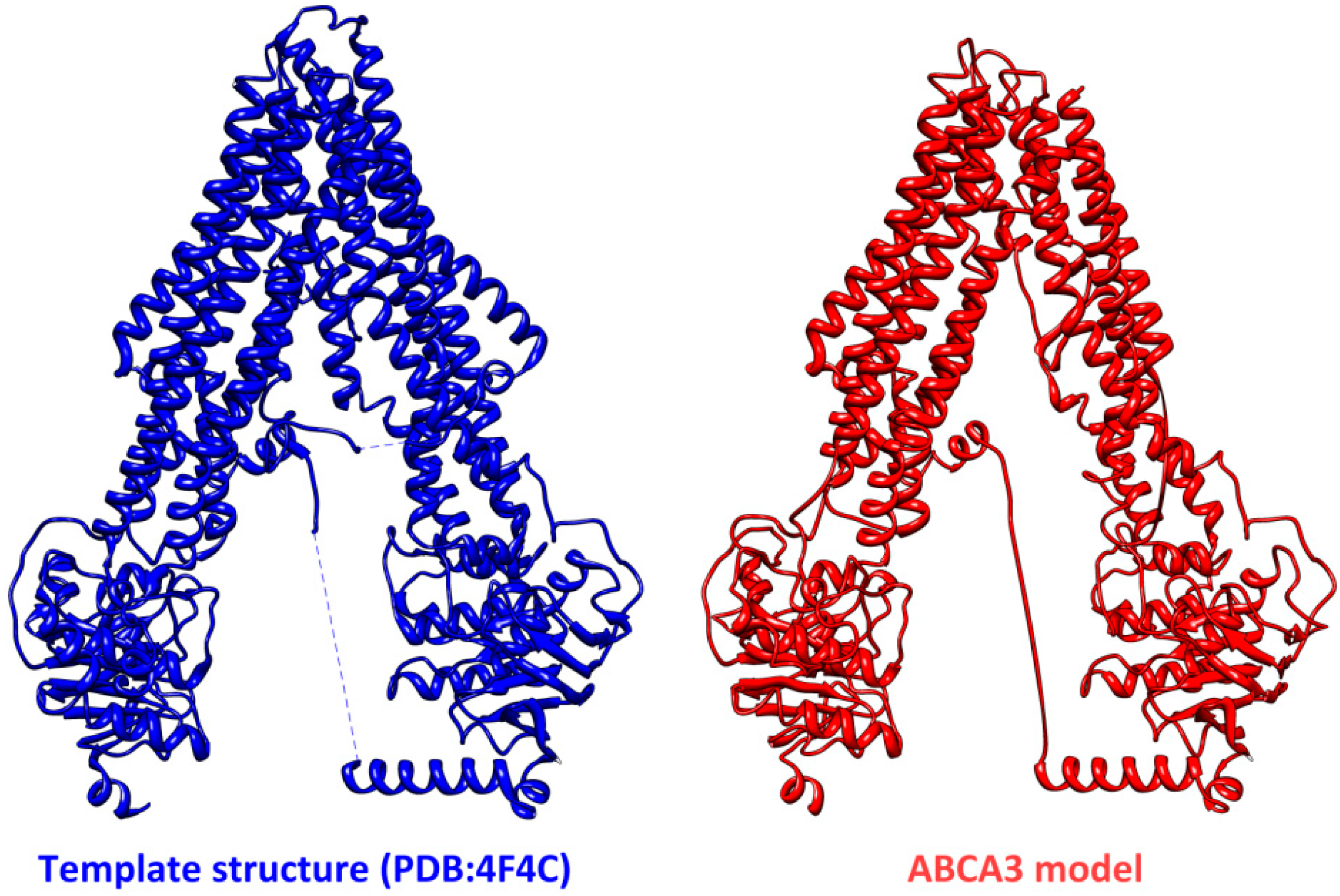
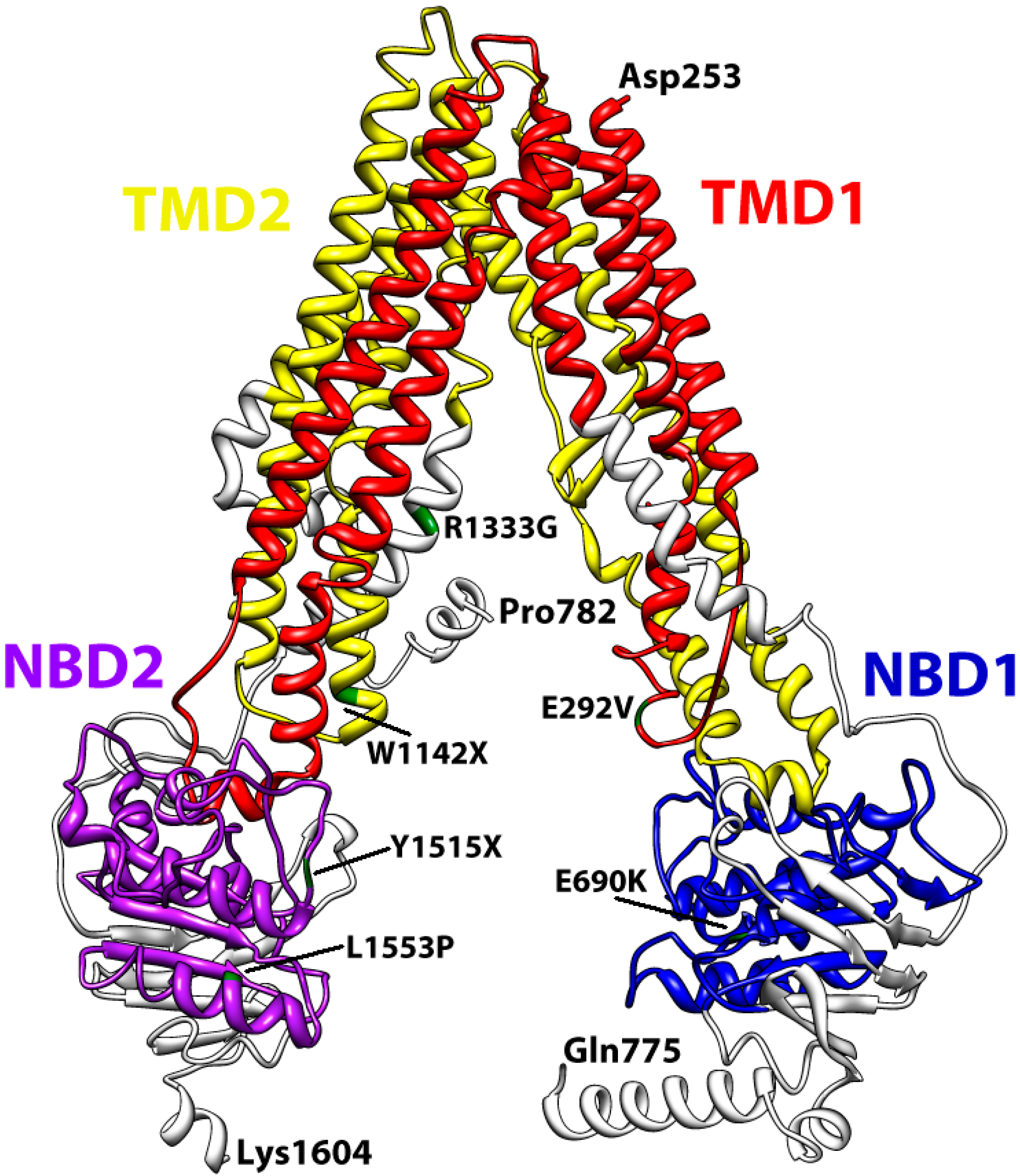
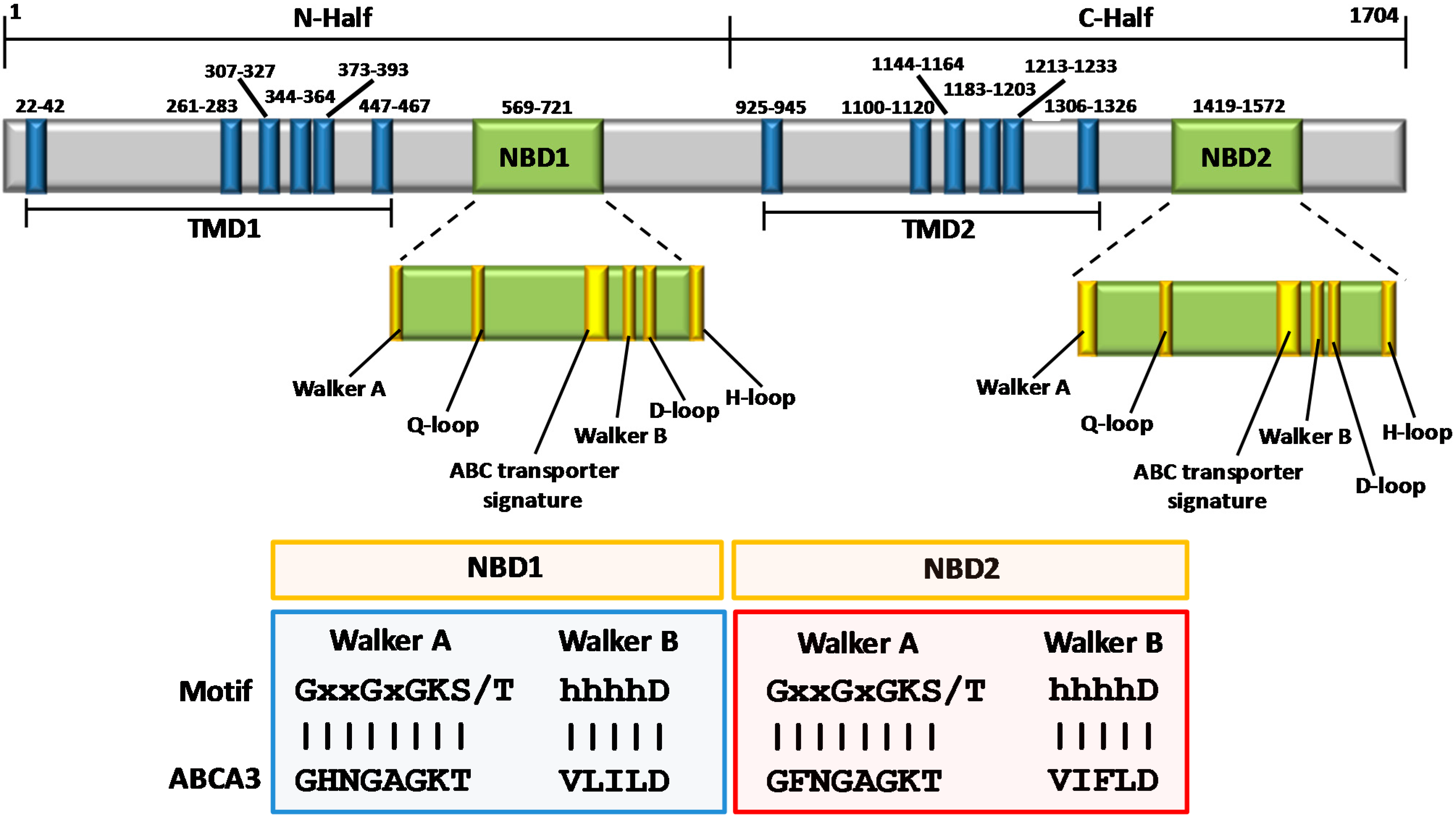
3. Structural Features of ABCA3
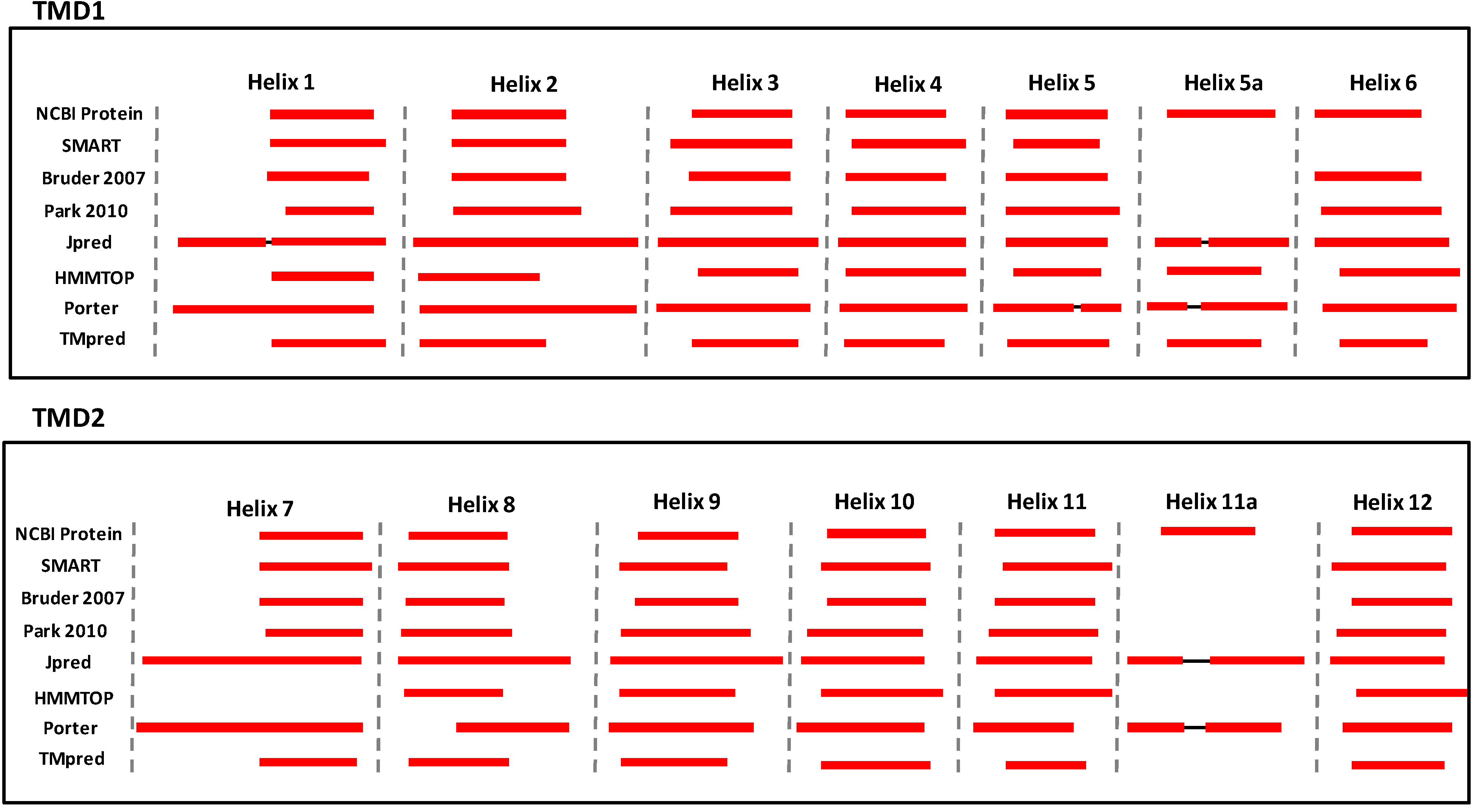
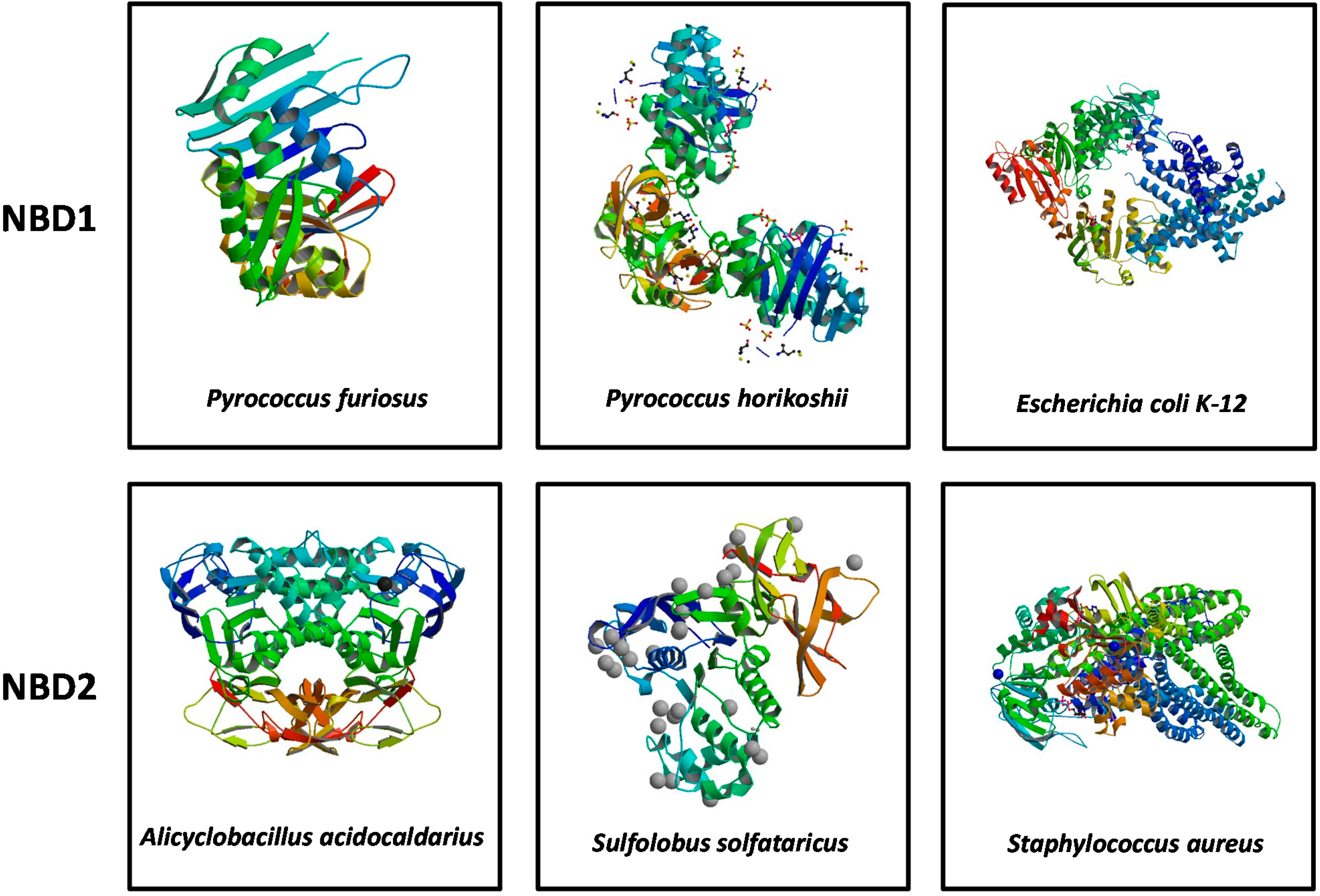
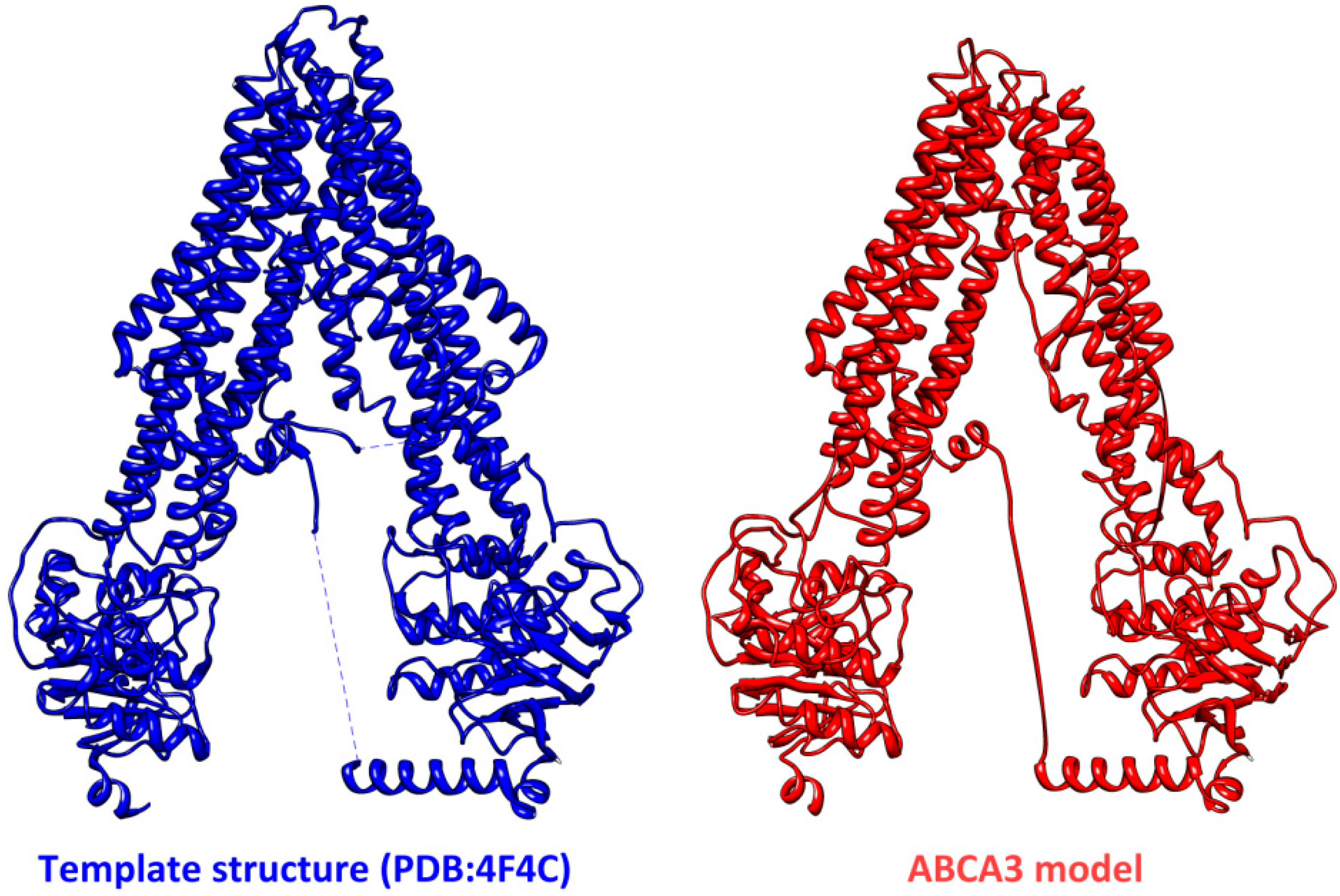
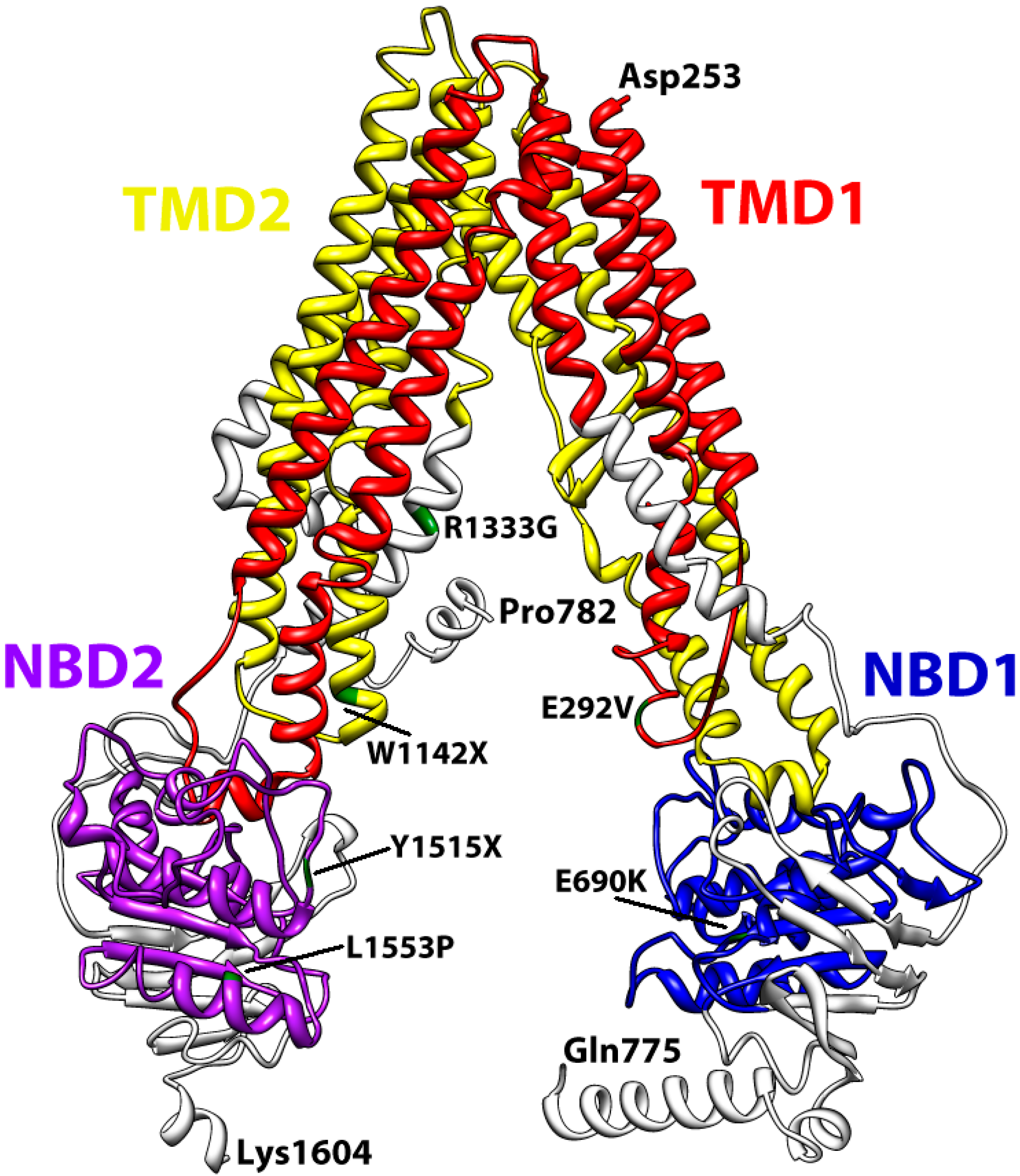
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pohl, A.; Devaux, P.F.; Herrmann, A. Function of prokaryotic and eukaryotic ABC proteins in lipid transport. Biochim. Biophys. Acta 2005, 1733, 29–52. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genom. Hum. Genet. 2005, 6, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta 2009, 1791, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Peelman, F.; Labeur, C.; Vanloo, B.; Roosbeek, S.; Devaud, C.; Duverger, N.; Denefle, P.; Rosier, M.; Vandekerckhove, J.; Rosseneu, M. Characterization of the ABCA transporter subfamily: Identification of prokaryotic and eukaryotic members, phylogeny and topology. J. Mol. Biol. 2003, 325, 259–274. [Google Scholar] [CrossRef]
- Tarling, E.J.; de Aguiar Vallim, T.Q.; Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 2013, 24, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, C.; Viturro, E. The ABCA subfamily—Gene and protein structures, functions and associated hereditary diseases. Pflug. Arch. 2007, 453, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Peca, D.; Boldrini, R.; Johannson, J.; Shieh, J.T.; Citti, A.; Petrini, S.; Salerno, T.; Cazzato, S.; Testa, R.; Messina, F.; et al. Clinical and ultrastructural spectrum of diffuse lung disease associated with surfactant protein C mutations. Eur. J. Hum. Genet. 2015, 23, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.B.; Blight, M.A. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol. 1999, 293, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Linton, K.J. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 2004, 11, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.L.; Davidson, A.L.; Chen, J. Structural insights into ABC transporter mechanism. Curr. Opin. Struct. Biol. 2008, 18, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Linton, K.J.; Higgins, C.F. Structure and function of ABC transporters: The ATP switch provides flexible control. Pflug. Arch. 2007, 453, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Ter Beek, J.; Guskov, A.; Slotboom, D.J. Structural diversity of ABC transporters. J. Gen. Physiol. 2014, 143, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Yamamoto, A.; Ban, N.; Tanaka, A.R.; Matsuo, M.; Kioka, N.; Inagaki, N.; Ueda, K. Human ABCA3, a product of a responsible gene for ABCA3 for fatal surfactant deficiency in newborns, exhibits unique ATP hydrolysis activity and generates intracellular multilamellar vesicles. Biochem. Biophys. Res. Commun. 2004, 324, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Yamano, G.; Funahashi, H.; Kawanami, O.; Zhao, L.X.; Ban, N.; Uchida, Y.; Morohoshi, T.; Ogawa, J.; Shioda, S.; Inagaki, N. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett. 2001, 508, 221–225. [Google Scholar] [CrossRef]
- Shulenin, S.; Nogee, L.M.; Annilo, T.; Wert, S.E.; Whitsett, J.A.; Dean, M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N. Engl. J. Med. 2004, 350, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Gray, J.M.; Notarfrancesco, K.L.; Gonzales, L.W.; Koval, M.; Feinstein, S.I.; Ballard, P.L.; Fisher, A.B.; Shuman, H. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J. Biol. Chem. 2002, 277, 22147–22155. [Google Scholar] [CrossRef] [PubMed]
- Stahlman, M.T.; Besnard, V.; Wert, S.E.; Weaver, T.E.; Dingle, S.; Xu, Y.; von Zychlin, K.; Olson, S.J.; Whitsett, J.A. Expression of ABCA3 in developing lung and other tissues. J. Histochem. Cytochem. 2007, 55, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Wert, S.E.; Whitsett, J.A.; Nogee, L.M. Genetic disorders of surfactant dysfunction. Pediatr. Dev. Pathol. 2009, 12, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Doan, M.L.; Guillerman, R.P.; Dishop, M.K.; Nogee, L.M.; Langston, C.; Mallory, G.B.; Sockrider, M.M.; Fan, L.L. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax 2008, 63, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Garmany, T.H.; Moxley, M.A.; White, F.V.; Dean, M.; Hull, W.M.; Whitsett, J.A.; Nogee, L.M.; Hamvas, A. Surfactant composition and function in patients with ABCA3 mutations. Pediatr. Res. 2006, 59, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 2010, 61, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Wambach, J.A.; Casey, A.M.; Fishman, M.P.; Wegner, D.J.; Wert, S.E.; Cole, F.S.; Hamvas, A.; Nogee, L.M. Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am. J. Respir. Crit. Care Med. 2014, 189, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Sakai, H.; Sasaki, M.; Ban, N.; Inagaki, N. ABCA3-mediated choline-phospholipids uptake into intracellular vesicles in A549 cells. FEBS Lett. 2007, 581, 3139–3144. [Google Scholar] [CrossRef] [PubMed]
- Cheong, N.; Madesh, M.; Gonzales, L.W.; Zhao, M.; Yu, K.; Ballard, P.L.; Shuman, H. Functional and trafficking defects in ATP binding cassette A3 mutants associated with respiratory distress syndrome. J. Biol. Chem. 2006, 281, 9791–9800. [Google Scholar] [CrossRef] [PubMed]
- Cheong, N.; Zhang, H.; Madesh, M.; Zhao, M.; Yu, K.; Dodia, C.; Fisher, A.B.; Savani, R.C.; Shuman, H. ABCA3 is critical for lamellar body biogenesis in vivo. J. Biol. Chem. 2007, 282, 23811–23817. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Xavier, R.; Haley, K.J.; Welti, R.; Goss, J.L.; Brown, C.E.; Zhuang, D.Z.; Bell, S.A.; Lu, N.; McKee, M.; et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J. Lipid Res. 2007, 48, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Herber-Jonat, S.; Mittal, R.; Huppmann, M.; Hammel, M.; Liebisch, G.; Yildirim, A.O.; Eickelberg, O.; Schmitz, G.; de Angelis, M.H.; Flemmer, A.W.; et al. Abca3 haploinsufficiency is a risk factor for lung injury induced by hyperoxia or mechanical ventilation in a murine model. Pediatr. Res. 2013, 74, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Michel, G.; Hoefer, C.; Klaften, M.; Muller-Hocker, J.; de Angelis, M.H.; Holzinger, A. Targeted inactivation of the murine Abca3 gene leads to respiratory failure in newborns with defective lamellar bodies. Biochem. Biophys. Res. Commun. 2007, 359, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Matsumura, Y.; Sakai, H.; Takanezawa, Y.; Sasaki, M.; Arai, H.; Inagaki, N. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J. Biol. Chem. 2007, 282, 9628–9634. [Google Scholar] [CrossRef] [PubMed]
- Tryka, A.F.; Wert, S.E.; Mazursky, J.E.; Arrington, R.W.; Nogee, L.M. Absence of lamellar bodies with accumulation of dense bodies characterizes a novel form of congenital surfactant defect. Pediatr. Dev. Pathol. 2000, 3, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Besnard, V.; Matsuzaki, Y.; Clark, J.; Xu, Y.; Wert, S.E.; Ikegami, M.; Stahlman, M.T.; Weaver, T.E.; Hunt, A.N.; Postle, A.D.; et al. Conditional deletion of Abca3 in alveolar type II cells alters surfactant homeostasis in newborn and adult mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L646–L659. [Google Scholar] [CrossRef] [PubMed]
- Dean, M. The genetics of ATP-binding cassette transporters. Methods Enzymol. 2005, 400, 409–429. [Google Scholar] [PubMed]
- Piehler, A.P.; Wenzel, J.J.; Olstad, O.K.; Haug, K.B.; Kierulf, P.; Kaminski, W.E. The human ortholog of the rodent testis-specific ABC transporter Abca17 is a ubiquitously expressed pseudogene (ABCA17P) and shares a common 5ʹ end with ABCA3. BMC Mol. Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Bruder, E.; Hofmeister, J.; Aslanidis, C.; Hammer, J.; Bubendorf, L.; Schmitz, G.; Rufle, A.; Buhrer, C. Ultrastructural and molecular analysis in fatal neonatal interstitial pneumonia caused by a novel ABCA3 mutation. Mod. Pathol. 2007, 20, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Amos, L.; Rao, A.; Quasney, M.W.; Matsumura, Y.; Inagaki, N.; Dahmer, M.K. Identification and characterization of a novel ABCA3 mutation. Physiol. Genom. 2010, 40, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Vedhachalam, C.; Duong, P.T.; Nickel, M.; Nguyen, D.; Dhanasekaran, P.; Saito, H.; Rothblat, G.H.; Lund-Katz, S.; Phillips, M.C. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J. Biol. Chem. 2007, 282, 25123–25130. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Roth, C.B. Structure of MsbA from E. coli: A homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 2001, 293, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P.; Lee, A.T.; Rees, D.C. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science 2002, 296, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Pollastri, G.; McLysaght, A. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21, 1719–1720. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Stoffel, W. Tmbase-A database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler 1993, 374, 166. [Google Scholar]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.W.; Peng, X.H.; Sauna, Z.E.; FitzGerald, P.C.; Xia, D.; Muller, M.; Nandigama, K.; Ambudkar, S.V. The conserved Tyrosine residues 401 and 1044 in ATP sites of human P-glycoprotein are critical for ATP binding and hydrolysis: Evidence for a conserved subdomain, the A-loop in the ATP-binding cassette. Biochemistry 2006, 45, 7605–7616. [Google Scholar] [CrossRef] [PubMed]
- Linton, K.J. Structure and function of ABC transporters. Physiology 2007, 22, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Karcher, A.; Shin, D.S.; Craig, L.; Arthur, L.M.; Carney, J.P.; Tainer, J.A. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 2000, 101, 789–800. [Google Scholar] [CrossRef]
- De la Rosa, M.B.; Nelson, S.W. An interaction between the Walker A and D-loop motifs is critical to ATP hydrolysis and cooperativity in bacteriophage T4 Rad50. J. Biol. Chem. 2011, 286, 26258–26266. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ojeda-May, P.; Pu, J. H-loop histidine catalyzes ATP hydrolysis in the E. coli ABC-transporter HlyB. Phys. Chem. Chem. Phys. 2013, 15, 15811–15815. [Google Scholar] [CrossRef] [PubMed]
- Kurashima-Ito, K.; Ikeya, T.; Senbongi, H.; Tochio, H.; Mikawa, T.; Shibata, T.; Ito, Y. Heteronuclear multidimensional NMR and homology modelling studies of the C-terminal nucleotide-binding domain of the human mitochondrial ABC transporter ABCB6. J. Biomol. NMR 2006, 35, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Ecker, G.F.; Stockner, T.; Chiba, P. Computational models for prediction of interactions with ABC-transporters. Drug Discov. Today 2008, 13, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Shintre, C.A.; Pike, A.C.W.; Li, Q.; Kim, J.I.; Barr, A.J.; Goubin, S.; Shrestha, L.; Yang, J.; Berridge, G.; Ross, J.; et al. Structures of ABCB10, a human ATP-binding cassette transporter in apo-and nucleotide-bound states. Proc. Natl. Acad. Sci. USA 2013, 110, 9710–9715. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Nguyen, P.T.; Yeates, T.O.; Rees, D.C. Inward facing conformations of the MetNI methionine ABC transporter: Implications for the mechanism of transinhibition. Protein Sci. 2012, 21, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Scheffel, F.; Demmer, U.; Warkentin, E.; Hulsmann, A.; Schneider, E.; Ermler, U. Structure of the ATPase subunit CysA of the putative sulfate ATP-binding cassette (ABC) transporter from Alicyclobacillus acidocaldarius. FEBS Lett. 2005, 579, 2953–2958. [Google Scholar] [CrossRef] [PubMed]
- Verdon, G.; Albers, S.V.; van Oosterwijk, N.; Dijkstra, B.W.; Driessen, A.J.; Thunnissen, A.M. Formation of the productive ATP-Mg2+-bound dimer of GlcV, an ABC-ATPase from Sulfolobus solfataricus. J. Mol. Biol. 2003, 334, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.; Locher, K.P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Oldham, M.L.; Zhang, Q.; Chen, J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 2012, 490, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.E.; Wert, S.E.; Whitsett, J.A.; Dean, M.; Nogee, L.M. ABCA3 mutations associated with pediatric interstitial lung disease. Am. J. Respir. Crit. Care Med. 2005, 172, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolini, A.; Baldassarre, A.; Del Gaudio, I.; Masotti, A. Structural Features of the ATP-Binding Cassette (ABC) Transporter ABCA3. Int. J. Mol. Sci. 2015, 16, 19631-19644. https://doi.org/10.3390/ijms160819631
Paolini A, Baldassarre A, Del Gaudio I, Masotti A. Structural Features of the ATP-Binding Cassette (ABC) Transporter ABCA3. International Journal of Molecular Sciences. 2015; 16(8):19631-19644. https://doi.org/10.3390/ijms160819631
Chicago/Turabian StylePaolini, Alessandro, Antonella Baldassarre, Ilaria Del Gaudio, and Andrea Masotti. 2015. "Structural Features of the ATP-Binding Cassette (ABC) Transporter ABCA3" International Journal of Molecular Sciences 16, no. 8: 19631-19644. https://doi.org/10.3390/ijms160819631