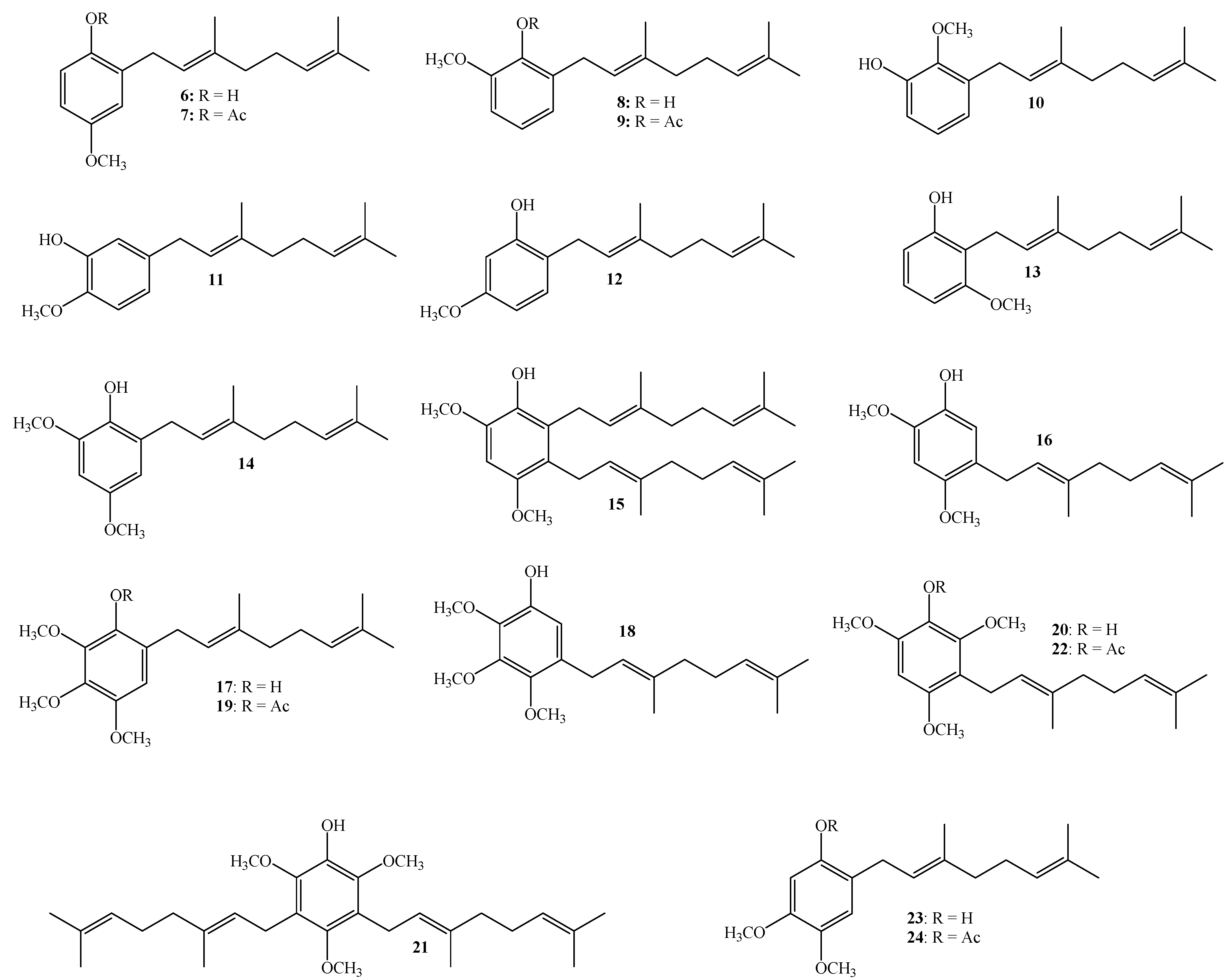
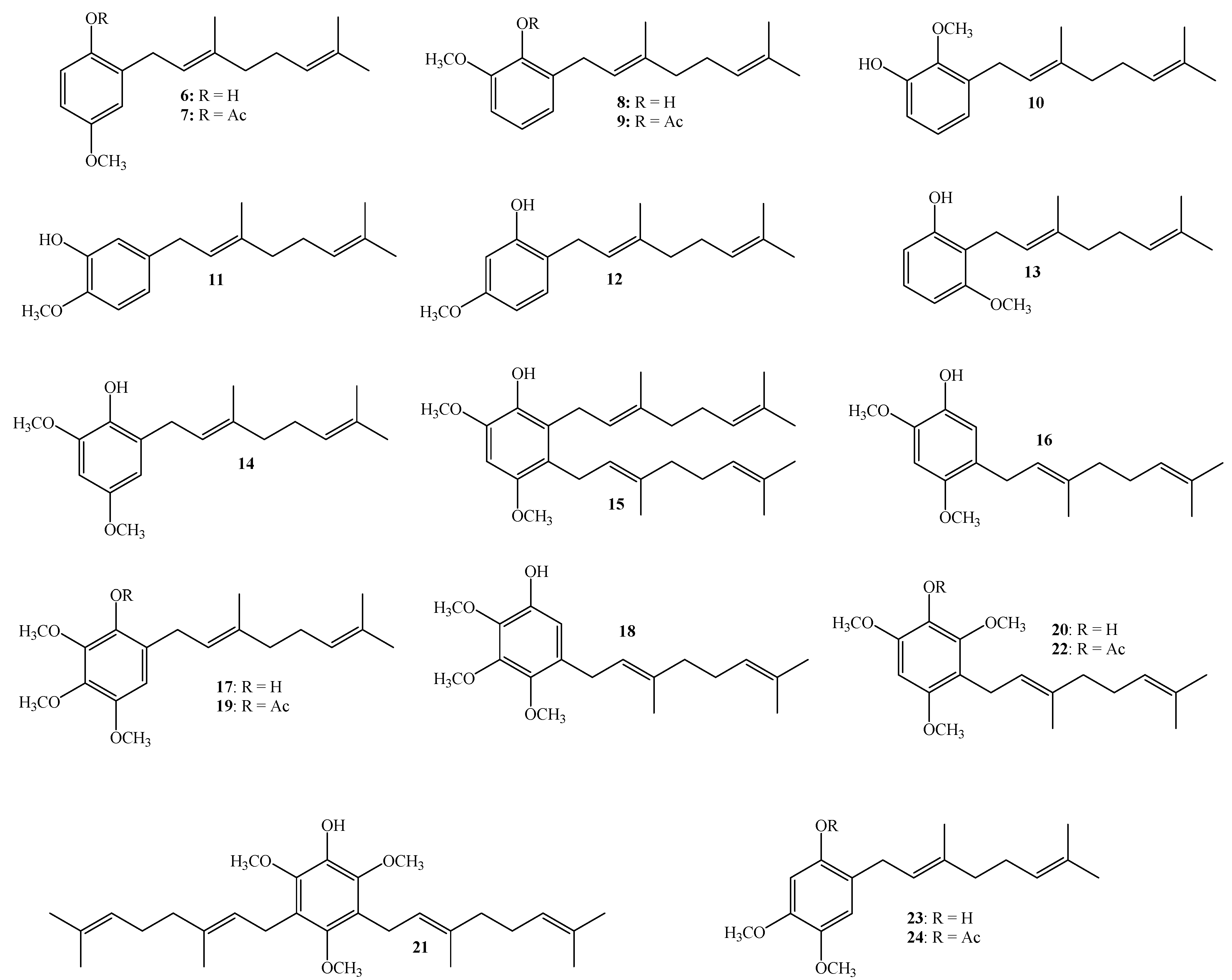
3.2.3. Synthesis of Geranylated Methoxyphenol/Phenylacetate Derivatives
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4-methoxyphenol (6)
Compound
6 was obtained as colorless viscous oil (375.5 mg, 18%) by coupling of 4-methoxyphenol (1.00 g, 8.0 mmol) and geraniol (1.2 g, 7.9 mmol) in acetonitrile (15 mL), saturated with AgNO
3, and BF
3·OEt
2 (0.2 g 1.6 mmol) as catalyst. NMR data of
10 was consistent with that reported in literature [
15].
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4-methoxyphenyl Acetate (7)
Compound
7 was obtained by standard acetylation of compound
10 (90 mg, 0.346 mmol) with Ac
2O (1.0 mL), DMAP (5.0 mg) and pyridine (2.0 mL) in dichloromethane (30 mL). Compound
7 was obtained as colorless viscous oil (99.3 mg, 95%). NMR data of compound
7 was consistent with that found in literature [
15].
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-6-methoxyphenyl Acetate (9)
Compound 9 was obtained by standard acetylation of compound 8 (50 mg, 0.192 mmol) with Ac2O (1.0 mL), DMAP (2.0 mg) and pyridine (1.0 mL) in dichloromethane (20 mL). Compound 13 was obtained as a pale yellow viscous oil (53.9 mg, 93%). Compound 9: IR (cm−1) 2924, 1762, 1496, 1195, 1041; 1H NMR (CDCl3, 400.1 MHz) δ 7.12 (1H, dd, J = 8.0 and 8.0 Hz, H-4), 6.98 (1H, s, H-5), 6.82 (1H, d, J = 8.0 Hz, H-3), 5.23 (1H, t, J = 7.8 Hz, H-2ʹ), 5.09 (1H, t, J = 6.3 Hz, H-6ʹ), 3.81 (3H, s, CH3O), 3.24 (2H, d, J = 7.2 Hz, H-1ʹ), 2.32 (3H, s, CH3CO), 2.10–2.07 (2H, m, H-5ʹ), 2.04–2.03 (2H, m, H-4ʹ), 1.72 (3H, s, CH3-C3ʹ), 1.59 (3H, s, H-8ʹ, 1.56 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 168.9 (C, CH3CO), 151.5 (C, C-6), 145.4 (C, C-1), 135.8 (C, C-3ʹ), 131.5 (C, C-2), 128.3 (C, C-7ʹ), 126.3 (CH, C-4), 125.5 (CH, C-6ʹ), 124.2 (CH, C-2ʹ), 121.5 (CH, C-3), 109.9 (CH, C-5), 55.9 (CH3, CH3O), 39.7 (CH2, C-4ʹ), 29.7 (CH2, C-1ʹ), 28.5 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 20.5 (CH3, CH3CO), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ).
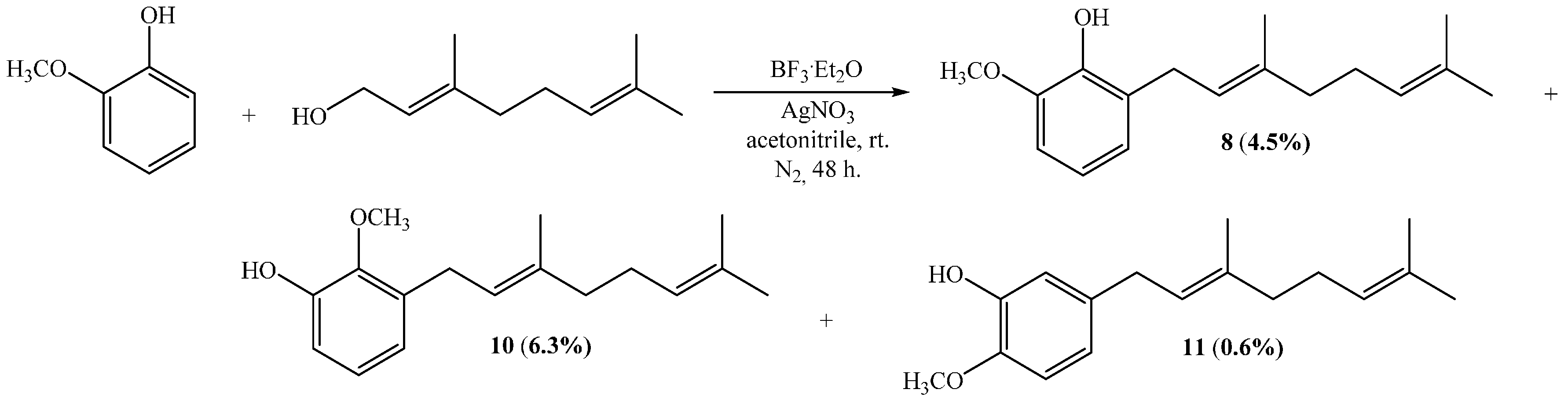
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-6-methoxyphenol (8), (E)-3-(3,7-Dimethylocta-2,6-dienyl)-2-methoxyphenol (10), (E)-5-(3,7-Dimethylocta-2,6-dien-1-yl)-2-methoxyphenol (11)
Guayacol (1.5 g, 12.1 mmol) and geraniol (0.9 g, 6.0 mmol) were reacted in acetonitrile (15 mL), saturated with AgNO
3, using BF
3·OEt
2 (0.45 g, 3.2 mmol) as catalyst. By following the general procedure described above, three fractions were obtained: Fraction I, 70.6 mg of yellow viscous oil (4.5% yield, compound
8); Fraction II, 98.5 mg of yellow viscous oil (6.3% yield, compound
10); and Fraction III, 9.3 mg of yellow viscous oil (0.6% yield, compound
11). NMR data of compounds
8,
10 and
11 were consistent with those reported in literature [
11].
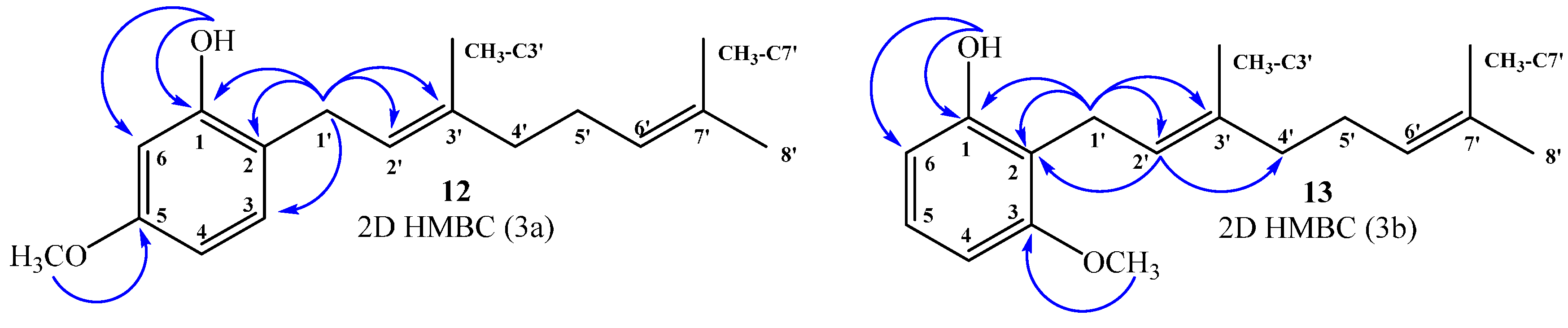
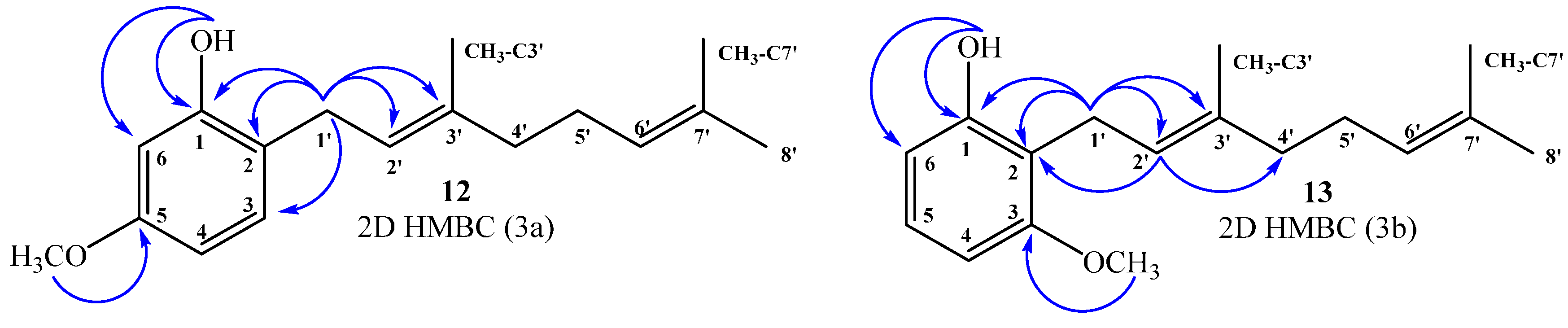
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-5-methoxyphenol (12), (E)-2-(3,7-Dimethylocta-2,6-dienyl)-3-methoxyphenol (13)
Reaction of 3-methoxyphenol (0.8 g, 6.5 mmol) and geraniol (1.0 g, 6.5 mmol) was carried out in acetonitrile (20 mL), saturated with AgNO3, using BF3·OEt2 (0.46 g 3.2 mmol) as catalyst. By following the general procedure described above, two fractions were obtained: Fraction I, 125 mg of reddish viscous oil (7.4% yield, compound 12); and Fraction II, 71.1 mg of reddish viscous oil (4.2% yield, compound 13). Compound 12: 1H NMR (CDCl3, 400.1 MHz) δ 6.99 (1H, d, J = 8.0 Hz, H-3), 6.44 (1H, dd, J = 2.5 and 8.0 Hz, H-4), 6.42 (1H, d, J = 2.5 Hz, H-6), 5.31 (1H, t, J = 7.2 Hz, H-2ʹ), 5.25 (1H, s, OH), 5.08 (1H, t, J = 5.9 Hz, H-6ʹ), 3.76 (3H, s, CH3O), 3.31 (2H, d, J = 7.2 Hz, H-1ʹ), 2.13–2.12 (2H, m, H-5ʹ), 2.09–2.06 (2H, m, H-4ʹ), 4.77 (3H, s, CH3-C3ʹ), 1.69 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 159.4 (C, C-5), 155.4 (C, C-1), 138.4 (C, C-3ʹ), 132.0 (C, C-7ʹ), 130.3 (CH, C-3), 123.8 (CH, C-6ʹ), 122.0 (CH, C-2ʹ), 118.9 (C, C-2), 106.1 (CH, C-4), 102.0 (CH, C-6), 55.3 (CH3, CH3O), 39.7 (CH2, C-4ʹ), 29.2 (CH2, C-1ʹ), 26.4 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ); HRMS m/z 261.1785 (calcd for C17H24O2, 261.1776). Compound 13: 1H NMR (CDCl3, 400.1 MHz) δ 7.06 (1H, dd, J = 8.2 and 8.2 Hz, H-5), 6.49 (2H, d, J = 8.2 Hz, H-4 and H-6), 5.32 (1H, s, OH), 5.24 (1H, t, J = 7.0 Hz, H-2ʹ), 5.06 (1H, t, J = 6.6 Hz, H-6ʹ), 3.81 (3H, s, CH3O), 3.43 (2H, d, J = 7.0 Hz, H-1ʹ), 2.11–2.08 (2H, m, H-5ʹ), 2.06–2.04 (2H, m, H-4ʹ), 1.81 (3H, s, CH3-C3ʹ), 1.68 (3H, s, H-8ʹ), 1.59 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 157.9 (C, C-3), 155.6 (C, C-1), 138.1 (C, C-3ʹ), 131.8 (C, C-7ʹ), 127.1 (CH, C-5), 123.9 (CH, C-6ʹ), 121.9 (CH, C-2ʹ), 115.2 (C, C-2), 108.1 (CH, C-6), 103.1 (CH, C-4), 55.8 (CH3, CH3O), 39.7 (CH2, C-4ʹ), 26.4 (CH2, C-5ʹ), 25.6 (CH3, C-8ʹ), 22.2 (CH2, C-1ʹ), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ); HRMS m/z 259.17151 (calcd for C17H24O2, 259.1776).
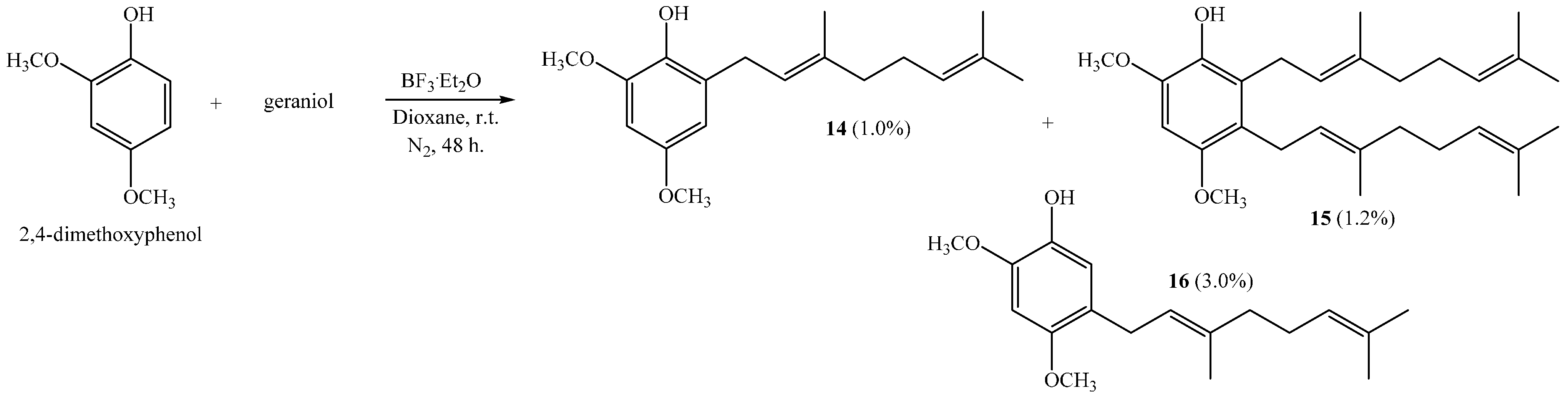
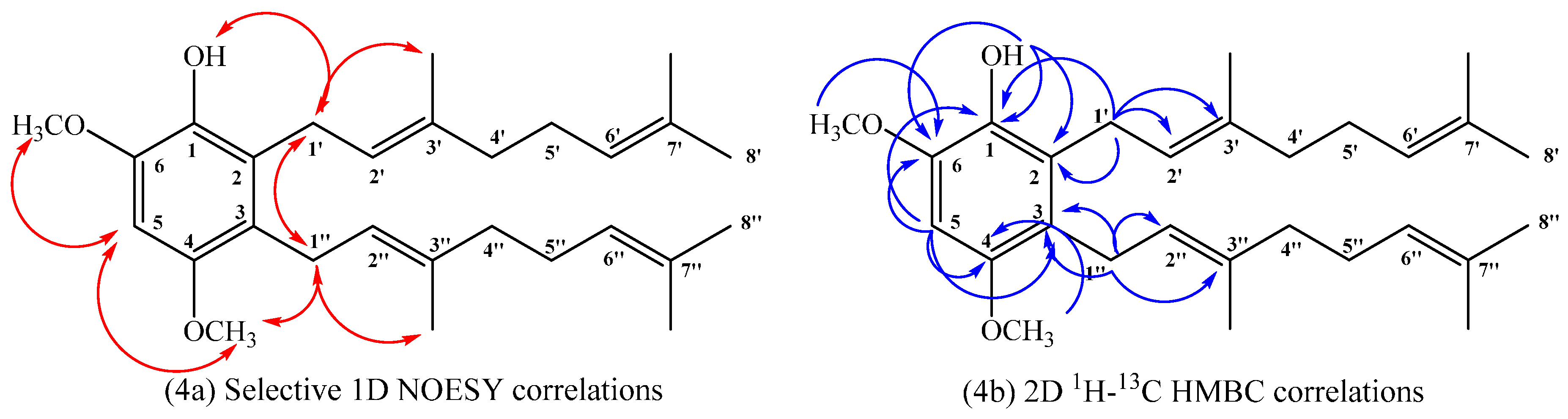
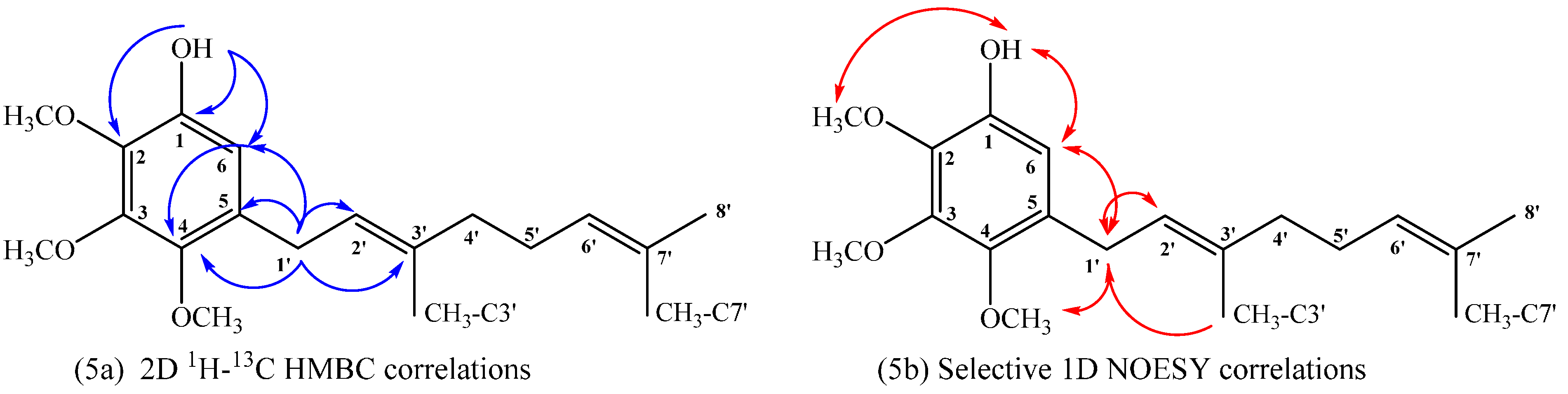
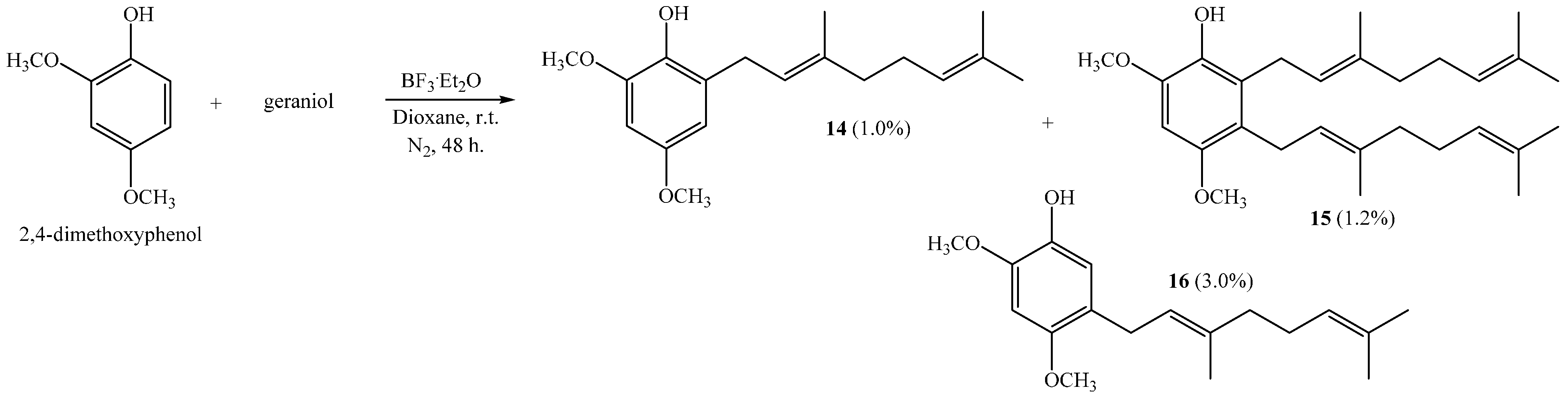
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4,6-dimethoxyphenol (14), 2,3-bis((E)-3,7-Dimethylocta-2,6-dienyl)-4,6-dimethoxyphenol (15), (E)-5-(3,7-Dimethylocta-2,6-dien-1-yl)-2,4-dimethoxyphenol (16)
Reaction of 2,4-dimethoxyphenol (0.807 g, 5.2 mmol) and geraniol (0.802 g, 5.2 mmol) was carried out in dioxane (20 mL) with BF
3·OEt
2 (0.31 g, 2.2 mmol) as catalyst. By following the general procedure described above, three fractions were obtained: Fraction I, 15.0 mg of viscous light brown oil (1.0% yield, compound
14); Fraction II, 28.0 mg of viscous light brown oil (1.2% yield, compound
15); Fraction III, 45.0 mg of viscous dark brown oil (3.0% yield, compound
16). Compound
14: IR (cm
−1): 3556, 2964, 2919, 2854, 1613, 1497, 1466, 1432, 1376, 1227, 1197, 1148, 1089, 1055, 939, 829;
1H NMR (CDCl
3, 400.1 MHz) δ 6.36 (1H, d,
J = 2.6 Hz, H-5), 6.30 (1H, d,
J = 2.6 Hz, H-3), 5.33 (1H, t,
J = 7.2 Hz, H-2ʹ), 5.28 (1H, s, OH), 5.11 (1H, t,
J = 7.7 Hz, H-6ʹ), 3.86 (3H, s, C
H3O-C6), 3.75 (3H, s, C
H3O-C4), 3.35 (2H, d,
J = 7.2 Hz, H-1ʹ), 2.11–2.08 (2H, m, H-5ʹ), 2.06–2.03 (2H, m, H-4ʹ), 1.72 (3H, s, CH
3 C-3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s, CH
3 C-7ʹ);
13C NMR (CDCl
3, 100.6 MHz) δ 152.9 (C, C-4), 146.7 (C, C-6), 137.4 (C, C-1), 136.5 (C, C-3ʹ), 131.4 (C, C-7ʹ), 127.5 (C, C-2), 124.3 (CH, C-6ʹ), 122.0 (CH, C-2ʹ), 105.5 (CH, C-3), 96.7 (CH, C-5), 56.0 (CH
3,
CH
3O-C6), 55.7 (CH
3,
CH
3O-C4), 39.8 (CH
2, C-4ʹ), 28.1 (CH
2, C-1ʹ), 26.7 (CH
2, C-5ʹ), 25.7 (CH
3, C-8ʹ), 17.7 (CH
3,
CH
3-C7ʹ), 16.1 (CH
3,
CH
3-C3ʹ); MS
m/
z 290 (M
+) (100), 205 (24), 189 (19), 168 (65) ((M
+ + H − 123), (C
9H
15·)), 167 (39), 166 (45), 161 (29), 69 (18), 41 (21). NMR data of compound
14 was consistent with that in the literature [
6]. Compound
15: IR (cm
−1): 3556, 2965, 2925, 2854, 1616, 1485, 1451, 1437, 1376, 1341, 1237, 1202, 1089, 941, 855;
1H NMR (CDCl
3, 400.1 MHz) δ 6.42 (1H, s, H-5), 5.31 (1H, s, OH), 5.12–5.06 (4H, m, H-2ʹ, H-2ʹʹ, H-6ʹ and H-6ʹʹ), 3.87 (3H, s, C
H3O-C6), 3.77 (3H, s, C
H3O-C4), 3.39 (2H, d,
J = 6.0 Hz, H-1ʹ), 3.31 (2H, d,
J = 6.0 Hz, H-1ʹʹ), 2.07–2.05 (4H, m, H-5ʹ and H-5ʹʹ), 2.03–1.98 (4H, m, H-4ʹ and H-4ʹʹ), 1.76 (3H, s, C
H3-C3ʹ), 1.74 (3H, s, C
H3-C3ʹ), 1.66 (6H, s, H-8ʹ and H-8ʹʹ), 1.58 (6H, s, C
H3-C7ʹ and C
H3-C7ʹʹ);
13C NMR (CDCl
3, 100.6 MHz) δ 150.7 (C, C-4), 144.4 (C, C-6), 137.9 (C, C-1), 135.2 (C, C-3ʹ), 134.4 (C, C-3ʹʹ), 131.2 (C, C-7ʹʹ and C-7ʹ), 127.3 (C, C-2), 124.4 (CH, C-6ʹ and C-6ʹʹ), 123.8 (CH, C-2ʹ), 122.8 (CH, C-2ʹʹ), 122.1 (C, C-3), 95.3 (C, C-5), 56.9 (CH
3,
CH
3O-C4), 56.1 (CH
3,
CH
3O-C6), 39.7 (CH
2, C-4ʹ and C-4ʹʹ), 26.7 (CH
2, C-5ʹ and C-5ʹʹ), 25.6 (CH
3, C-8ʹ and C-8ʹʹ), 25.3 (CH
2, C-1ʹʹ), 24.6 (CH
2, C-1ʹ), 17.6 (CH
3,
CH
3-C7ʹ and
CH
3-C7ʹʹ), 16.2 (CH
3,
CH
3-C3ʹ), 16.1 (CH
3,
CH
3-C3ʹʹ); MS
m/
z 426 (M
+) (74), 302 (16), 259 (32), 220 (40), 219 (100), 201 (41), 187 (63), 167 (19), 69 (67), 41 (48); HRMS
m/
z 427.3141 (calcd for C
28H
42O
3, 427.3134). Compound
16: IR (cm
−1) 2924, 1602, 1512, 1194, 1042;
1H NMR (CDCl
3, 400.1 MHz) δ 6.68 (1H, s, H-6), 6.54 (1H, s, H-3), 5.51 (1H, s, OH), 5.28 (1H, t,
J = 7.1 Hz, H-2ʹ), 5.11 (1H, t,
J = 6.9 Hz, H-6ʹ), 3.82 (3H, s, C
H3O-C2), 3.76 (3H, s, C
H3O-C4), 3.26 (2H, d,
J = 7.2 Hz, H-1ʹ), 2.12–2.08 (2H, m, H-5ʹ), 2.06–2.02 (2H, m, H-4ʹ), 1.70 (3H, s,
CH
3-C3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s,
CH
3-C7ʹ);
13C NMR (CDCl
3, 100.6 MHz) δ 151.7 (C, C-4), 144.1 (C, C-1), 140.1 (C, C-2), 136.0 (C, C-3ʹ), 131.4 (C, C-7ʹ), 124.3 (CH, C-6ʹ), 122.8 (CH, C-2ʹ), 120.9 (C, C-5), 112.8 (CH, C-6), 99.3 (CH, C-3), 56.8 (CH
3,
CH
3O-C2), 56.1 (CH
3,
CH
3O-C4), 39.8 (CH
2, C-4ʹ), 27.7 (CH
2, C-1ʹ), 26.8 (CH
2, C-5ʹ), 25.7 (CH
3, C-8ʹ), 17.7 (CH
3,
CH
3-C7ʹ), 16.1 (CH
3,
CH
3-C3ʹ); MS
m/
z 290 (M
+) (100), 221 (15), 207 (12), 189 (39), 178 (22), 167 (84) ((M
+ − 123), (C
9H
15·)), 161 (68), 129 (26), 69 (24), 41 (27).
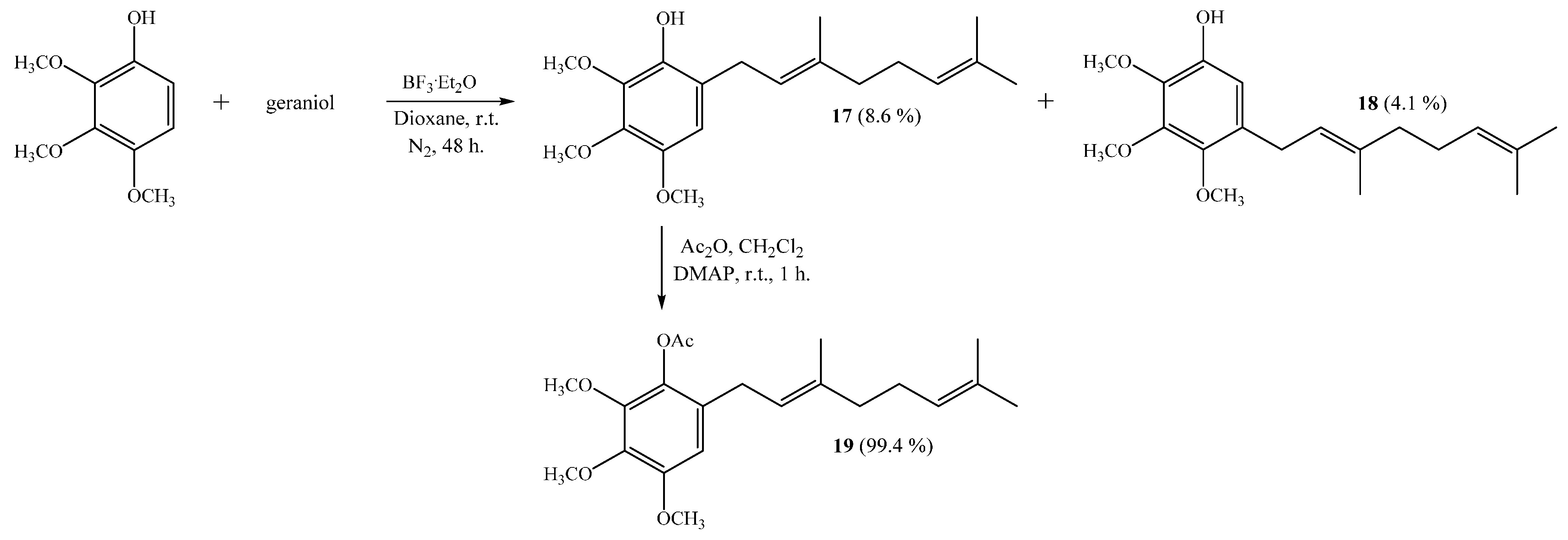
(E)-6-(3,7-Dimethylocta-2,6-dienyl)-2,3,4-trimethoxyphenol (17) and (E)-5-(3,7-Dimethylocta-2,6-dien-1-yl)-2,3,4-trimethoxyphenol (18)
Coupling of 2,3,4-trimethoxyphenol (1.0 g, 5.5 mmol) and geraniol (0.85 g, 5.5 mmol) was carried out in dioxane (20 mL) with BF
3·OEt
2 (0.46 g, 3.2 mmol) as catalyst. By following the general procedure described above, two fractions were obtained: Fraction I, 150.5 mg of viscous light brown oil (8.6% yield, compound
17); Fraction II, 71.0 mg of viscous dark brown oil (4.1% yield, compound
18). Compound
17: IR (cm
−1) 3446; 2966; 1497; 1464; 1125; 1072;
1H NMR (CDCl
3, 400.1 MHz) δ 6.44 (1H, s, H-5), 5.47 (1H, s, OH), 5.32 (1H, t,
J = 6.8 Hz, H-2ʹ), 5.11 (1H, t,
J = 6.6 Hz, H-6ʹ), 3.95 (3H, s, C
H3O-C2), 3.87 (3H, s, C
H3O-C3), 3.79 (3H, s, C
H3O-C4), 3.31 (2H, d,
J = 7.2 Hz, H-1ʹ), 2.11–2.09 (2H, m, H-5ʹ), 2.07–2.04 (2H, m, H-4ʹ), 1.72 (3H, s, CH
3 C-3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s, CH
3 C-7ʹ);
13C NMR (CDCl
3, 100.6 MHz) δ 146.1 (C, C-4), 140.8 (C, C-1), 140.0 (C, C-2 and C-3), 136.6 (C, C-3ʹ), 131.4 (C, C-7ʹ), 124.2 (CH, C-6ʹ), 122.0 (CH, C-2ʹ), 121.6 (CH, C-6), 108.2 (C, C-5), 61.2 (CH
3,
CH
3O-C2), 60.9 (CH
3,
CH
3O-C3), 56.6 (CH
3,
CH
3O-C4), 39.7 (CH
2, C-4ʹ), 27.9 (CH
2, C-1ʹ), 26.7 (CH
2, C-5ʹ), 25.7 (CH
3, C-8ʹ), 17.7 (CH
3,
CH
3-C7ʹ), 16.1 (CH
3,
CH
3-C3ʹ); MS
m/
z 320 (M
+) (71), 235 (15), 198 (18), 197 (100) ((M
+ − 123), (C
9H
15·)), 181 (12), 159 (12), 69 (9), 41 (12). NMR data of compound
17 was consistent with that reported in the literature [
6].
Compound 18: IR (cm−1) 3421, 2966, 1590, 1487, 1465, 1198, 1110; 1H NMR (CDCl3, 400.1 MHz) δ 6.50 (1H, s, H-6), 5.46 (1H, s, OH), 5.25 (1H, t, J = 6.2 Hz, H-2ʹ),5.10 (1H, bt, J = 6.8 Hz, H-6ʹ), 3.93 (3H, s, CH3O-C3), 3.92 (3H, s, CH3O-C2), 3.78 (3H, s, CH3O-C4), 3.27 (2H, d, J = 7.2 Hz, H-1ʹ), 2.11–2.08 (2H, m, H-5ʹ), 2.05–2.02 (2H, m, H-4ʹ), 1.70 (3H, s, CH3-C3ʹ), 1.68 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 145.9 (C, C-3), 144.9 (C, C-4), 144.5 (C, C-1), 138.0 (C, C-2), 136.2 (C, C-3ʹ), 131.5 (C, C-7ʹ), 130.6 (C, C-5), 124.2 (CH, C-6ʹ), 122.5 (CH, C-2ʹ), 109.5 (CH, C-6), 61.2 (CH3, CH3O-C3), 61.0 (CH3, CH3O-C2), 60.7 (CH3, CH3O-C4), 39.7 (CH2, C-4ʹ), 27.8 (CH2, C-1ʹ), 26.6 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.0 (CH3, CH3-C3ʹ); MS m/z 320 (M+) (100), 219 (33), 206 (26), 197 (75) ((M+ − 123), (C9H15·)), 183 (18), 159 (49), 69 (25), 41 (27); HRMS m/z 321.1994 (calcd for C19H28O4, 321.1988).
(E)-6-(3,7-Dimethylocta-2,6-dienyl)-2,3,4-trimethoxyphenyl Acetate (19)
Standard acetylation of compound 17 (48 mg, 0.150 mmol) with Ac2O (1.08 g, 10.6 mmol), DMAP (3.0 mg) and pyridine (1.0 mL) in dichloromethane (20 mL) gives compound 19 as a viscous light brown oil (54 mg, 99.4% yield). Compound 19: IR (cm−1) 2935, 1766, 1587, 1492, 1463, 1124, 1071; 1H NMR (CDCl3, 400.1 MHz) δ 6.49 (1H, s, H-5), 5.22 (1H, t, J = 7.0 Hz, H-2ʹ), 5.10 (1H, t, J = 6.4 Hz, H-6ʹ), 3.87 (3H, s, CH3O-C3), 3.86 (3H, s, CH3O-C2), 3.83 (3H, s, CH3O-C4), 3.18 (2H, d, J = 7.2 Hz, H-1ʹ), 2.31 (3H, s, CH3CO), 2.11–2.07 (2H, m, H-5ʹ), 2.05–2.01 (2H, m, H-4ʹ), 1.69 (3H, s, CH3-C3ʹ), 1.67 (3H, s, H-8ʹ), 1.61 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 169.4 (C, COCH3), 151.2 (C, C-4), 145.6 (C, C-2), 140.9 (C, C-3), 136.9 (C, C-1), 135.8 (C, C-6), 131.5 (C, C-7ʹ), 128.9 (C, C-3ʹ), 124.1 (CH, C-6ʹ), 121.4 (CH, C-2ʹ), 107.1 (CH, C-5), 61.0 (CH3, CH3O-C2), 60.9 (CH3, CH3O-C3), 56.1 (CH3, CH3O-C4), 39.7 (CH2, C-4ʹ), 28.6 (CH2, C-1ʹ), 26.7 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 20.4 (CH3, COCH3), 17.7 (CH3, CH3-C7ʹ), 16.2 (CH3, CH3-C3ʹ); MS m/z 362 (M+) (11), 320 (32), 235 (16), 221 (10), 197 (100) ((M+ − 123), (C9H15·)), 181 (9), 159 (10), 69 (10), 41 (13).
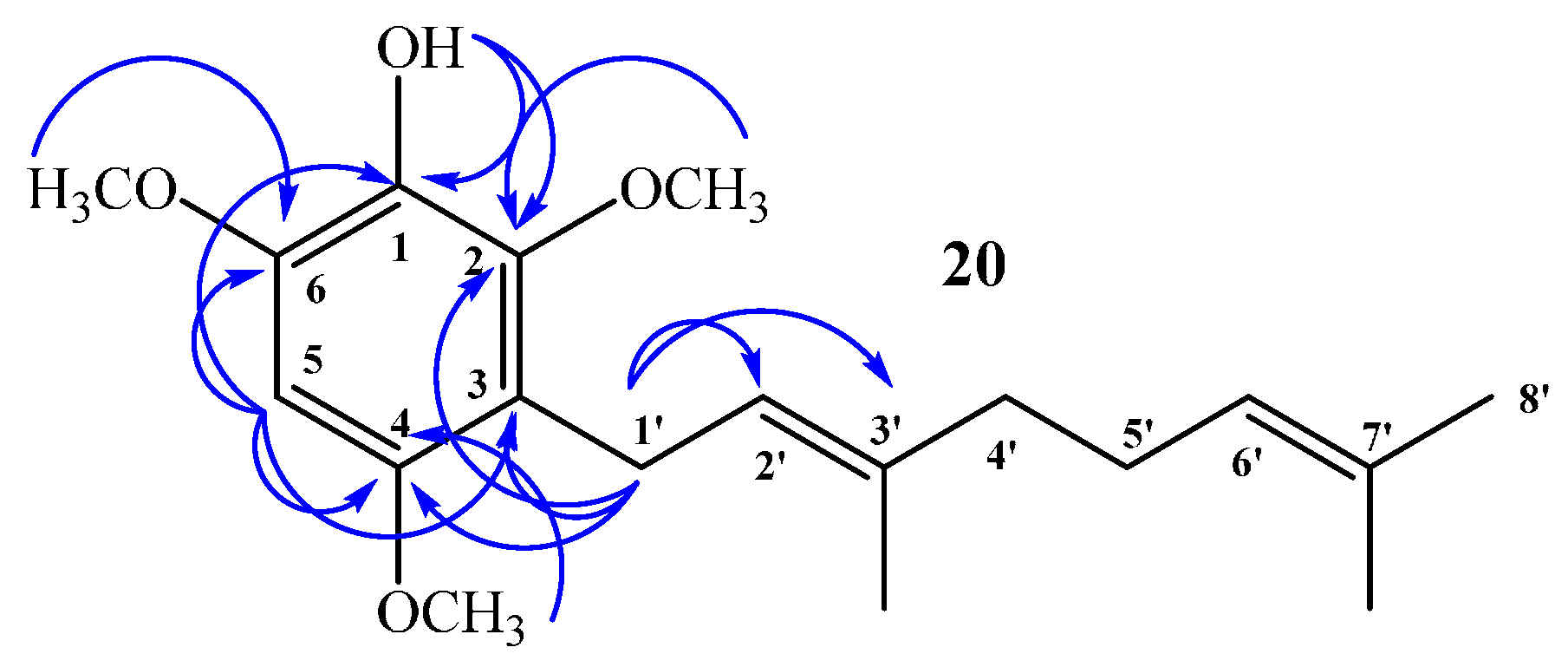
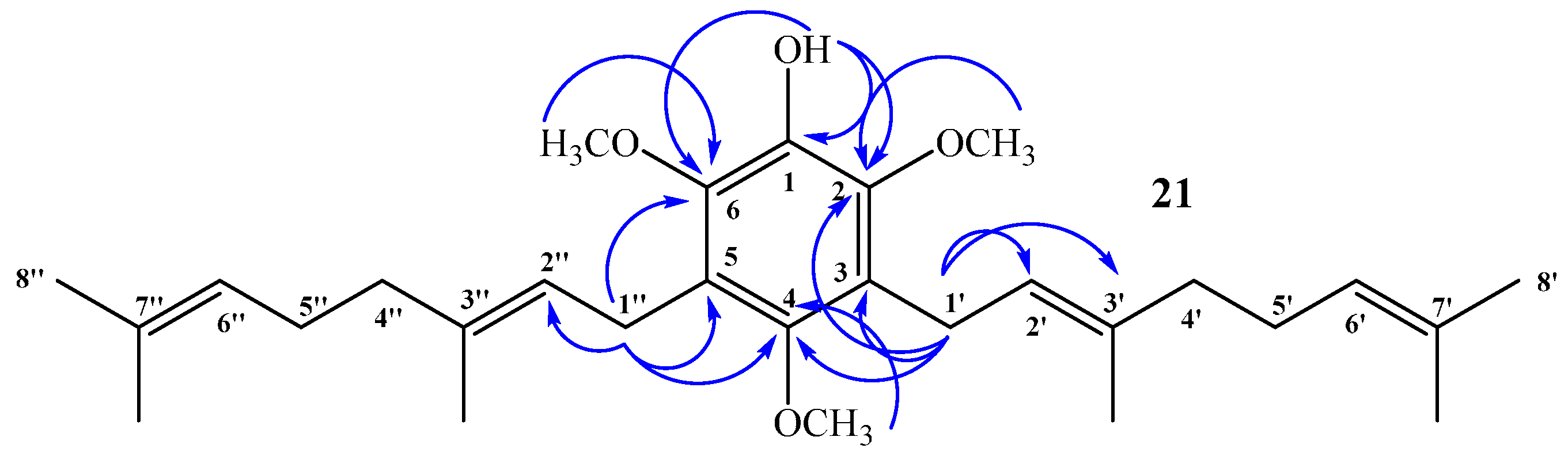
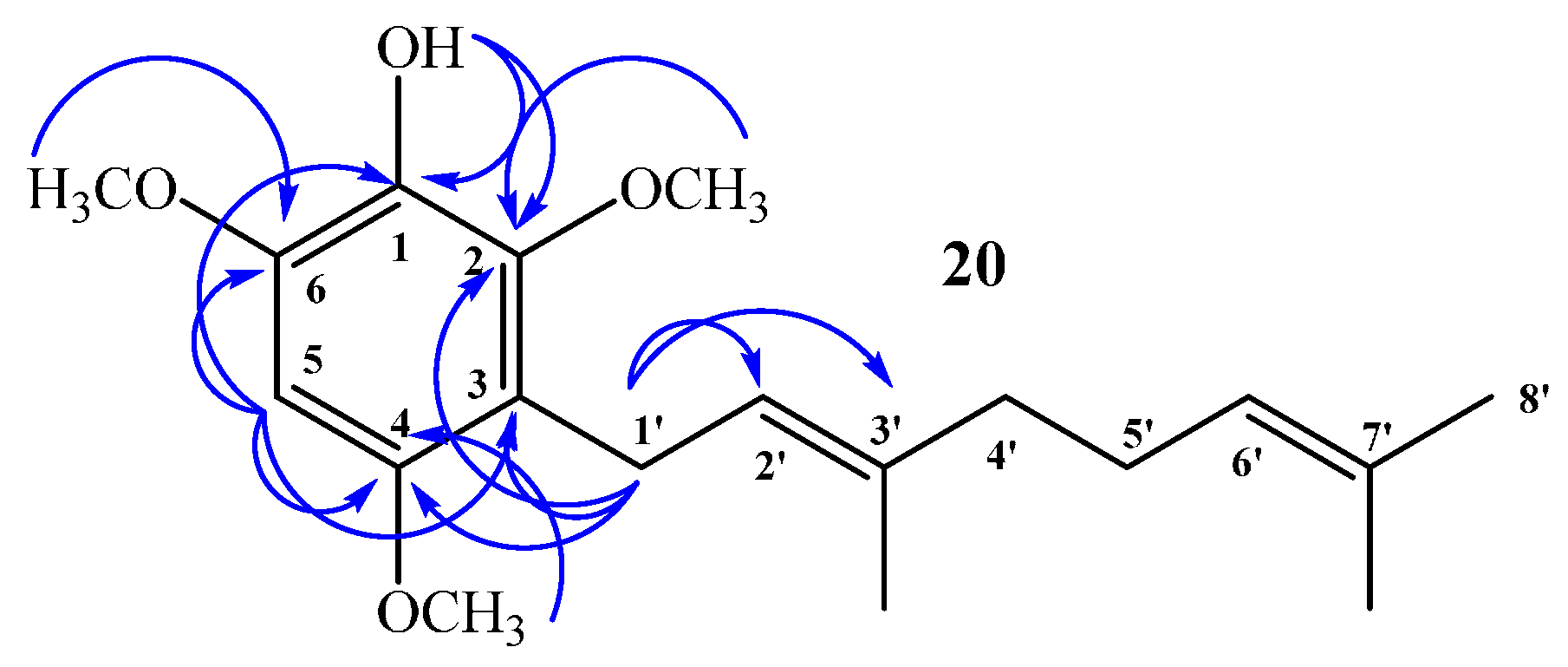
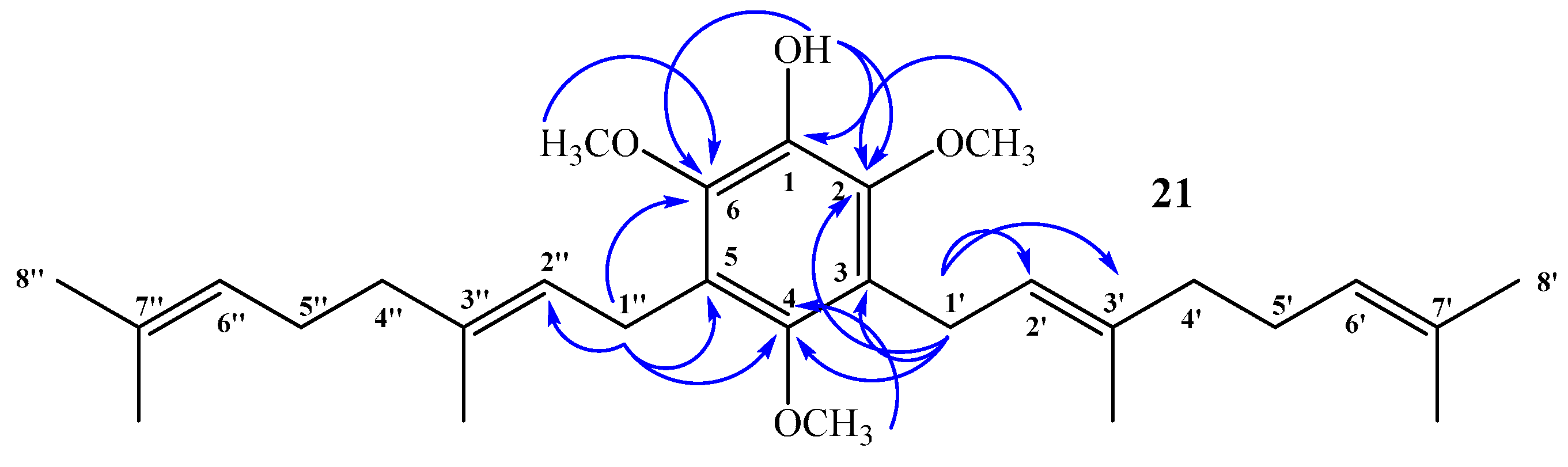
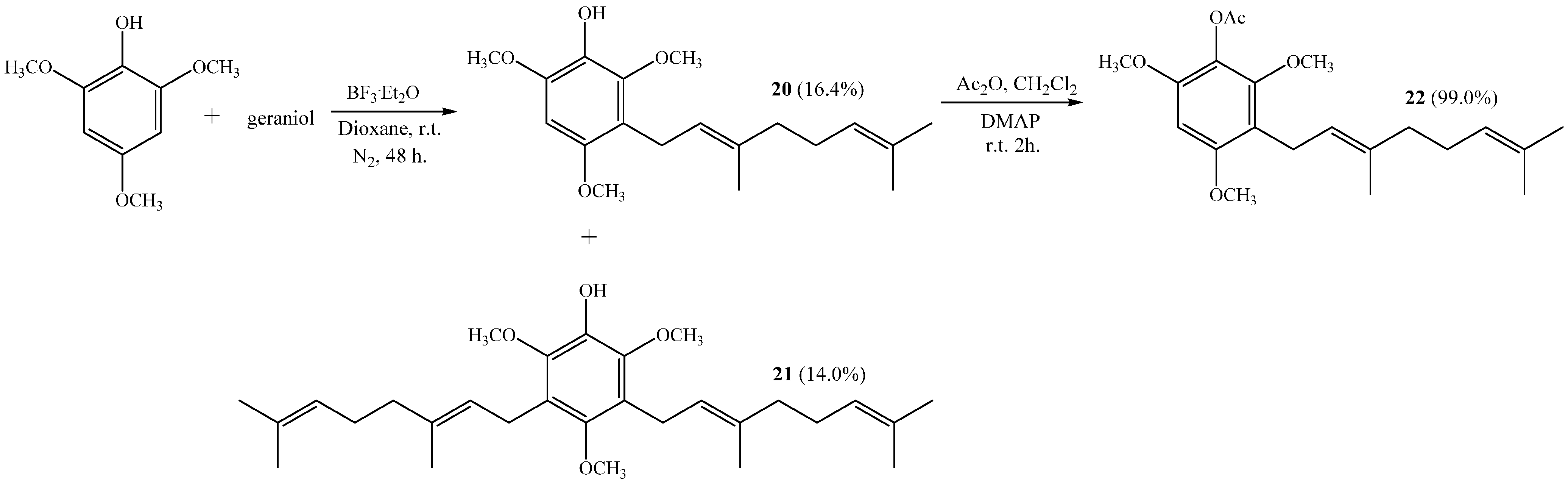
(E)-3-(3,7-Dimethylocta-2,6-dien-1-yl)-2,4,6-trimethoxyphenol (20) and 3,5-bis((E)-3,7-Dimethylocta-2,6-dien-1-yl)-2,4,6-trimethoxyphenol (21)
Coupling of 2,4,6-trimethoxyphenol (2.58 g, 14.0 mmol) and geraniol (2.49 g, 16.0 mmol) was carried out in dioxane (20 mL) with BF3·OEt2 (0.90 g, 6.3 mmol) as catalyst. Two fractions were obtained: Fraction I, 735.9 mg of viscous brown oil (16.4% yield, compound 20); Fraction II, 901.8 mg of viscous light brown oil (14.0% yield, compound 21). Compound 20: IR (cm−1) 3447, 2965, 2929, 2838, 1668, 1615, 1500, 1454, 1377, 1345, 1245, 1199, 1105, 910, 872, 801; 1H NMR (CDCl3, 400.1 MHz) δ 6.32 (1H, s, H-5), 5.17 (1H, t, J = 6.2 Hz, H-2ʹ), 5.15 (1H, s, OH), 5.06 (1H, t, J = 6.7 Hz, H-6ʹ), 3.88 (3H, s, CH3O-C6), 3.83 (3H, s, CH3O-C2), 3.78 (3H, s, CH3O-C4), 3.30 (2H, d, J = 6.8 Hz, H-1ʹ), 2.06–2.02 (2H, m, H-5ʹ), 1.98–1.94 (2H, m, H-4ʹ), 1.76 (3H, s, CH3-C3ʹ), 1.64 (3H, s, H-8ʹ), 1.57 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 150.6 (C, C-4), 145.9 (C, C-2), 145.3 (C, C-6), 134.4 (C, C-3ʹ), 133.0 (C, C-1), 131.1 (C, C-7ʹ), 124.4 (CH, C-6ʹ), 123.5 (CH, C-2ʹ), 116.8 (C, C-3), 93.0 (CH, C-5), 60.8 (CH3, CH3O-C2), 56.5 (CH3, CH3O-C4), 56.3 (CH3, CH3O-C6), 39.7 (CH2, C-4ʹ), 26.7 (CH2, C-5ʹ), 25.6 (CH3, C-8ʹ), 22.5 (CH2, C-1ʹ), 17.6 (CH3, CH3-C7ʹ), 16.0 (CH3, CH3-C3ʹ); MS m/z 320 (M+) (77), 251 (22), 237 (16), 221 (31), 219 (33), 197 (100) ((M+ − 123), (C9H15·)), 186 (27), 183 (24), 159 (17), 69 (18), 41 (23). Compound 21: IR (cm−1) 3447, 3086, 2968, 2926, 2856, 1675, 1642, 1604, 1460, 1422, 1376, 1235, 1106, 1031, 920, 835; 1H NMR (CDCl3, 400.1 MHz) δ 5.34 (1H, s, OH), 5.24–5.20 (2H, m, H-2ʹ and H-2ʹʹ), 5.08 (2H, m, H-6ʹ and H-6ʹʹ), 3.81 (6H, s, CH3O-C6 and CH3O-C2), 3.66 (3H, s, CH3O-C4), 3.34 (4H, d, J = 6.6 Hz, H-1ʹ and H-1ʹʹ), 2.07–2.04 (4H, m, H-5ʹ and H-5ʹʹ), 2.01–1.97 (4H, m, H-4ʹ and H-4ʹʹ), 1.77 (6H, s, CH3-C3ʹ and CH3-C3ʹʹ), 1.64 (6H, s, H-8ʹ and H-8ʹʹ), 1.57 (6H, s, CH3-C7ʹ and CH3-C7ʹʹ); 13C NMR (CDCl3, 100.6 MHz) δ 149.5 (C, C-4), 144.4 (C, C-2 and C-6), 139.0 (C, C-1), 134.8 (C, C-3ʹ and C-3ʹʹ), 131.3 (C, C-7ʹ and C-7ʹʹ), 124.4 (C, C-3 and C-5), 124.3 (C, C-6ʹ and C-6ʹʹ), 123.8 (CH, C-2ʹ and C-2ʹʹ), 61.7 (CH3, CH3O-C4), 60.9 (CH3, CH3O-C2 and CH3O-C6), 39.7 (CH2, C-4ʹ and C-4ʹʹ), 26.6 (CH2, C-5ʹ and C-5ʹʹ), 25.6 (CH3, C-8ʹ and C-8ʹʹ), 23.5 (CH2, C-1ʹ and C-1ʹʹ), 17.6 (CH3, CH3-C7ʹ and CH3-C7ʹʹ), 16.2 (CH3, CH3-C3ʹ and CH3-C3ʹʹ); MS m/z 456 (M+) (33), 333 (22) ((M+ − 123), (C9H15·)), 301 (11), 263 (100), 211 (34), 69 (54), 41 (38); HRMS m/z 457.3247 (calcd for C29H44O4, 457.3240).
(E)-3-(3,7-Dimethylocta-2,6-dienyl)-2,4,6-trimethoxyphenyl Acetate (22)
Standard acetylation of compound 20 (65 mg, 0.18 mmol) with Ac2O (0.5 mL, 5.3 mmol), DMAP (5.0 mg) and pyridine (1.0 mL) in dichloromethane (30 mL) gives compound 22 as a viscous yellow oil (72 mg, 99% yield). Compound 22: IR (cm−1) 3402, 2961, 2970, 2843, 1767, 1607, 1496, 1458, 1375, 1201, 1108, 1024, 927; 1H NMR (CDCl3, 400.1 MHz) δ 6.32 (1H, s, H-5), 5.16 (1H, t, J = 6.4 Hz, H-2ʹ), 5.06 (1H, t, J = 6.8 Hz, H-6ʹ), 3.81 (6H, s, CH3O-C4 and CH3O-C6), 3.74 (3H, s, CH3O-C2), 3.29 (2H, d, J = 6.7 Hz, H-1ʹ), 2.33 (3H, s, COCH3), 2.05–2.02 (2H, m, H-5ʹ), 1.98–1.94 (2H, m, H-4ʹ), 1.75 (3H, s, CH3-C3ʹ), 1.64 (3H, s, H-8ʹ), 1.57 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 169.1 (C, COCH3), 155.8 (C, C-4), 151.3 (C, C-2), 150.2 (C, C-6), 134.5 (C, C-3ʹ), 131.1 (C, C-7ʹ), 127.0 (C, C-1), 124.4 (CH, C-6ʹ), 123.2 (CH, C-2ʹ), 116.4 (C, C-3), 92.5 (CH, C-5), 61.3 (CH3, CH3O-C2), 56.1 (CH3, CH3O-C4), 55.8 (CH3, CH3O-C6), 39.7 (CH2, C-4ʹ), 26.6 (CH2, C-5ʹ), 25.6 (CH3, C-8ʹ), 22.5 (CH2, C-1ʹ), 20.4 (CH3, COCH3), 17.6 (CH3, CH3-C7ʹ), 15.9 (CH3, CH3-C3ʹ).
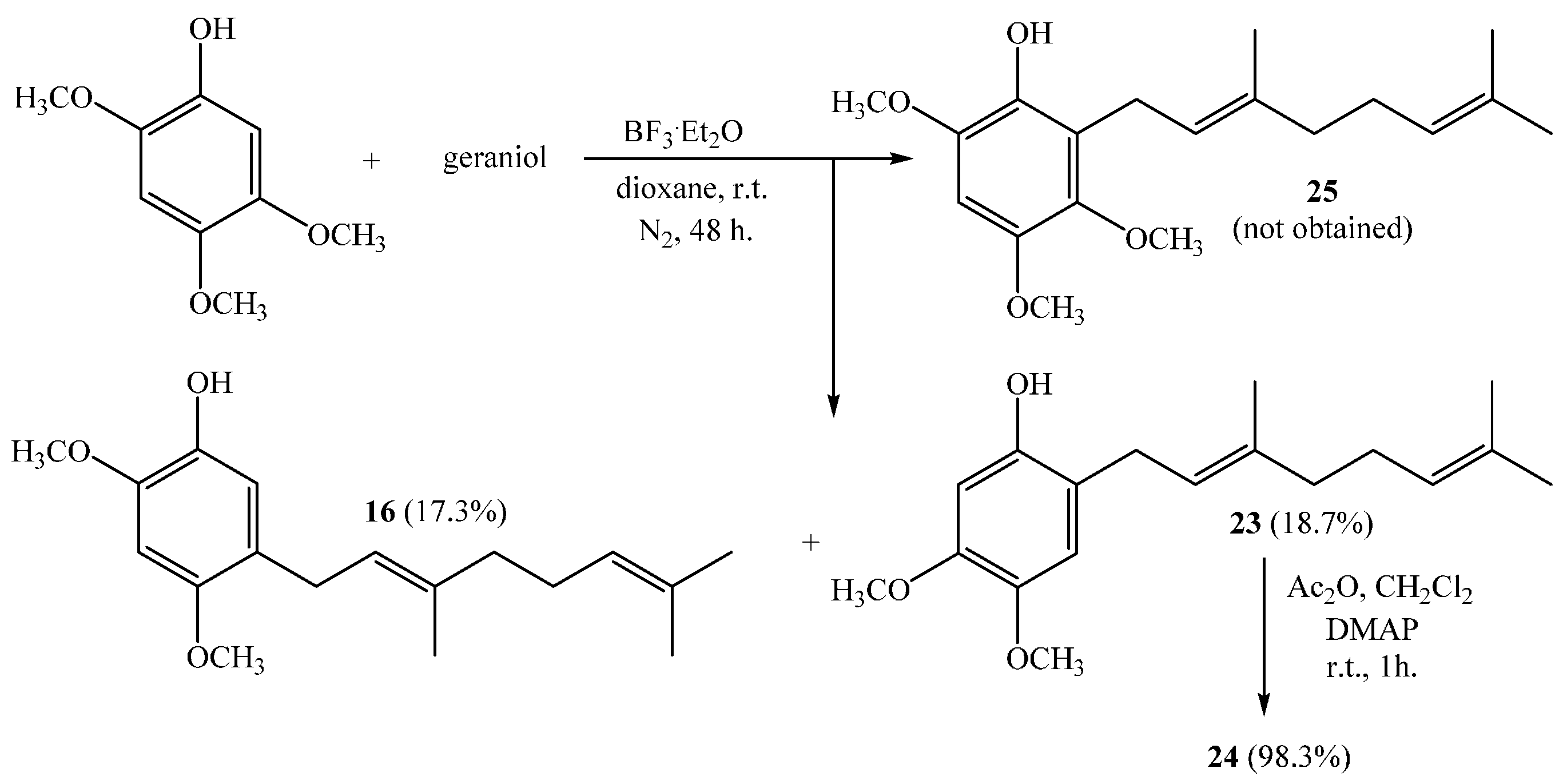
(E)-5-(3,7-Dimethylocta-2,6-dienyl)-2,4-dimethoxyphenol (16), (E)-2-(3,7-Dimethylocta-2,6-dienyl)-4,5-dimethoxyphenol (23)
Reaction of 2,4,5-trimethoxyphenol (2.46 g, 13.3 mmol) and geraniol (2.07 g, 13.3 mmol) was carried out in dioxane (20 mL) with BF3·OEt2 (0.62g, 4.4 mmol) as catalyst. By following the described general procedure of geranylation reaction, two fractions were obtained: Fraction I, 719.6 mg of viscous brown oil (18.7% yield, compound
23); Fraction II, 677.5 mg of viscous light brown oil (17.3% yield, compound
16). Spectroscopic data of compound
16 was consistent with those found above. Compound
23: IR (cm
−1) 2924, 1619, 1512, 1451, 1195;
1H NMR (CDCl
3, 400.1 MHz) δ 6.62 (1H, s, H-3), 6.45 (1H, s, H-6), 5.30 (1H, t,
J = 7.1 Hz, H-2ʹ), 5.07 (1H, t,
J = 7.1 Hz, H-6ʹ), 4.93 (1H, s, OH), 3.82 (6H, s, OC
H3 × 2), 3.30 (2H, d,
J = 7.0 Hz, H-1ʹ), 2.09 (4H, m, H-5ʹ and H-4ʹ), 1.78 (3H, s,
CH
3-C3ʹ), 1.68 (3H, s, H-8ʹ), 1.60 (3H, s,
CH
3-C7ʹ);
13C NMR (CDCl
3, 100.6 MHz) δ 148.4 (C, C-1), 148.3 (C, C-5), 142.8 (C, C-4), 138.6 (C, C-3ʹ), 132.0 (C, C-7ʹ), 123.7 (CH, C-6ʹ), 121.9 (CH, C-2ʹ), 117.3 (C, C-2), 113.7 (CH, C-3), 101.2 (CH, C-6), 56.6 (CH
3,
CH
3O-C5), 55.9 (CH
3,
CH
3O-C4), 39.6 (CH
2, C-4ʹ), 29.6 (CH
2, C-1ʹ), 26.4 (CH
2, C-5ʹ), 25.6 (CH
3, C-8ʹ), 17.7 (CH
3,
CH
3-C7ʹ), 16.2 (CH
3,
CH
3-C3ʹ); MS
m/
z 290 (M
+) (45), 205 (19), 167 (100) ((M
+ − 123), (C
9H
15·)), 69 (11), 41 (13). NMR data of compound
27 was consistent with those in the literature [
6,
10].
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4,5-dimethoxyphenyl Acetate (24)
Standard acetylation of compound 23 (50.8 mg, 0.175 mmol) with Ac2O (100 µL), DMAP (3.0 mg) and pyridine (1.0 mL) in dichloromethane (20 mL) gives compound 24 as a dark brown oil (57.2 mg, 98.3% yield). Compound 24: IR (cm−1) 2973, 2929, 2854, 1765, 1616, 1514, 1448, 1369, 1206, 1101, 1015, 907, 855; 1H NMR (CDCl3, 400.1 MHz) δ 6.70 (1H, s, H-3), 6.56 (1H, s, H-6), 5.23 (1H, t, J = 6.9 Hz, H-2ʹ), 5.10 (1H, t, J = 6.4 Hz, H-6ʹ), 3.84 (3H, s, CH3O-C4), 3.83 (3H, s, CH3O-C5), 3.17 (2H, d, J = 7.0 Hz, H-1ʹ), 2.29 (3H, s, COCH3), 2.09–2.05 (2H, m, H-5ʹ), 2.05–2.04 (2H, m, H-4ʹ), 1.69 (3H, s, CH3-C3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 169.8 (C, COCH3), 147.5 (C, C-5), 146.9 (C, C-4), 141.8 (C, C-1), 136.8 (C, C-3ʹ), 131.5 (C, C-7ʹ), 124.8 (C, C-2), 124.1 (CH, C-6ʹ), 121.8 (CH, C-2ʹ), 112.3 (CH, C-3), 106.1 (CH, C-6), 56.1 (CH3, CH3O-C4), 56.0 (CH3, CH3O-C5), 39.7 (CH2, C-4ʹ), 28.2 (CH2, C-1ʹ), 26.7 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 20.8 (CH3, COCH3), 17.7 (CH3, CH3-C7ʹ), 16.2 (CH3, CH3-C3ʹ); MS m/z 332 (M+) (39), 290 (34), 221 (16), 205 (25), 167 (100) ((M+ − CH3CO − 123), (C9H15·)), 69 (13), 41 (13).
3.2.4. Synthesis of Trimethoxyphenols
In a typical reaction, m-chloroperoxybenzoic acid (mCPBA) is added to a stirred solution of trimethoxybenzaldehyde and sodium bicarbonate in dichloromethane (70 mL). The mixture is stirred at room temperature until the end of the reaction is reached (3 h). Then, the reaction mixture is filtered in a vacuum; the organic phase is washed with NaHCO3 (3 × 50 mL) and water (2 × 50 mL), and dried over anhydrous Na2SO4. The solvent is evaporated under reduced pressure, whereas the crude is re-dissolved in CH2Cl2 (5 mL) and chromatographed on silica gel with petroleum ether/EtOAc of increasing polarity mixtures (50:0→30:20). Trimethoxyphenyl formate is obtained as a dark brown solid. Then, triethylamine (2 mL) is added to a solution of trimethoxyphenyl formate in methanol (50 mL), and the mixture is stirred at room temperature until the end of reaction is verified by TLC (3 h). The solvent is evaporated under reduced pressure; the crude is diluted with ethyl acetate (20 mL) and the organic phase is washed with 5% HCl (2 × 30 mL) and water (2 × 15 mL), and dried over anhydrous Na2SO4. The solvent is evaporated under reduced pressure; the crude is re-dissolved in CH2Cl2 (5 mL) and chromatographed on silica gel with petroleum ether/EtOAc of increasing polarity mixtures (50:0→30:20).
2,3,4-Trimethoxyphenol. mCPBA (3.30 g, 19 mmol) was added to a solution of 2,3,4-trimethoxybenzaldehyde (2.0 g, 10 mmol) and sodium bicarbonate (1.82 g, 21 mmol) in dichloromethane (70 mL). 2,3,4-trimethoxyphenyl formate was obtained as a dark brown solid (1.81 g, 89.2% yield). Triethylamine (2 mL; 1.45 g; 14.0 mmol) was added to a solution of 2,3,4-trimethoxyphenyl formate (1.7 g, 8.0 mmol) in methanol (50 mL). 2,3,4-trimethoxyphenol was obtained as a dark brown oil (1.80 g, 99.6% yield). IR (cm−1) 3421, 2995, 2940, 1601, 1479, 1427, 1360, 1052, 955, 797; 1H NMR (CDCl3, 400.1 MHz) δ 6.62 (1H, d, J = 9.0 Hz, H-6), 6.55 (1H, d, J = 9.0 Hz, H-5), 3.95 (3H, s, CH3O), 3.89 (3H, s, CH3O), 3.80 (3H, s, CH3O); 13C NMR (CDCl3, 100.6 MHz) δ 146.9 (C, C-1), 143.3 (C, C-2), 142.2 (C, C-3), 140.4(C, C-4), 108.5 (CH, C-6), 107.6 (CH, C-5), 61.2 (CH3, CH3O), 60.9 (CH3, CH3O), 55.5 (CH3, CH3O).
2,4,6-Trimethoxyphenol. mCPBA (4.45 g, 25.8 mmol) was added to a solution of 2,4,6-trimethoxybenzaldehyde (3.03 g, 15.4 mmol) and sodium bicarbonate (2.70 g, 32.1 mmol) in dichloromethane (70 mL). 2,4,6-trimethoxyphenyl formate was obtained as a dark brown solid (2.95 g, 90.2% yield). Triethylamine (6 mL, 42.0 mmol) was added to a solution of 2,4,6-trimethoxyphenyl formate (2.95 g, 13.9 mmol) in methanol (75 mL). 2,4,6-trimethoxyphenol was obtained as a dark brown solid (2.44 g, 95.0% yield). mp 60.5–62.9 °C; IR (cm−1) 3422, 3010, 2964, 1626, 1469, 1439, 1361, 1236, 1053, 942, 815; 1H NMR (CDCl3, 400.1 MHz) δ 6.16 (2H, s, H-3 and H-5), 3.83 (6H, s, 2 × CH3O), 3.74 (3H, s, CH3O); 13C NMR (CDCl3, 100.6 MHz) δ 153.0 (C, C-1), 147.2 (C, C-2 and C-6), 128.9 (C, C-4), 91.7 (CH, C-3 and C-5), 56.1 (CH3, 2 × CH3O), 55.6 (CH3, CH3O).
2,4,5-Trimethoxyphenol. mCPBA (5.0 g, 29.0 mmol) was added to a solution of 2,4,5-trimethoxybenzaldehyde (3.01 g, 15.0 mmol) and sodium bicarbonate (1.8 g, 32.0 mmol) in dichloromethane (70 mL). 2,4,5-trimethoxyphenyl formate was obtained as a dark brown solid (2.99 g, 91.6% yield). Triethylamine (4 mL, 28.0 mmol) was added to a solution of 2,4,5-trimethoxyphenyl formate (2.99 g, 14.0 mmol) in methanol (75 mL). 2,4,5-trimethoxyphenol was obtained as a dark brown solid (2.46 g, 94.3% yield). mp 83.5–87.0 °C; IR (cm−1) 3480, 3067, 2996, 1621, 1479, 1441, 1375, 1209, 1027, 823M; 1H NMR (CDCl3, 400.1 MHz) δ 6.61 (1H, s, H-6), 6.57 (1H, s, H-3), 5.29 (1H, s, OH), 3.87 (3H, s, CH3O), 3.84 (6H, s, 2 × CH3O); 13C NMR (CDCl3, 100.6 MHz) δ 144.2 (C, C-2), 143.6 (C, C-5), 143.1 (C, C-1), 141.9 (C, C-4), 104.8 (CH, C-3), 100.1 (CH, C-6).


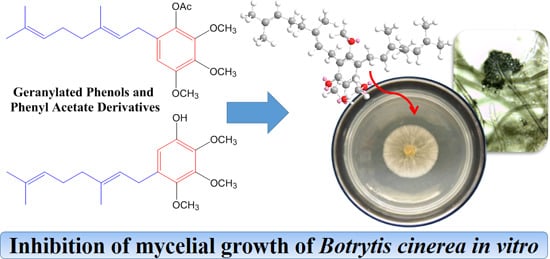
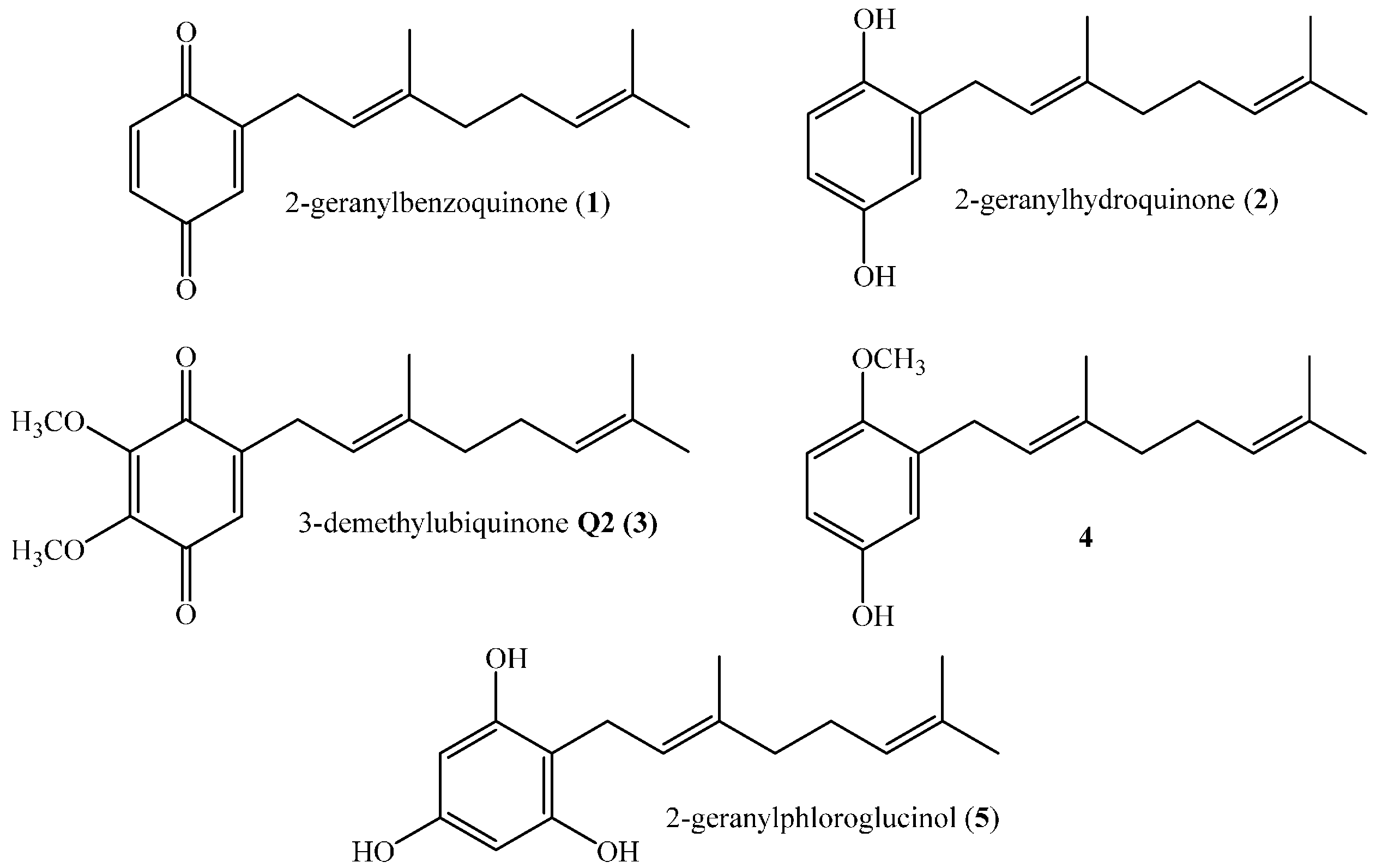
 ,
, 

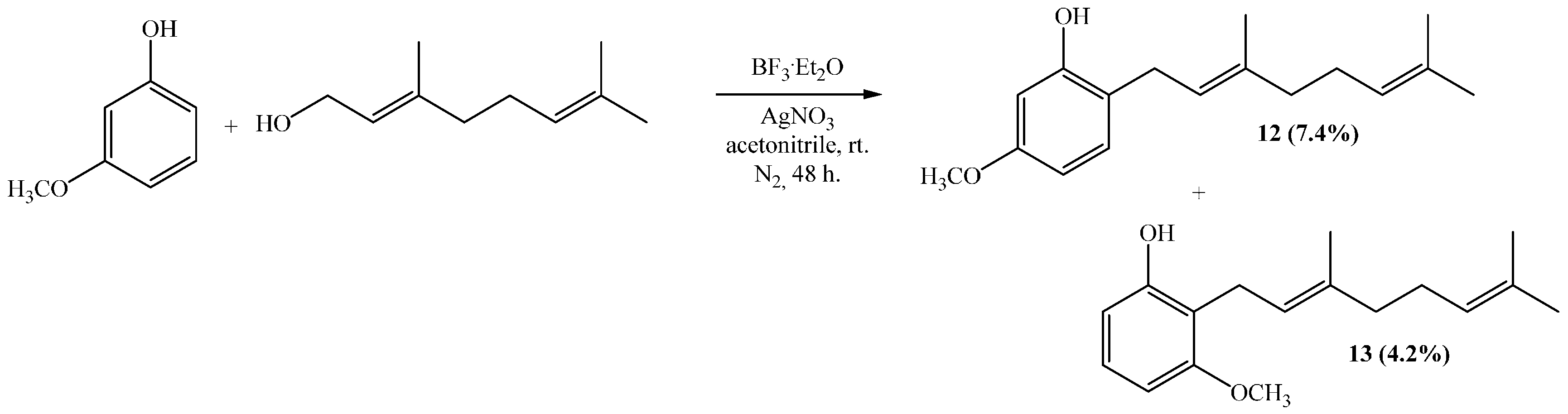



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
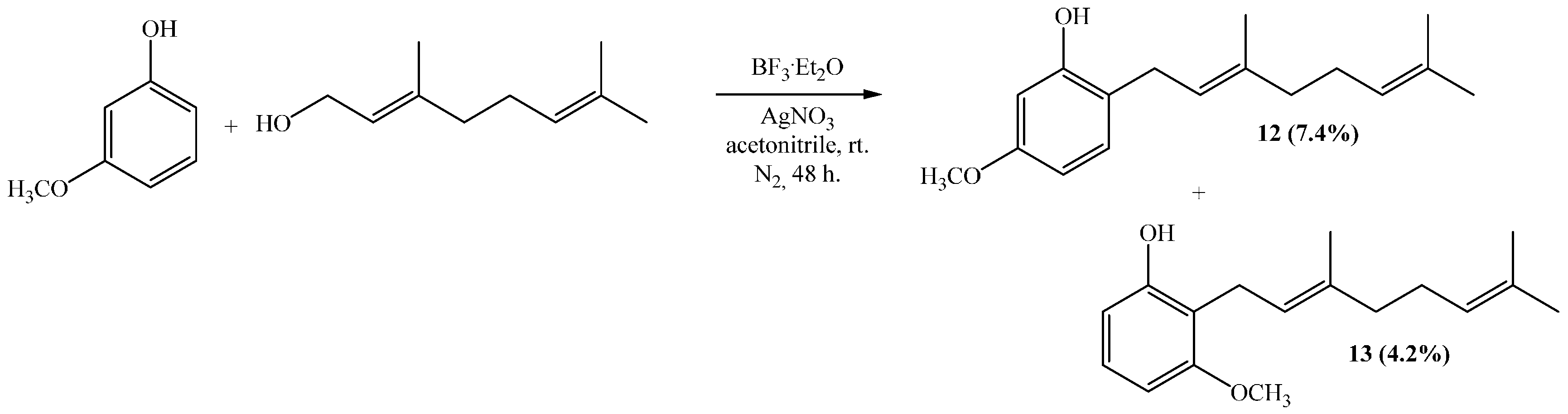{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}