
Overview of Nrf2 as Therapeutic Target in Epilepsy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Overview of Oxidative Stress and Nrf2
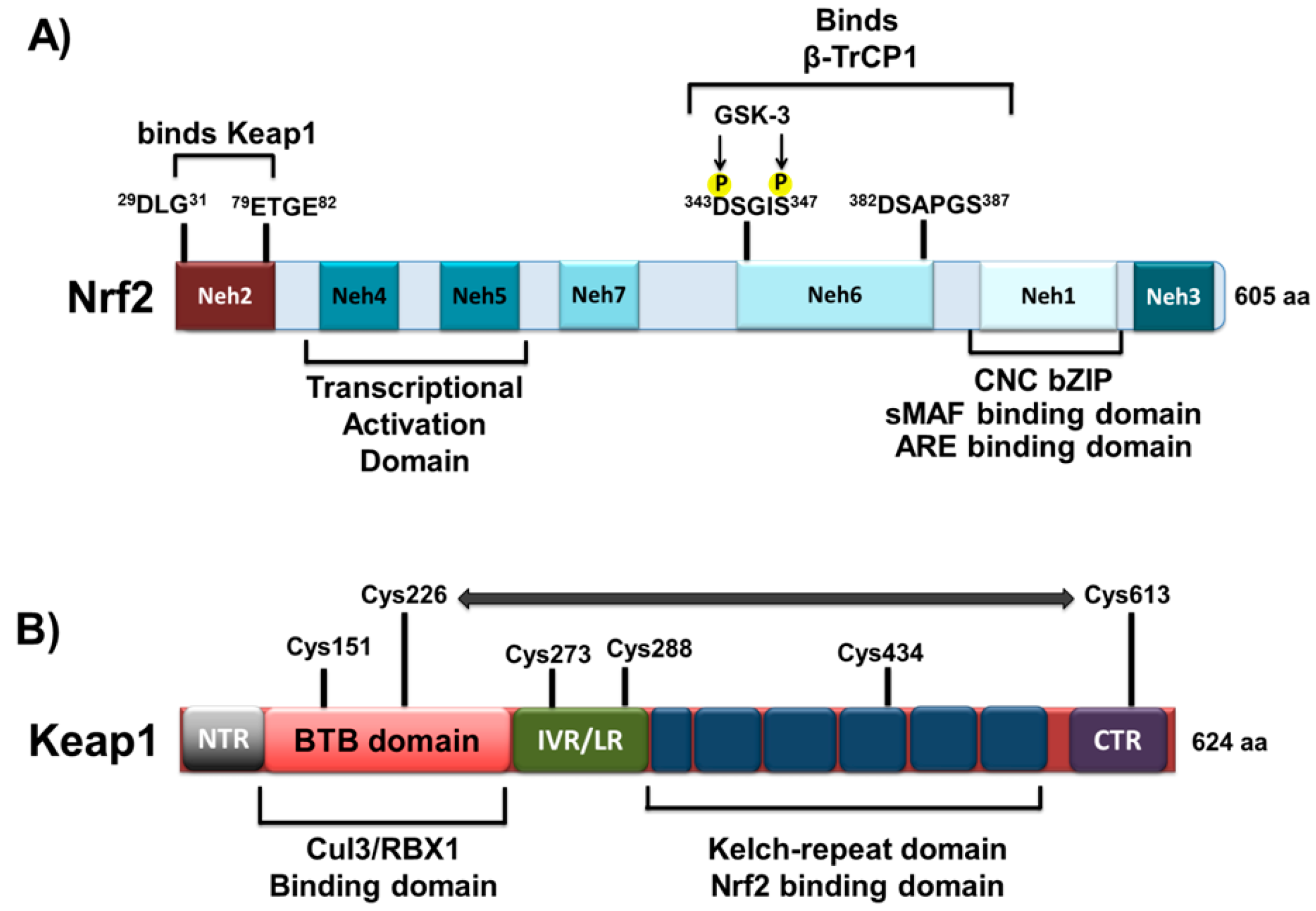
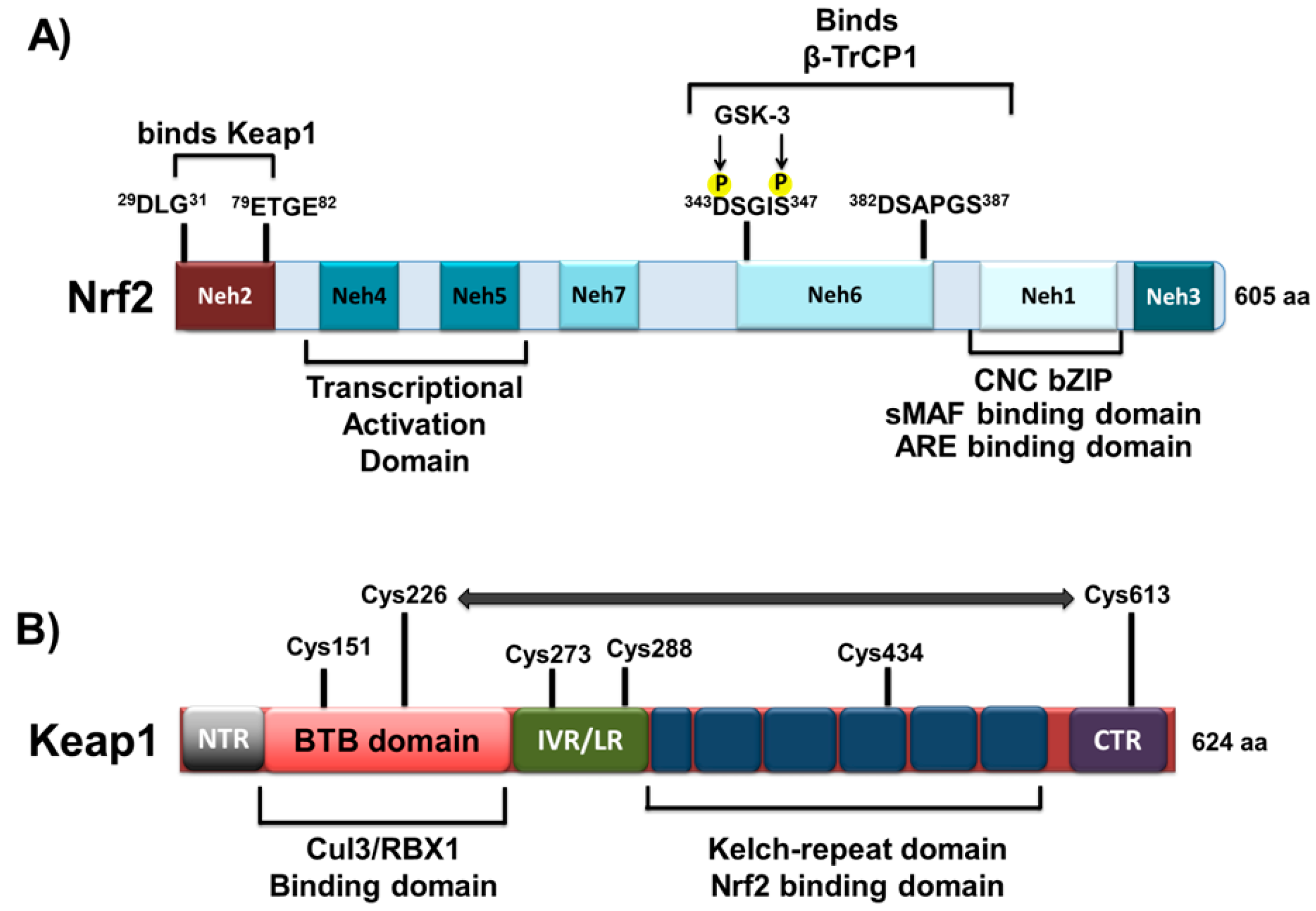
1.1. Structural Characteristics of Nrf2 Protein
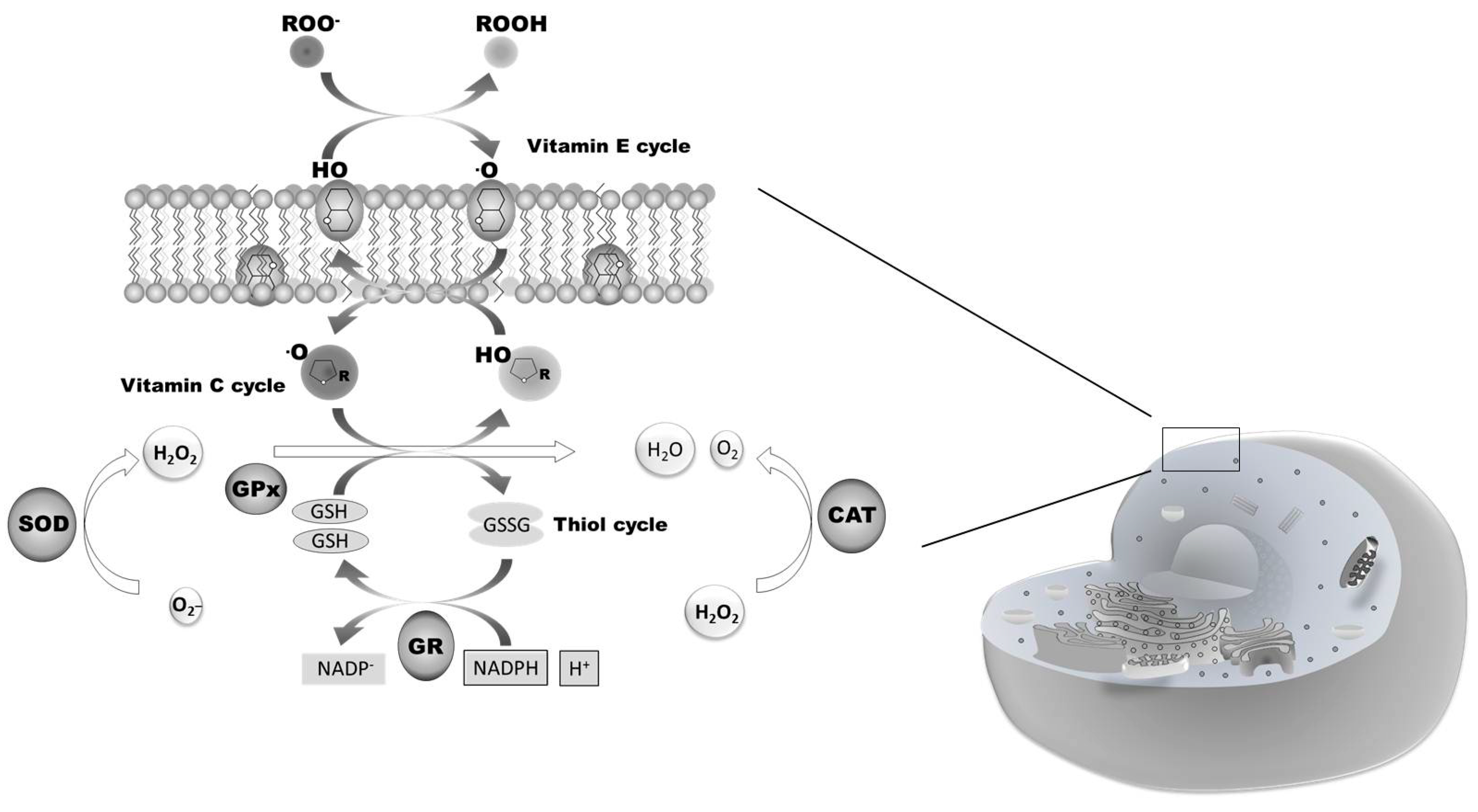
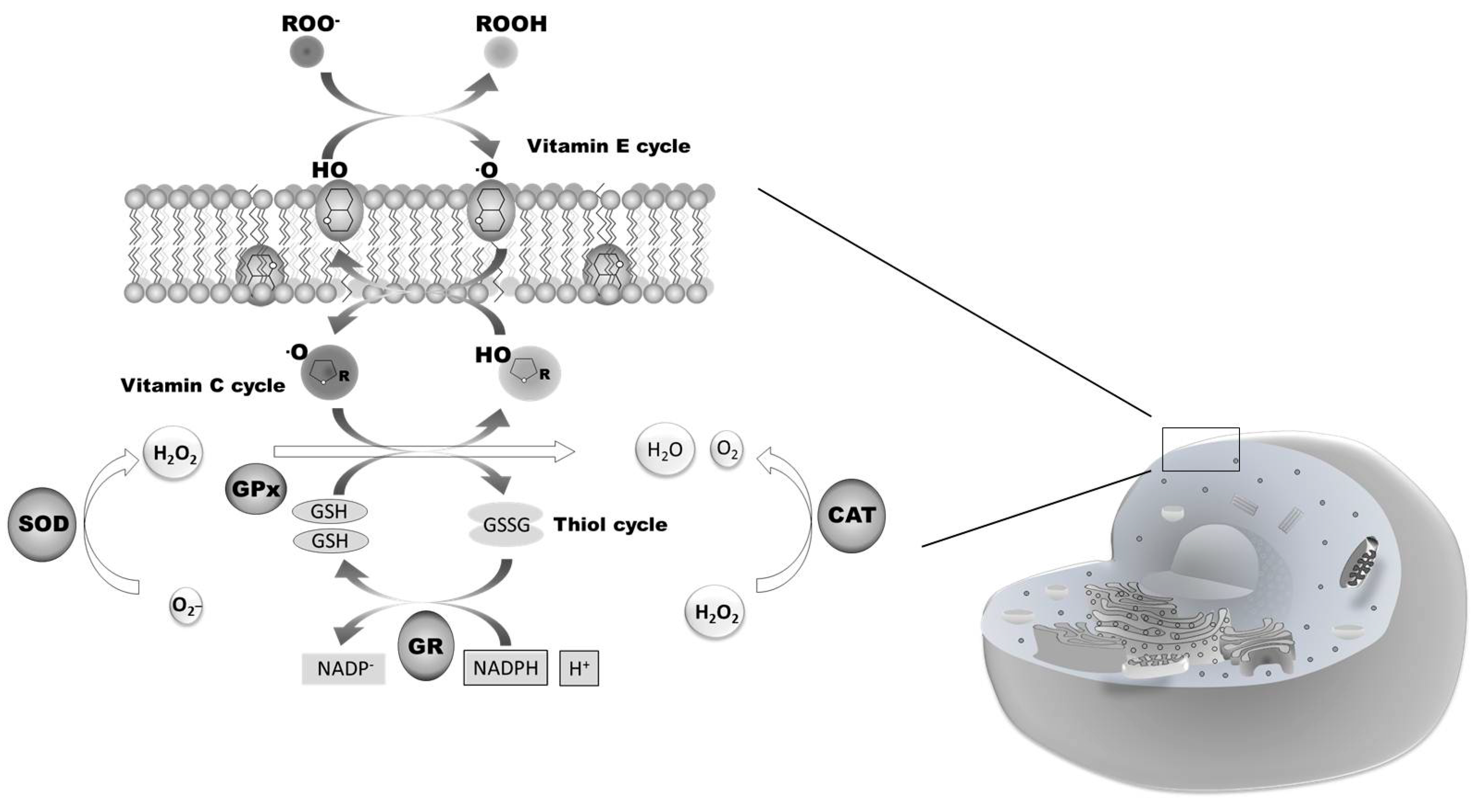
1.2. Nrf2 and Oxidative Stress: Signal Transduction and Gene Regulation
2. Epilepsy and Nrf2
2.1. Generalities of Epilepsy
2.2. Mechanisms Involved in Epilepsy
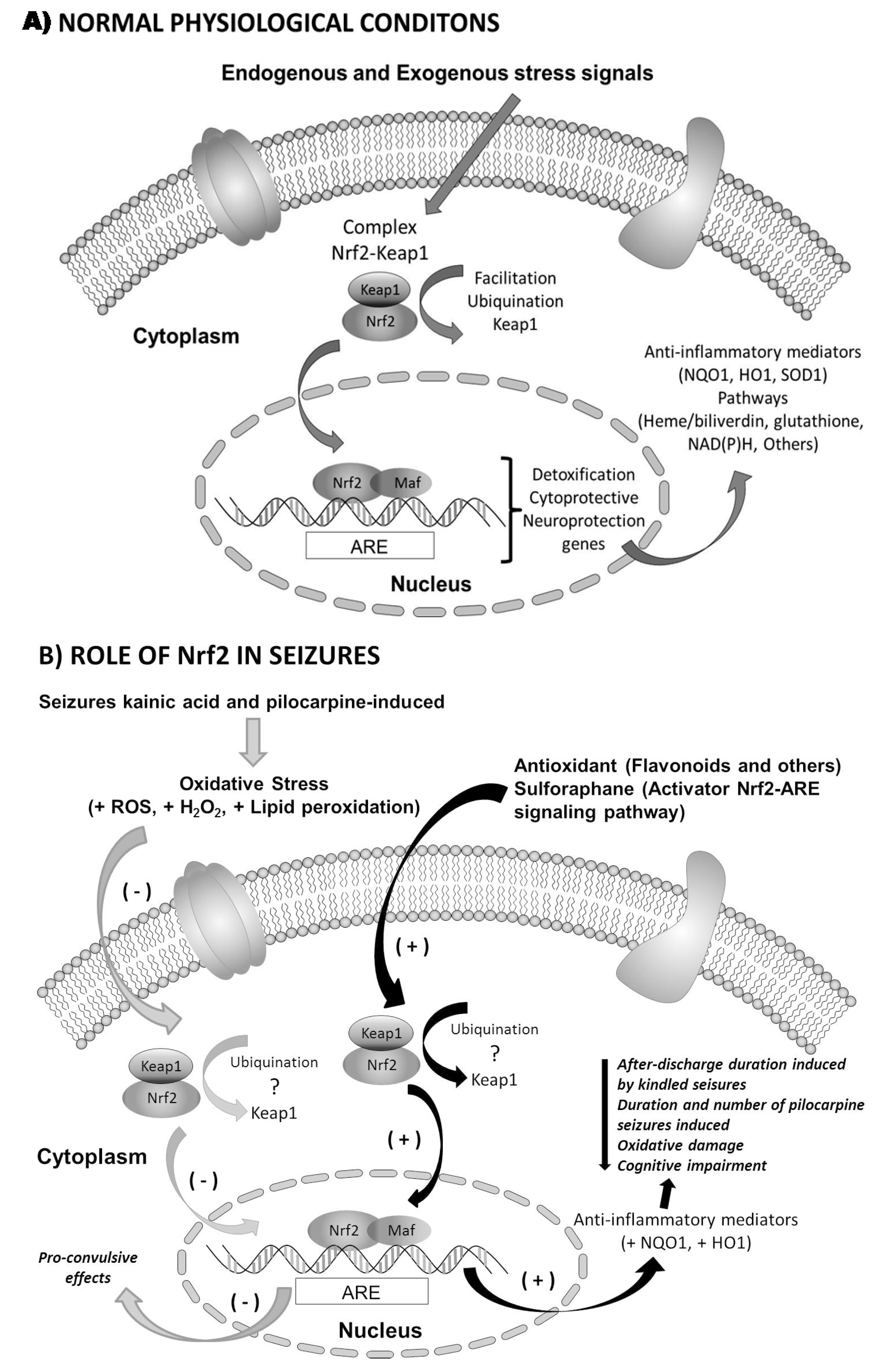
2.3. Nrf2 in Epilepsy
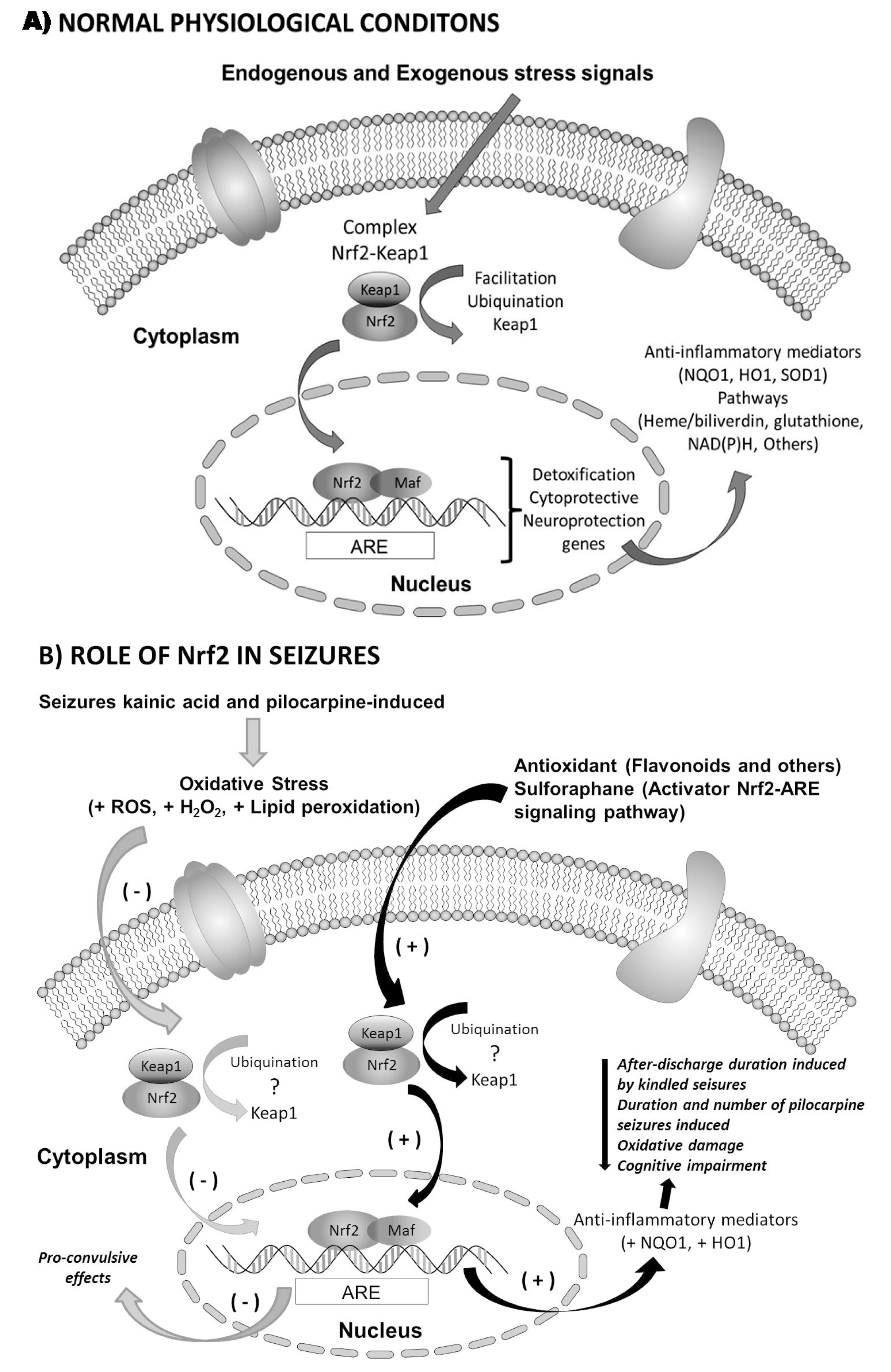
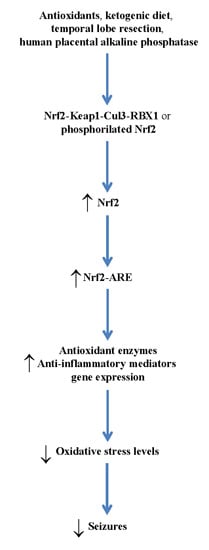
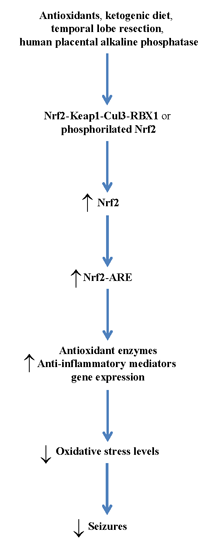
3. Therapeutic Relevance
Acknowledgments
Author Contributions
Conflict of Interest
References
- Marques-da-Silva, D.; Gutierrez-Merino, C. Caveolin-rich lipid rafts of the plasma membrane of mature cerebellar granule neurons are microcompartments for calcium/reactive oxygen and nitrogen species cross-talk signaling. Cell Calcium 2014, 56, 108–123. [Google Scholar] [PubMed]
- Bonini, M.G.; Consolaro, M.E.; Hart, P.C.; Mao, M.; de Abreu, A.L.; Master, A.M. Redox control of enzymatic functions: The electronics of life’s circuitry. IUBMB Life 2014, 63, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. Biol. Med. Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Carney, J.M. Free radical damage to protein and DNA: Mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol. 1992, 32, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free radicals in biology and medicine. Free Radic. Biol. Med. 1985, 1, 331–332. [Google Scholar] [CrossRef]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Kosiakov, K.S. Blood catalase activity in brain tumors during the postoperative period. Vopr. Neirokhir. 1975, 6, 41–44. [Google Scholar] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Gandhi, S.; Wood, N.W. Understanding the molecular causes of Parkinson’s disease. Trends Mol. Med. 2006, 12, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Yang, C.S.; Pickett, C.B. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic. Biol. Med. 2004, 37, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Edeas, M.A. SOD, oxidative stress and human pathologies: A brief history and a future vision. Biomed. Pharmacother. 2005, 59, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.; van Beurden, H.E.; von den Hoff, J.W.; Adema, G.J.; Figdor, C.G. The heme-heme oxygenase system: A molecular switch in wound healing. Blood 2003, 102, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jaiswal, A.K. Regulation of human NAD(P)H: Quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J. Biol. Chem. 1992, 267, 15097–15104. [Google Scholar] [PubMed]
- Long, D.J.; Jaiswal, A.K. NRH: Quinone oxidoreductase2 (NQO2). Chem. Biol. Interact. 2000, 129, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Iskander, K.; Li, J.; Han, S.; Zheng, B.; Jaiswal, A.K. NQO1 and NQO2 regulation of humoral immunity and autoimmunity. J. Biol. Chem. 2006, 281, 30917–30924. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H: Quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [PubMed]
- Kimura, M.; Yamamoto, T.; Zhang, J.; Itoh, K.; Kyo, M.; Kamiya, T.; Aburatani, H.; Katsuoka, F.; Kurokawa, H.; Tanaka, T.; et al. Molecular basis distinguishing the DNA binding profile of Nrf2-Maf heterodimer from that of Maf homodimer. J. Biol. Chem. 2015, 290, 10644–10644. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H: Quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Antioxidant-induced INrf2 (Keap1) tyrosine 85 phosphorylation controls the nuclear export and degradation of the INrf2-Cul3-Rbx1 complex to allow normal Nrf2 activation and repression. J. Cell Sci. 2012, 125, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Jaiswal, A.K. Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB J. 2011, 25, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Jaiswal, A.K. Antioxidant-induced phosphorylation of tyrosine 486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to the antioxidant response element and activate defensive gene expression. J. Biol. Chem. 2010, 285, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Van Roon-Mom, W.M.; Pepers, B.A.; AC’t Hoen, P.A.; Verwijmeren, C.A.; den Dunnen, J.T.; Dorsman, J.C.; van Ommen, G.B. Mutant huntingtin activates Nrf2-responsive genes and impairs dopamine synthesis in a PC12 model of Huntington’s disease. BMC Mol. Biol. 2008, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Tong, K.I.; Mio, K.; Maruyama, Y.; Kurokawa, H.; Sato, C.; Yamamoto, M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl. Acad. Sci. USA 2010, 107, 2842–2847. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Fainstein, M.K. Nrf2: La historia de un nuevo factor de transcripción que responde a estrés oxidativo. REB 2007, 26, 18–25. [Google Scholar]
- Riley, R.J.; Workman, P. DT-diaphorase and cancer chemotherapy. Biochem. Pharmacol. 1992, 43, 1657–1669. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.; Limonciel, A.; Felice, L.; Leonard, M.O. An overview of transcriptional regulation in response to toxicological insult. Arch. Toxicol. 2013, 87, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Yamaguchi, Y.; Kondo, N.; Masutani, H.; Yodoi, J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene 2003, 22, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Gasiorek, J.J.; Blank, V. Regulation and function of the NFE2 transcription factor in hematopoietic and non-hematopoietic cells. Cell Mol. Life Sci. 2015, 72, 2323–2335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues and energy status, and pathways through which it attenuates degenerative disease. Free Radic Biol. Med. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Yehiely, F.; Bamborough, P.; Da Costa, M.; Perry, B.J.; Thinakaran, G.; Cohen, F.E.; Carlson, G.A.; Prusiner, S.B. Identification of candidate proteins binding to prion protein. Neurobiol. Dis. 1997, 3, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; van Horssen, J.; van Rossum, S.; Dijkstra, C.D.; Drukarch, B.; de Vries, H.E. Therapeutic potential and biological role of endogenous antioxidant enzymes in multiple sclerosis pathology. Brain Res. Rev. 2007, 56, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Vargas, M.R.; Cassina, P.; Barbeito, A.G.; Beckman, J.S.; Barbeito, L. Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Lee, J.M.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J. Neurochem. 2006, 98, 1852–1865. [Google Scholar] [CrossRef] [PubMed]
- Engel, J. Concepts of epilepsy. Epilepsia 1995, 36, S23–S29. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Starkman, S. Overview of seizures. Emerg. Med. Clin. N. Am. 1994, 12, 895–923. [Google Scholar]
- Engel, J.J.; Pedley, A.T. Introduction to the epilepsies. In Epilepsy: A Comprehensive Textbook, 1st ed.; Engel, J.J., Pedley, A.T., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1997; pp. 765–772. [Google Scholar]
- Sander, J.W. ILAE Commission Report. The epidemiology of the epilepsies: Future directions. International League against epilepsy. Epilepsia 1997, 38, 614–618. [Google Scholar]
- Guidelines for Epidemiologic Studies on Epilepsy. Commission on epidemiology and prognosis, International League against epilepsy. Epilepsia 1993, 34, 592–596. [Google Scholar]
- Hauser, W.A.; Annegers, J.F.; Kurland, L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993, 34, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Dichter, M.A. Emerging insights into mechanisms of epilepsy: Implications for new antiepileptic drug development. Epilepsia 1994, 35, S51–S57. [Google Scholar] [CrossRef] [PubMed]
- Téllez-Zenteno, J.F.; Hernández-Ronquillo, L. A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res. Treat. 2012, 2012, 630853. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.R.; Garcia, H.H. Epilepsy in poor regions of the world. Lancet 2012, 380, 1193–1201. [Google Scholar] [CrossRef]
- Neligan, A.; Hauser, W.A.; Sander, J.W. The epidemiology of the epilepsies. Handb. Clin. Neurol. 2012, 107, 113–133. [Google Scholar] [PubMed]
- Ghofrani, M.; Akhondian, J. Intractable epilepsy in children. Iran. J. Child Neurol. 2010, 4, 7–14. [Google Scholar]
- Huang, H.; Zhou, H.; Wang, N. Recent advances in epilepsy management. Cell Biochem. Biophys. 2015, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Definition of refractory epilepsy: Defining the indefinable? Lancet Neurol. 2010, 9, 27–29. [Google Scholar] [CrossRef]
- Weaver, D.F.; Pohlmann-Eden, B. Pharmacoresistant epilepsy: Unmet needs in solving the puzzle(s). Epilepsia 2013, 54, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Téllez-Zenteno, J.F.; Hernández-Ronquillo, L.; Buckley, S.; Zahagun, R.; Rizvi, S. A validation of the new definition of drug-resistant epilepsy by the International League against Epilepsy. Epilepsia 2014, 55, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Bralowsky, S. Algunas definiciones. In Epilepsia, Enfermedad Sagrada del Cerebro; Secretaría de Educación Publica: México, 1999. [Google Scholar]
- Armijo, J.A. Which drugs should be chosen for the different types of epilepsy? Rev. Neurol. 1997, 25, 356–366. [Google Scholar] [PubMed]
- Dudek, F.E. Epileptogenesis: A new twist on the balance of excitation and inhibition. Epilepsy Curr. 2009, 9, 174–176. [Google Scholar] [PubMed]
- Banerjee, J.; Chandra, S.P.; Kurwale, N.; Tripathi, M. Epileptogenic networks and drug-resistant epilepsy: Present and future perspectives of epilepsy research-utility for the epileptologist and the epilepsy surgeon. Ann. Indian Acad. Neurol. 2014, 17, S134–S140. [Google Scholar] [PubMed]
- Kaila, K.; Ruusuvuori, E.; Seja, P.; Voipio, J.; Puskarjov, M. GABA actions and ionic plasticity in epilepsy. Curr. Opin. Neurobiol. 2014, 26, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B. Anal. Technol. Biomed. Life Sci. 2005, 827, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.A.; Oliveira, M.S.; Furian, A.F.; Rambo, L.M.; Ribeiro, L.R.; Lima, F.D.; Dalla Corte, L.C.; Silva, L.F.; Retamoso, L.T.; Dalla Corte, C.L.; et al. Swimming training prevents pentylenetetrazol-induced inhibition of Na+, K+-ATPase activity, seizures, and oxidative stress. Epilepsia 2009, 50, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Jeong, J.H.; Chung, Y.H.; Kim, W.K.; Ko, K.H.; Bach, J.H.; Hong, J.S.; Yoneda, Y.; Kim, H.C. Role of oxidative stress in epileptic seizures. Neurochem. Int. 2011, 59, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H. Oxygen, antioxidants and brain dysfunction. Yonsei Med. J. 1993, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bondy, S.C. The relation of oxidative stress and hyperexcitation to neurological disease. Proc. Soc. Exp. Biol. Med. 1995, 208, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Armstead, W.M.; Mirro, R.; Leffler, C.W.; Busija, D.W. Cerebral superoxide anion generation during seizures in newborn pigs. J. Cereb. Blood Flow Metab. 1989, 9, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.; Pazdernik, T.L.; Wagner, J.; Samson, F.; Andrews, G.K. Temporal spatial patterns of expression of metallothionein-I and -III and other stress related genes in rat brain after kainic acid-induced seizures. Neurochem. Int. 1995, 27, 59–71. [Google Scholar] [CrossRef]
- Dal-Pizzol, F.; Klamt, F.; Vianna, M.M.; Schröder, N.; Quevedo, J.; Benfato, M.S.; Moreira, J.C.; Walz, R. Lipid peroxidation in hippocampus early and late after status epilepticus induced by pilocarpine or kainic acid in Wistar rats. Neurosci. Lett. 2000, 291, 179–182. [Google Scholar] [CrossRef]
- Frantseva, M.V.; Perez Velazquez, J.L.; Tsoraklidis, G.; Mendonca, A.J.; Adamchik, Y.; Mills, L.R.; Carlen, P.L.; Burnham, M.W. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience 2000, 97, 431–435. [Google Scholar] [CrossRef]
- Martinc, B.; Grabnar, I.; Vovk, T. Antioxidants as a preventive treatment for epileptic process: A review of the current status. Curr. Neuropharmacol. 2014, 12, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.P.; Ho, Y.S.; Patel, M. Mitochondrial superoxide production in kainate- induced hippocampal damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Kovács, R.; Schuchmann, S.; Gabriel, S.; Kann, O.; Kardos, J.; Heinemann, U. Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J. Neurophysiol. 2002, 88, 2909–2918. [Google Scholar] [CrossRef] [PubMed]
- Rowley, S.; Patel, M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic. Biol. Med. 2013, 62, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Folbergrová, J. Oxidative stress in immature brain following experimentally-induced seizures. Physiol. Res. 2013, 62, S39–S48. [Google Scholar] [PubMed]
- Cárdenas-Rodríguez, N.; González-Trujano, M.E.; Aguirre-Hernández, E.; Ruíz-García, M.; Sampieri, A.; Coballase-Urrutia, E.; Carmona-Aparicio, L. Anticonvulsant and antioxidant effects of Tilia americana var. Mexicana and flavonoids constituents in pentylenetetrazole-induced seizures. Oxid. Med. Cell Longev. 2014, 2014, 329172. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Rodríguez, N.; Coballase-Urrutia, E.; Pérez-Cruz, C.; Montesinos-Correa, H.; Rivera-Espinosa, L.; Sampieri, A.; Carmona-Aparicio, L. Relevance of the glutathione system in temporal lobe epilepsy: Evidence in human and experimental models. Oxid. Med. Cell Longev. 2014, 2014, 759293. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Rodríguez, N.; Coballase-Urrutia, E.; Rivera-Espinosa, L.; Romero-Toledo, A.; Sampieri, A.; Ortega-Cuellar, D.; Montesinos-Correa, H.; Floriano-Sánchez, E.; Carmona-Aparicio, L. Modulation of antioxidant enzymatic activities by certain antiepileptic drugs (valproic acid, oxcarbazepine, and topiramate): Evidence in humans and experimental models. Oxid. Med. Cell Longev. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. Biol. Med. Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef] [PubMed]
- Mahle, C.; Dasgupta, A. Decreased total antioxidant capacity and elevated lipid hydroperoxide concentrations in sera of epileptic patients receiving phenytoin. Life Sci. 1997, 61, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.S.; Wu, H.M.; Kao, S.H.; Wei, Y.H. Phenytoin-mediated oxidative stress in serum of female epileptics: A possible pathogenesis in the fetal hydantoin syndrome. Hum. Exp. Toxicol. 1997, 16, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Sakamoto, A.; Sakura, N. Plasma total glutathione concentrations in epileptic patients taking anticonvulsants. Clin. Chim. Acta 2000, 298, 135–143. [Google Scholar] [CrossRef]
- Sudha, K.; Rao, A.V.; Rao, A. Oxidative stress and antioxidants in epilepsy. Clin. Chim. Acta 2001, 303, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.G.; Trabesinger, A.H.; Boesiger, P.; Wieser, H.G. Brain glutathione levels in patients with epilepsy measured by in vivo (1) H-MRS. Neurology 2001, 57, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Abuhandan, M.; Calik, M.; Taskin, A.; Yetkin, I.; Selek, S.; Iscan, A. The oxidative and antioxidative status of simple febrile seizure patients. J. Pak. Med. Assoc. 2013, 63, 594–597. [Google Scholar] [PubMed]
- Shih, A.Y.; Li, P.; Murphy, T.H. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J. Neurosci. 2005, 25, 10321–10335. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kobori, N.; Aronowski, J.; Dash, P.K. Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci. Lett. 2006, 393, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, Y.; Zhang, G.; Fang, H.; Wang, H.; Zang, H.; Xie, T.; Wang, W. Activation of Nrf2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 2014, 1544, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, W.P.; Zhang, G.L.; Wu, Y.F.; Xie, T.; Kan, M.C.; Fang, H.B.; Wang, H.C. Activation of Nrf2-ARE signal pathway in hippocampus of amygdala kindling rats. Neurosci. Lett. 2013, 543, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Mazzuferi, M.; Kumar, G.; van Eyll, J.; Danis, B.; Foerch, P.; Kaminski, R.M. Nrf2 defense pathway: Experimental evidence for its protective role in epilepsy. Ann. Neurol. 2013, 74, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jeong, G.S.; Kang, D.G.; Lee, H.S.; Kim, Y.C. Cytoprotective effects of lindenenyl acetate isolated from Linderastrychnifolia on mouse hippocampal HT22 cells. Eur. J. Pharmacol. 2009, 614, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Samoylenko, A.; Immenschuh, S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J. Biol. Chem. 2003, 278, 17927–17936. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Nrf2 to the rescue. Epilepsy Curr. 2015, 15, 39–40. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona-Aparicio, L.; Pérez-Cruz, C.; Zavala-Tecuapetla, C.; Granados-Rojas, L.; Rivera-Espinosa, L.; Montesinos-Correa, H.; Hernández-Damián, J.; Pedraza-Chaverri, J.; Sampieri, A.I.; Coballase-Urrutia, E.; et al. Overview of Nrf2 as Therapeutic Target in Epilepsy. Int. J. Mol. Sci. 2015, 16, 18348-18367. https://doi.org/10.3390/ijms160818348
Carmona-Aparicio L, Pérez-Cruz C, Zavala-Tecuapetla C, Granados-Rojas L, Rivera-Espinosa L, Montesinos-Correa H, Hernández-Damián J, Pedraza-Chaverri J, Sampieri AI, Coballase-Urrutia E, et al. Overview of Nrf2 as Therapeutic Target in Epilepsy. International Journal of Molecular Sciences. 2015; 16(8):18348-18367. https://doi.org/10.3390/ijms160818348
Chicago/Turabian StyleCarmona-Aparicio, Liliana, Claudia Pérez-Cruz, Cecilia Zavala-Tecuapetla, Leticia Granados-Rojas, Liliana Rivera-Espinosa, Hortencia Montesinos-Correa, Jacqueline Hernández-Damián, José Pedraza-Chaverri, Aristides III Sampieri, Elvia Coballase-Urrutia, and et al. 2015. "Overview of Nrf2 as Therapeutic Target in Epilepsy" International Journal of Molecular Sciences 16, no. 8: 18348-18367. https://doi.org/10.3390/ijms160818348