
Molecular Classification and Pharmacogenetics of Primary Plasma Cell Leukemia: An Initial Approach toward Precision Medicine

 , , ,
, , ,
Abstract
:1. Introduction
2. Molecular Classification and Prognostic Risk Stratification

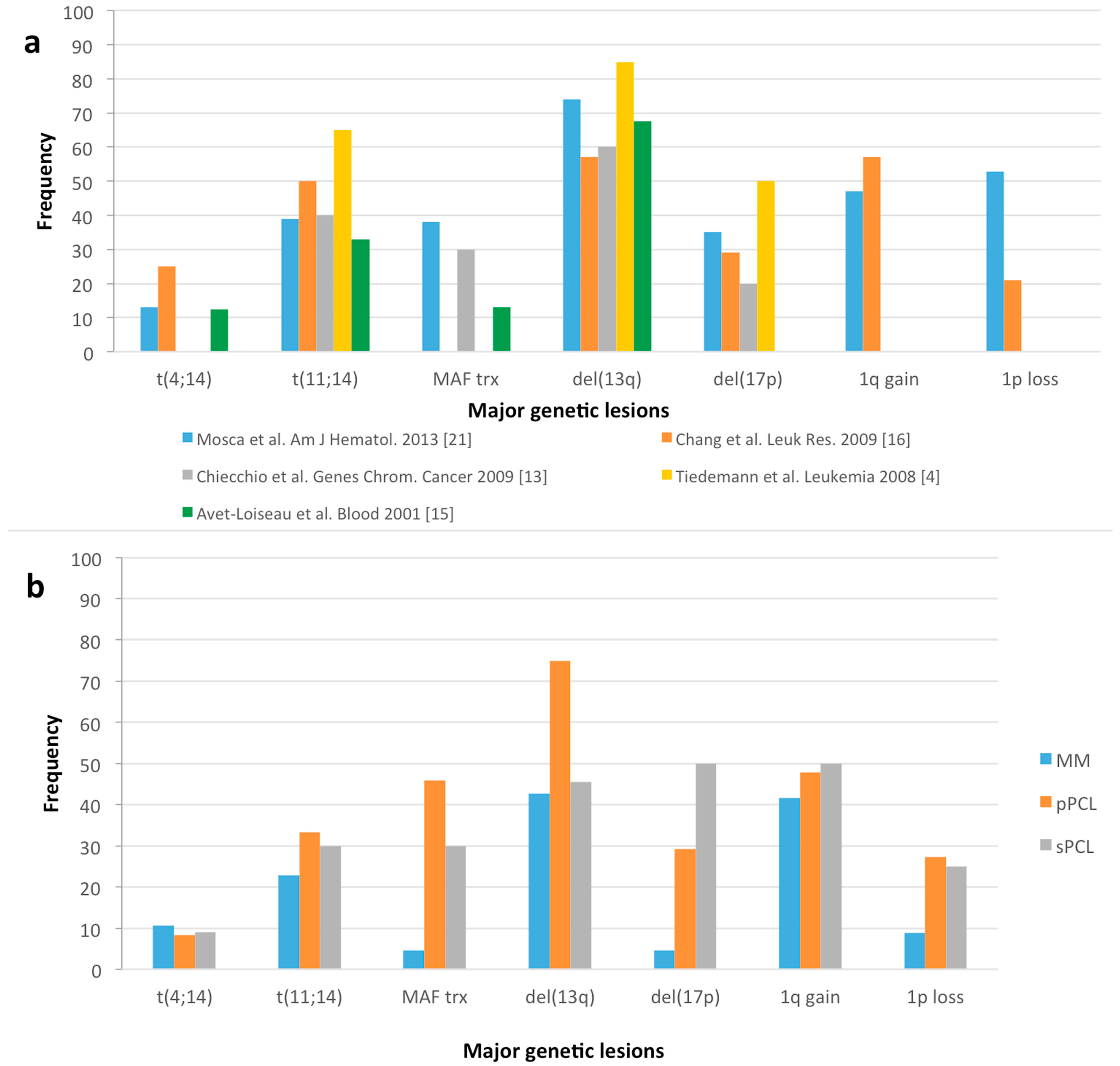
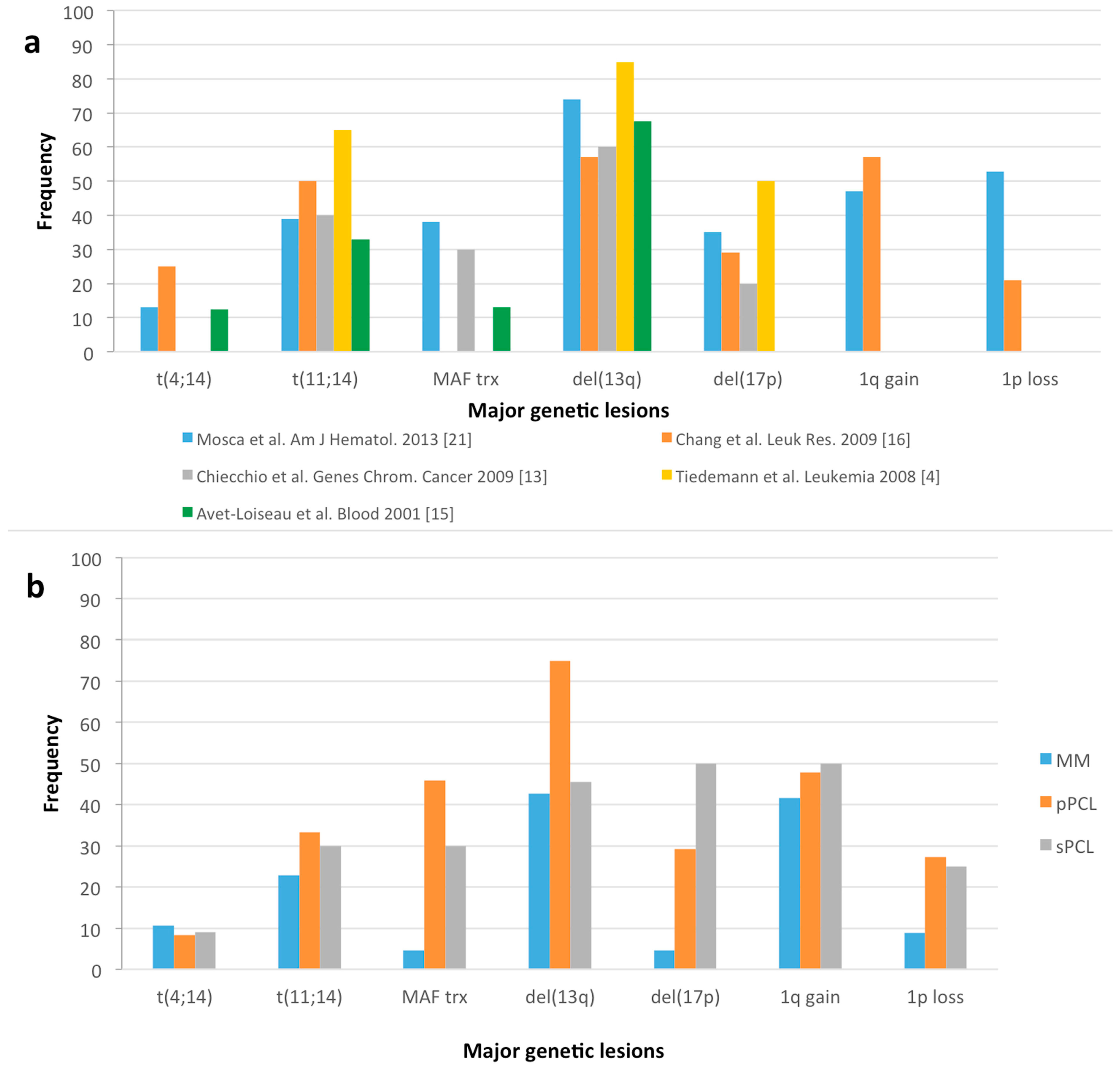{kind=link}
| Reference | pPCL Patients | Molecular Classification | Integrated Analysis | Gene/miRNA Signature (pPCL vs. MM) | Clinical Outcome Signature | Gene/miRNA List |
|---|---|---|---|---|---|---|
| Usmani et al., 2012 [20] | 13 | GEP | - | 203 DE genes | - | - |
| Mosca L. et al., 2013 [21] | 23 | FISH | GEP | - | - | - |
| 17 | SNP-array | |||||
| Todoerti et al., 2013 [23] | 21 | GEP | - | 503 DE genes | 3 genes-response rate | YIPF6, EDEM3, YB5D2 |
| 27 genes-OS | PECAM1, MKX, FAM111B, MCTP1, CALCRL, C10orf10, FNBP1, EFEMP1, C3orf14, ALDH1L2, WARS, SLC15A2, FAIM3, CPEB4, EDN1, PVALB, LY86, LAPTM5, RNU5D, PARP15, PLEKHF2, PDK4, TNFAIP3, FAM105A, CTH, HOOK1, TCN2 | |||||
| Lionetti et al., 2013 [24] | 18 | miRNA | SNP-array/GEP | 83 DE miRNAs | 4 miRNAs-treatment response | miR-106b, miR-497, miR-181b, miR-181a |
| 4 miRNAs-PFS/OS | miR-92a, miR-330-3p, miR-22, miR-146a | |||||
| Cifola et al., 2015 [25] | 12 | WES | SNP-array/GEP | - | - | - |
3. Pharmacogenetics of New and Old Agents: Adverse Drug Reaction and Efficacy
| Drug | Gene | SNP | Alleles | Amino Acid Translation | Annotation | Reference |
|---|---|---|---|---|---|---|
| Melphalan | ERCC2 | rs13181 | T>G | Lys751Gln or K751Q | longer time to treatment failure | Vangsted et al., 2007 [37] |
| XRCC3 | rs861539 | G>A | Thr241Met T241M | |||
| ALDH2 | rs440 | T>C | - | response to HDM | Dumontet et al., 2010 [38] | |
| rs4646777 | G>A | - | ||||
| GSTT2 | rs1622002 | G>A | Met139Ile | |||
| BRCA1 | rs799917 | C>G; C>T; C>A | Pro824Gln; Pro824Leu | |||
| rs4986850 | G>A; G>T; G>C | Asp646Tyr; Asp646Asn | ||||
| CYP1A1 | rs1048943 | T>G; T>C; T>A | Ile462Val; Ile462Leu; Ile462Phe | disease progression | ||
| RAD51 | rs1801320 | G>C | - | |||
| PARP4 | rs13428 | G>A; G>C | Gly1280Arg; Gly1280Cys | |||
| ALDH2 | rs886205 | A>G | - | OS | ||
| CYP1A1 | rs1048943 | T>G; T>C; T>A | Ile462Val; Ile462Leu; Ile462Phe | |||
| BRCA1 | rs4986850 | G>A; G>T; G>C | Asp646Tyr; Asp646Asn | severe mucositis after HDM | ||
| CDKN1A | rs1801270 | C>A | Ser31Arg | |||
| XRCC1 | rs25487 | T>C | Gln399Arg | |||
| SLC7A5 | rs4240803 | G>A | - | gastrointestinal side effects | Giglia et al., 2014 [39] | |
| IL-1β | rs1143627 | -31 C/T | - | OS | Vangsted et al., 2009 [40] | |
| Vincristine | GLI1 | rs2228224 | G>A | Gly892Asp | early-onset peripheral neuropathy | Broyl et al., 2010 [41] |
| rs2242578 | G>C | - | ||||
| DPYD | rs1413239 | C>T | - | late-onset peripheral neuropathy | ||
| ABCC1 | rs3887412 | A>C | - | |||
| GSTP1 | rs1695 | A>G | Ile105Val | response after chemotherapy | Maggini et al., 2008 [42] | |
| TYMS or ENOSF1 | rs2790 | A>T; A>G | - | |||
| rs699517 | C>T | - | response after SCT | |||
| Dexamethasone | MRD1 or ABCB1 | rs2032582 | A>T; A>C | Ser893Ala; Ser893;Thr | OS | Maggini et al., 2008 [43] |
| rs1045642 | A>T; A>G | Ile1145Ile | ||||
| GSTP1 | rs1695 | A>G | Ile105Val | response after chemotherapy | Maggini et al., 2008 [42] | |
| TYMS or ENOSF1 | rs2790 | A>T; A>G | - | |||
| rs699517 | C>T | - | response after SCT | |||
| Bortezomib | NFKB1 | rs28362491 | ATTG>del | - | PFS | Varga et al., 2015 [44] |
| TRAF3 | rs11160707 | G>A | - | PFS | Du et al., 2011 [45] | |
| NFKB2 | rs12769316 | G>A | - | OS | ||
| PSD | rs1056890 | G>A | - | OS | ||
| MRP1 or ABCC1 | rs4148356 | G>A | Arg723Gln | PFS-OS | Buda et al., 2010 [46] | |
| CASP9 | rs2020895 | A>G | - | early-onset peripheral neuropathy | Broyl et al., 2010 [41] | |
| rs2020903 | G>A | - | ||||
| rs4646034 | T>C | - | ||||
| ALOX12 | rs1126667 | A>G | Gln261Arg | |||
| rs434473 | A>G | Asn322Ser | ||||
| IGF1R | rs1879612 | T>C | - | |||
| ERCC4 | rs1799800 | G>A | - | late-onset peripheral neuropathy | ||
| rs1799801 | T>C | Ser835Ser | ||||
| ERCC3 | rs2276583 | G>A | - | |||
| PPARD | rs2267668 | G>A | - | |||
| CTLA4 | rs4553808 | A>G | - | peripheral neuropathy | Favis et al., 2011 [47] | |
| CTSS | rs12568757 | G>A | - | |||
| PSMB1 | rs1474642 | A>G | - | |||
| TCF4 | rs1261134 | A>T | - | |||
| DYNC1I1 | rs916758 | A>G | - | |||
| Thalidomide | ABCA1 | rs363717 | C>T | - | peripheral neuropathy | Johnson et al., 2011 [48] |
| ICAM1 | rs1799969 | G>A | Gly241Arg | |||
| PPARD | rs2076169 | A>G | - | |||
| SERPINB2 | rs6103 | C>G | Asn404Lys | |||
| SLC12A6 | rs7164902 | C>G; C>A | Leu144Leu | |||
| ERCC1 or CD3EAP | rs735482 | A>C | Lys259Thr | response rate | Cibeira et al., 2011 [49] | |
| ERCC5 | rs17655 | G>C | Asp1104His | |||
| XRCC5 | rs1051685 | A>G | - | |||
| ERCC1 or CD3EAP | rs735482 | A>C | Lys259Thr | OS | ||
| XRCC5 | rs1051685 | A>G | - | |||
| GSTT1 | rs4630 | G>A | - | peripheral neuropathy | ||
| CDKN1A | rs3829963 | C>A | - | VTE | Almasi et al., 2011 [50] | |
| TNF-alpha | rs361525 | G>A | - | PFS-OS | Du et al., 2010 [51] | |
| CYP2C19 | Extensive metabolizers | - | OS | Li et al., 2007 [52] | ||
| Lenalidomide | NFKB1 | rs3774968 | A>G | - | VTE | Bagratuni et al., 2013 [53] |
3.1. Melphalan
3.2. Vincristine
3.3. Cyclophosphamide
3.4. Dexamethasone
3.5. Bortezomib
3.6. Thalidomide
3.7. Lenalidomide
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Albarracin, F.; Fonseca, R. Plasma cell leukemia. Blood Rev. 2011, 25, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, T.; Kryukov, F.; Rihova, L.; Hajek, R. Plasma cell leukemia: From biology to treatment. Eur. J. Haematol. 2015, 95, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.; Lokhorst, H.M.; Anderson, K.C.; Richardson, P.G. How I treat plasma cell leukemia. Blood 2012, 120, 2376–2389. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, R.E.; Gonzalez-Paz, N.; Kyle, R.A.; Santana-Davila, R.; Price-Troska, T.; van Wier, S.A.; Chng, W.J.; Ketterling, R.P.; Gertz, M.A.; Henderson, K.; et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008, 22, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Fernández de Larrea, C.; Kyle, R.A; Durie, B.G.M.; Ludwig, H.; Usmani, S.; Vesole, D.H.; Hajek, R.; San Miguel, J.F.; Sezer, O.; Sonneveld, P.; et al. Plasma cell leukemia: Consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia 2013, 27, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Sant, M.; Allemani, C.; Tereanu, C.; de Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadie, M.; Simonetti, A.; Lutz, J.-M.; et al. Incidence of hematological malignancies in Europe by morphological subtype: results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef] [PubMed]
- Musto, P.; Pagano, L.; Petrucci, M.T.; Morabito, F.; Caravita, T.; di Raimondo, F.; Baldini, L.; Tosi, P.; Bringhen, S.; Offidani, M.; et al. Primary plasma cell leukemia in the era of new drugs: Has something changed? Crit. Rev. Oncol. Hematol. 2012, 82, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, M.; Agnelli, L.; Fabris, S.; Baldini, L.; Morabito, F.; Bicciato, S.; Verdelli, D.; Intini, D.; Nobili, L.; Cro, L.; et al. Gene expression profiling of plasma cell dyscrasias reveals molecular patterns associated with distinct IGH translocations in multiple myeloma. Oncogene 2005, 24, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Pagano, L.; Valentini, C.G.; de Stefano, V.; Venditti, A.; Visani, G.; Petrucci, M.T.; Candoni, A.; Specchia, G.; Visco, C.; Pogliani, E.M.; et al. Primary plasma cell leukemia: A retrospective multicenter study of 73 patients. Ann. Oncol. 2011, 22, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Musto, P.; Simeon, V.; Martorelli, M.C.; Petrucci, M.T.; Cascavilla, N.; di Raimondo, F.; Caravita, T.; Morabito, F.; Offidani, M.; Olivieri, A.; et al. Lenalidomide and low-dose dexamethasone for newly diagnosed primary plasma cell leukemia. Leukemia 2014, 28, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Roussel, M.; Campion, L.; Leleu, X.; Marit, G.; Jardel, H.; Dib, M.; Decaux, O.; Lamy, T.; Tiab, M.; et al. Cytogenetic and therapeutic characterization of primary plasma cell leukemia: the IFM experience. Leukemia 2012, 26, 158–159. [Google Scholar] [CrossRef] [PubMed]
- Chiecchio, L.; Protheroe, R.K.M.; Ibrahim, A.H.; Cheung, K.L.; Rudduck, C.; Dagrada, G.P.; Cabanas, E.D.; Parker, T.; Nightingale, M.; Wechalekar, A.; et al. Deletion of chromosome 13 detected by conventional cytogenetics is a critical prognostic factor in myeloma. Leukemia 2006, 20, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Chiecchio, L.; Dagrada, G.P.; White, H.E.; Towsend, M.R.; Protheroe, R.K.M.; Kan, L.C.; Stockley, D.M.; Orchard, K.H.; Cross, N.C.P.; Harrison, C.J.; et al. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes Cancer 2009, 48, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Sloan, S.; Li, D.; Patterson, B. Genomic aberrations in plasma cell leukemia shown by interphase fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2005, 156, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myelome and the Groupe Francais de Cytogenetique Hematolo. Blood 2001, 97, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Qi, X.; Yeung, J.; Reece, D.; Xu, W.; Patterson, B. Genetic aberrations including chromosome 1 abnormalities and clinical features of plasma cell leukemia. Leuk. Res. 2009, 33, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Gazzeri, S.; Brambilla, C.; Brambilla, E. Mdm2 overexpression and p14ARF inactivation are two mutually exclusive events in primary human lung tumors. Oncogene 2002, 2750–2761. [Google Scholar] [CrossRef] [PubMed]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Bezieau, S.; Devilder, M.C.; Avet-Loiseau, H.; Mellerin, M.P.; Puthier, D.; Pennarun, E.; Rapp, M.J.; Harousseau, J.L.; Moisan, J.P.; Bataille, R. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum. Mutat. 2001, 18, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Nair, B.; Qu, P.; Hansen, E.; Zhang, Q.; Petty, N.; Waheed, S.; Shaughnessy, J.D.; Alsayed, Y.; Heuck, C.J.; et al. Primary plasma cell leukemia: Clinical and laboratory presentation, gene-expression profiling and clinical outcome with Total Therapy protocols. Leukemia 2012, 26, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Musto, P.; Todoerti, K.; Barbieri, M.; Agnelli, L.; Fabris, S.; Tuana, G.; Lionetti, M.; Bonaparte, E.; Sirchia, S.M.; et al. Genome-wide analysis of primary plasma cell leukemia identifies recurrent imbalances associated with changes in transcriptional profiles. Am. J. Hematol. 2013, 88, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Barbieri, M.; Todoerti, K.; Agnelli, L.; Marzorati, S.; Fabris, S.; Ciceri, G.; Galletti, S.; Milesi, G.; Manzoni, M.; et al. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: implication for MEK-ERK pathway activation. Oncotarget 2015, in press. [Google Scholar]
- Todoerti, K.; Agnelli, L.; Fabris, S.; Lionetti, M.; Tuana, G.; Mosca, L.; Lombardi, L.; Grieco, V.; Bianchino, G.; D’Auria, F.; et al. Transcriptional characterization of a prospective series of primary plasma cell leukemia revealed signatures associated with tumor progression and poorer outcome. Clin. Cancer Res. 2013, 19, 3247–3258. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Musto, P.; di Martino, M.T.; Fabris, S.; Agnelli, L.; Todoerti, K.; Tuana, G.; Mosca, L.; Gallo Cantafio, M.E.; Grieco, V.; et al. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin. Cancer Res. 2013, 19, 3130–3142. [Google Scholar] [CrossRef] [PubMed]
- Cifola, I.; Lionetti, M.; Pinatel, E.; Todoerti, K.; Mangano, E.; Pietrelli, A.; Fabris, S.; Mosca, L.; Simeon, V.; Petrucci, M.T.; et al. Whole-exome sequencing of primary plasma cell leukemia discloses heterogeneous mutational patterns. Oncotarget 2015, in press. [Google Scholar]
- Jr, J.D.S.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar]
- Decaux, O.; Lodé, L.; Magrangeas, F.; Charbonnel, C.; Gouraud, W.; Jézéquel, P.; Attal, M.; Harousseau, J.-L.; Moreau, P.; Bataille, R.; et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: A study of the Intergroupe Francophone du Myélom. J. Clin. Oncol. 2008, 26, 4798–4805. [Google Scholar] [CrossRef] [PubMed]
- Dickens, N.J.; Walker, B.A.; Leone, P.E.; Johnson, D.C.; Brito, J.L.; Zeisig, A.; Jenner, M.W.; Boyd, K.D.; Gonzalez, D.; Gregory, W.M.; et al. Homozygous deletion mapping in myeloma samples identifies genes and an expression signature relevant to pathogenesis and outcome. Clin. Cancer Res. 2010, 16, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Biasiolo, M.; Agnelli, L.; Todoerti, K.; Mosca, L.; Fabris, S.; Sales, G.; Deliliers, G.L.; Bicciato, S.; Lombardi, L.; et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 2009, 114, e20–e26. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; di Martino, M.T.; Neri, A.; Tagliaferri, P.; Tassone, P. Non-coding RNA: A novel opportunity for the personalized treatment of multiple myeloma. Expert Opin. Biol. Ther. 2013, 13 (Suppl. 1), 125–137. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Amodio, N.; di Martino, M.T.; Tagliaferri, P.; Tassone, P.; Cho, W.C. MicroRNA and multiple myeloma: From laboratory findings to translational therapeutic approaches. Curr. Pharm. Biotechnol. 2014, 15, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Buscaglia, L.E.B.; Barker, J.R.; Li, Y. MicroRNAs in NF-κB signaling. J. Mol. Cell Biol. 2011, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J. Emerging roles of microRNA-22 in human disease and normal physiology. Curr. Mol. Med. 2012, 12, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, H.E.; Maitland, M.L.; Dolan, M.E.; Cox, N.J.; Ratain, M.J. Cancer pharmacogenomics: strategies and challenges. Nat. Rev. Genet. 2013, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Vangsted, A.; Klausen, T.W.; Vogel, U. Genetic variations in multiple myeloma II: Association with effect of treatment. Eur. J. Haematol. 2012, 88, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The pharmacogenetics and pharmacogenomics knowledge base. Methods Mol. Biol. 2005, 311, 179–191. [Google Scholar] [PubMed]
- Vangsted, A.; Gimsing, P.; Klausen, T.W.; Nexø, B.A.; Wallin, H.; Andersen, P.; Hokland, P.; Lillevang, S.T.; Vogel, U. Polymorphisms in the genes ERCC2, XRCC3 and CD3EAP influence treatment outcome in multiple myeloma patients undergoing autologous bone marrow transplantation. Int. J. Cancer 2007, 120, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Landi, S.; Reiman, T.; Perry, T.; Plesa, A.; Bellini, I.; Barale, R.; Pilarski, L.M.; Troncy, J.; Tavtigian, S.; et al. Genetic polymorphisms associated with outcome in multiple myeloma patients receiving high-dose melphalan. Bone Marrow Transplant. 2010, 45, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Giglia, J.L.; White, M.J.; Hart, A.J.; Toro, J.J.; Freytes, C.O.; Holt, C.C.; Cai, Y.; Williams, S.M.; Brandt, S.J. A single nucleotide polymorphism in SLC7A5 is associated with gastrointestinal toxicity after high-dose melphalan and autologous stem cell transplantation for multiple myeloma. Biol. Blood Marrow Transplant. 2014, 20, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Vangsted, A.J.; Klausen, T.W.; Ruminski, W.; Gimsing, P.; Andersen, N.F.; Gang, A.O.; Abildgaard, N.; Knudsen, L.M.; Nielsen, J.L.; Gregersen, H.; et al. The polymorphism IL-1β T-31C is associated with a longer overall survival in patients with multiple myeloma undergoing auto-SCT. Bone Marrow Transplant. 2009, 43, 539–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broyl, A.; Corthals, S.L.; Jongen, J.L.M.; van der Holt, B.; Kuiper, R.; de Knegt, Y.; van Duin, M.; el Jarari, L.; Bertsch, U.; Lokhorst, H.M.; et al. Mechanisms of peripheral neuropathy associated with bortezomib and vincristine in patients with newly diagnosed multiple myeloma: A prospective analysis of data from the HOVON-65/GMMG-HD4 trial. Lancet Oncol. 2010, 11, 1057–1065. [Google Scholar] [CrossRef]
- Maggini, V.; Buda, G.; Galimberti, S.; Conidi, E.; Giuliani, N.; Morabito, F.; Genestreti, G.; Iacopino, P.; Rizzoli, V.; Barale, R.; et al. Response to chemotherapy and tandem autologous transplantation of multiple myeloma patients and GSTP1 and TYMS polymorphisms. Leuk. Res. 2008, 32, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Maggini, V.; Buda, G.; Martino, A.; Presciuttini, S.; Galimberti, S.; Orciuolo, E.; Barale, R.; Petrini, M.; Rossi, A.M. MDR1 diplotypes as prognostic markers in multiple myeloma. Pharmacogenet. Genomics 2008, 18, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; Mikala, G.; Andrikovics, H.; Koszarska, M.; Balassa, K.; Ádám, E.; Kozma, A.; Tordai, A.; Masszi, T. NFKB1-94ins/delATTG polymorphism is a novel prognostic marker in first line-treated multiple myeloma. Br. J. Haematol. 2015, 168, 679–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Huo, J.; Shi, J.; Yuan, Z.; Zhang, C.; Fu, W.; Jiang, H.; Yi, Q.; Hou, J. Polymorphisms of nuclear factor-κB family genes are associated with development of multiple myeloma and treatment outcome in patients receiving bortezomib-based regimens. Haematologica 2011, 96, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Buda, G.; Ricci, D.; Huang, C.C.; Favis, R.; Cohen, N.; Zhuang, S.H.; Harousseau, J.L.; Sonneveld, P.; Bladé, J.; et al. Polymorphisms in the multiple drug resistance protein 1 and in P-glycoprotein 1 are associated with time to event outcomes in patients with advanced multiple myeloma treated with bortezomib and pegylated liposomal doxorubicin. Ann. Hematol. 2010, 89, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Favis, R.; Sun, Y.; van de Velde, H.; Broderick, E.; Levey, L.; Meyers, M.; Mulligan, G.; Harousseau, J.-L.; Richardson, P.G.; Ricci, D.S. Genetic variation associated with bortezomib-induced peripheral neuropathy. Pharmacogenet. Genom. 2011, 21, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Corthals, S.L.; Walker, B.A.; Ross, F.M.; Gregory, W.M.; Dickens, N.J.; Lokhorst, H.M.; Goldschmidt, H.; Davies, F.E.; Durie, B.G.M.; et al. Genetic factors underlying the risk of thalidomide-related neuropathy in patients with multiple myeloma. J. Clin. Oncol. 2011, 29, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Cibeira, M.T.; Fernández de Larrea, C.; Navarro, A.; Díaz, T.; Fuster, D.; Tovar, N.; Rosiñol, L.; Monzó, M.; Bladé, J. Impact on response and survival of DNA repair single nucleotide polymorphisms in relapsed or refractory multiple myeloma patients treated with thalidomide. Leuk. Res. 2011, 35, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Almasi, M.; Sevcikova, S.; Slaby, O.; Kaisarova, P.; Maisnar, V.; Penka, M.; Pika, T.; Pour, L.; Radocha, J.; Scudla, V.; et al. Association study of selected genetic polymorphisms and occurrence of venous thromboembolism in patients with multiple myeloma who were treated with thalidomide. Clin. Lymphoma Myeloma Leuk. 2011, 11, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yuan, Z.; Zhang, C.; Fu, W.; Jiang, H.; Chen, B.; Hou, J. Role of the TNF-α promoter polymorphisms for development of multiple myeloma and clinical outcome in thalidomide plus dexamethasone. Leuk. Res. 2010, 34, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hou, J.; Jiang, H.; Wang, D.; Fu, W.; Yuan, Z.; Chen, Y.; Zhou, L. Polymorphisms of CYP2C19 gene are associated with the efficacy of thalidomide-based regimens in multiple myeloma. Haematologica 2007, 92, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Bagratuni, T.; Kastritis, E.; Politou, M.; Roussou, M.; Kostouros, E.; Gavriatopoulou, M.; Eleutherakis-Papaiakovou, E.; Kanelias, N.; Terpos, E.; Dimopoulos, M.A. Clinical and genetic factors associated with venous thromboembolism in myeloma patients treated with lenalidomide-based regimens. Am. J. Hematol. 2013, 88, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Cavallo, F.; Gay, F.; di Raimondo, F.; Ben Yehuda, D.; Petrucci, M.T.; Pezzatti, S.; Caravita, T.; Cerrato, C.; Ribakovsky, E.; Genuardi, M.; et al. Autologous transplantation and maintenance therapy in multiple myeloma. N. Engl. J. Med. 2014, 371, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Nath, C.E.; Lazarus, H.M. Not too little, not too much—just right! (Better ways to give high dose melphalan). Bone Marrow Transplant. 2014, 49, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.; Ludeman, S.M.; Dolan, M.E. Drug focus: Pharmacogenetic studies related to cyclophosphamide-based therapy. Pharmacogenomics 2009, 10, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Vangsted, A.J.; Søeby, K.; Klausen, T.W.; Abildgaard, N.; Andersen, N.F.; Gimsing, P.; Gregersen, H.; Vogel, U.; Werge, T.; Rasmussen, H.B. No influence of the polymorphisms CYP2C19 and CYP2D6 on the efficacy of cyclophosphamide, thalidomide, and bortezomib in patients with Multiple Myeloma. BMC Cancer 2010, 10, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocio, E.M.; Richardson, P.G.; Rajkumar, S.V.; Palumbo, A.; Mateos, M.V.; Orlowski, R.; Kumar, S.; Usmani, S.; Roodman, D.; Niesvizky, R.; et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: A report from the International Myeloma Working Group (IMWG). Leukemia 2014, 28, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Morawska, M.; Grzasko, N.; Kostyra, M.; Wojciechowicz, J.; Hus, M. Therapy-related peripheral neuropathy in multiple myeloma patients. Hematol. Oncol. 2014, 25. [Google Scholar] [CrossRef] [PubMed]
- Neben, K.; Mytilineos, J.; Moehler, T.M.; Preiss, A.; Kraemer, A.; Ho, A.D.; Opelz, G.; Goldschmidt, H. Polymorphisms of the tumor necrosis factor-α gene promoter predict for outcome after thalidomide therapy in relapsed and refractory multiple myeloma. Blood 2002, 100, 2263–2265. [Google Scholar] [PubMed]
- Morabito, F.; Recchia, A.G.; Mazzone, C.; Gentile, M. Targeted therapy of multiple myeloma: the changing paradigm at the beginning of the new millennium. Curr. Cancer Drug Targets 2012, 12, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Attal, M.; Facon, T. Frontline therapy of multiple myeloma. Blood 2015, 125, 3076–3085. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Rajkumar, S.V.; San Miguel, J.F.; Larocca, A.; Niesvizky, R.; Morgan, G.; Landgren, O.; Hajek, R.; Einsele, H.; Anderson, K.C.; et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not eligible for standard autologous stem-cell transplantation. J. Clin. Oncol. 2014, 32, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Gozzetti, A.; Candi, V.; Papini, G.; Bocchia, M. Therapeutic advancements in multiple myeloma. Front. Oncol. 2014, 4, 241. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M. Personalized pharmacogenomics: Predicting efficacy and adverse drug reactions. Annu. Rev. Genom. Hum. Genet. 2013, 15, 349–370. [Google Scholar] [CrossRef] [PubMed]
- Jameson, J.L.; Longo, D.L. Precision medicine—Personalized, problematic, and promising. N. Engl. J. Med. 2015, 372, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Sameek, R.; Chinnaiyan, A.M. Translating genomics for precision cancer medicine. Annu. Rev. Genom. Hum. Genet. 2014, 15, 395–415. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Tassone, P.; Neri, A. Molecular profiling of multiple myeloma: from gene expression analysis to next-generation sequencing. Expert Opin. Biol. Ther. 2013, 13 (Suppl. 1), 55–68. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.A.; Zanger, U.M.; Schwab, M. Omics and drug response. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 475–502. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.F.; Alfirevic, A.; Pirmohamed, M. Pharmacogenomics: Current state-of-the-art. Genes 2014, 5, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Poste, G. Bring on the biomarkers. Nature 2011, 469, 156–157. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simeon, V.; Todoerti, K.; La Rocca, F.; Caivano, A.; Trino, S.; Lionetti, M.; Agnelli, L.; De Luca, L.; Laurenzana, I.; Neri, A.; et al. Molecular Classification and Pharmacogenetics of Primary Plasma Cell Leukemia: An Initial Approach toward Precision Medicine. Int. J. Mol. Sci. 2015, 16, 17514-17534. https://doi.org/10.3390/ijms160817514
Simeon V, Todoerti K, La Rocca F, Caivano A, Trino S, Lionetti M, Agnelli L, De Luca L, Laurenzana I, Neri A, et al. Molecular Classification and Pharmacogenetics of Primary Plasma Cell Leukemia: An Initial Approach toward Precision Medicine. International Journal of Molecular Sciences. 2015; 16(8):17514-17534. https://doi.org/10.3390/ijms160817514
Chicago/Turabian StyleSimeon, Vittorio, Katia Todoerti, Francesco La Rocca, Antonella Caivano, Stefania Trino, Marta Lionetti, Luca Agnelli, Luciana De Luca, Ilaria Laurenzana, Antonino Neri, and et al. 2015. "Molecular Classification and Pharmacogenetics of Primary Plasma Cell Leukemia: An Initial Approach toward Precision Medicine" International Journal of Molecular Sciences 16, no. 8: 17514-17534. https://doi.org/10.3390/ijms160817514