
Differential DNA Methylation in Relation to Age and Health Risks of Obesity


 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion

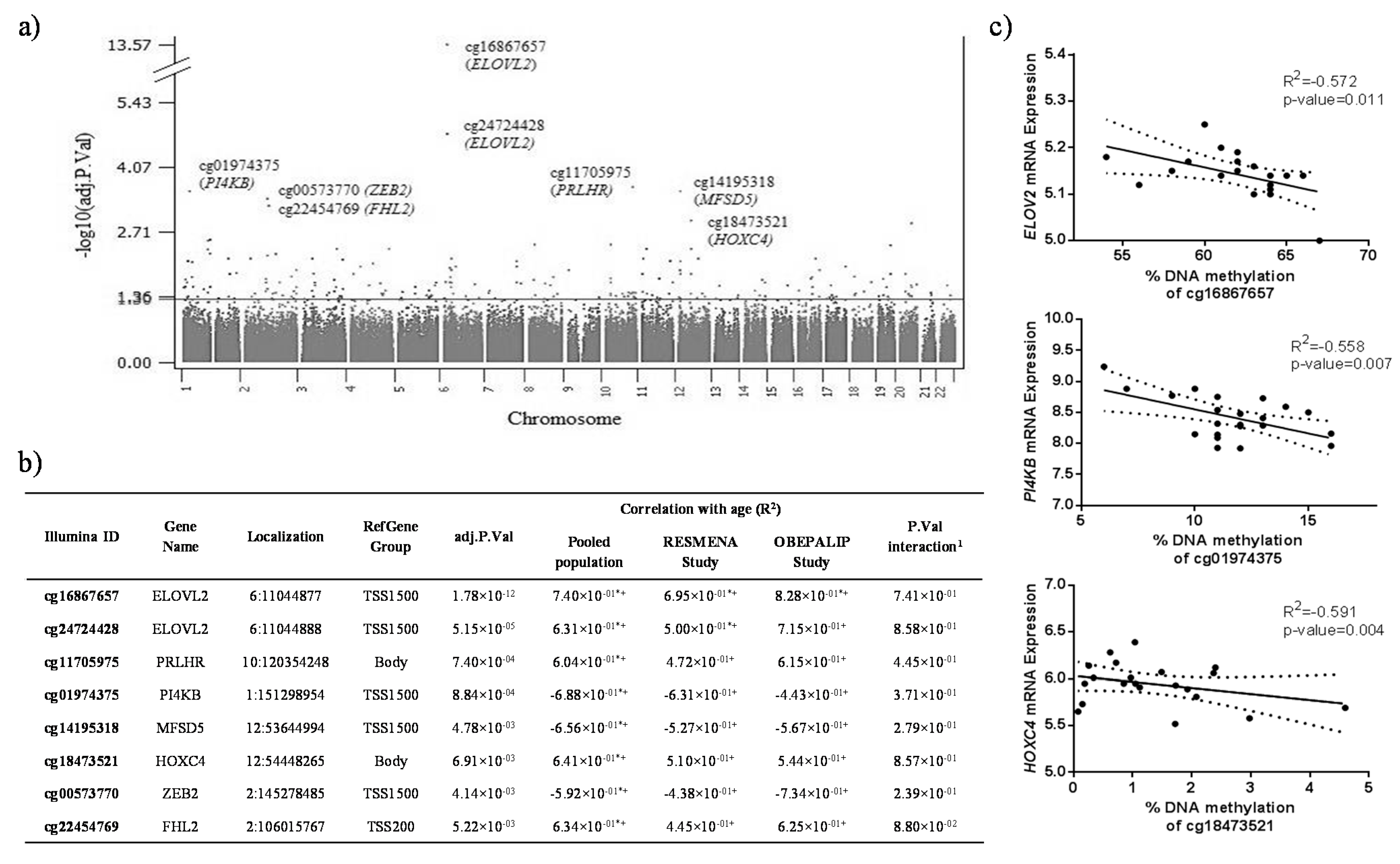{kind=link}
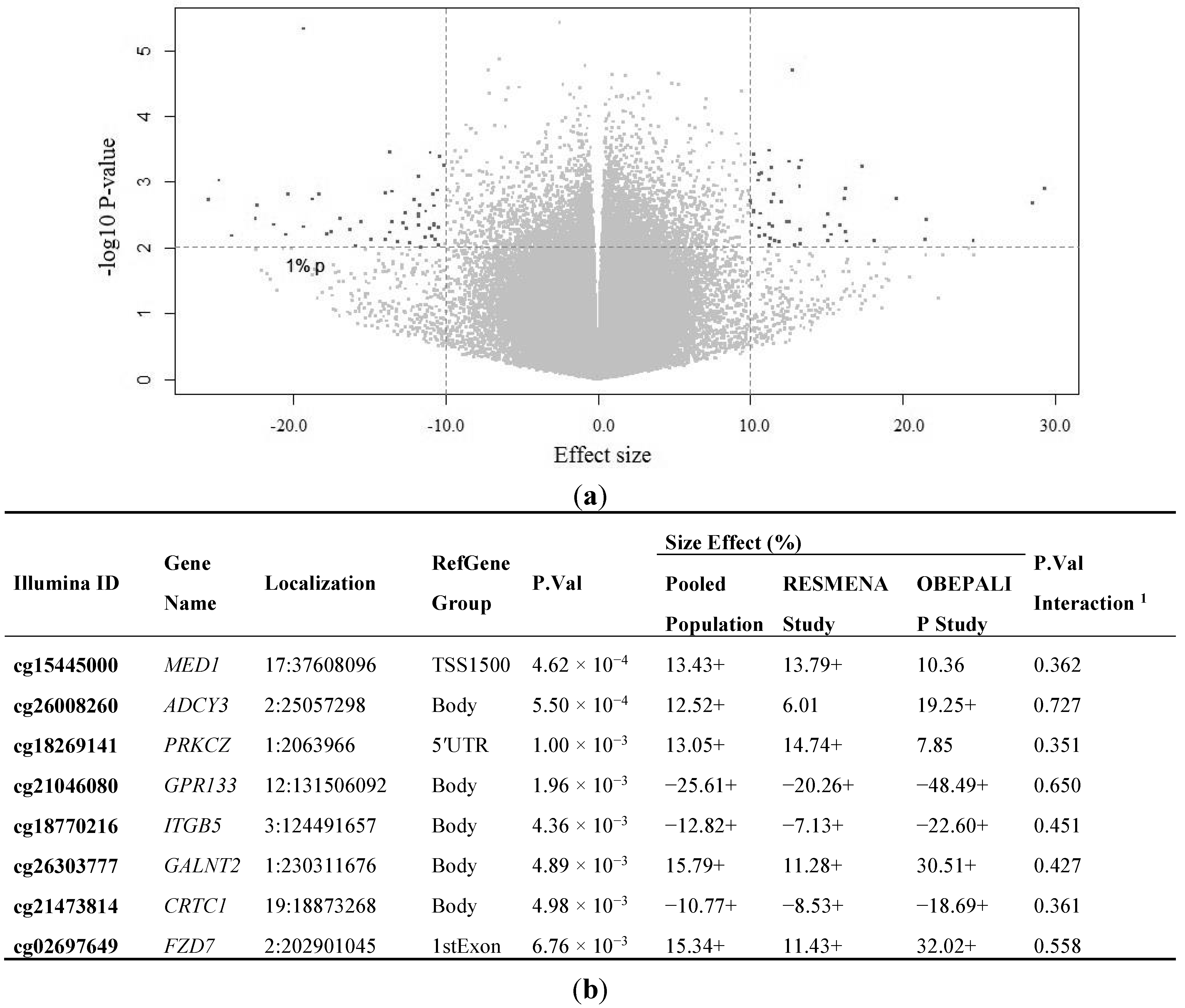{kind=link}
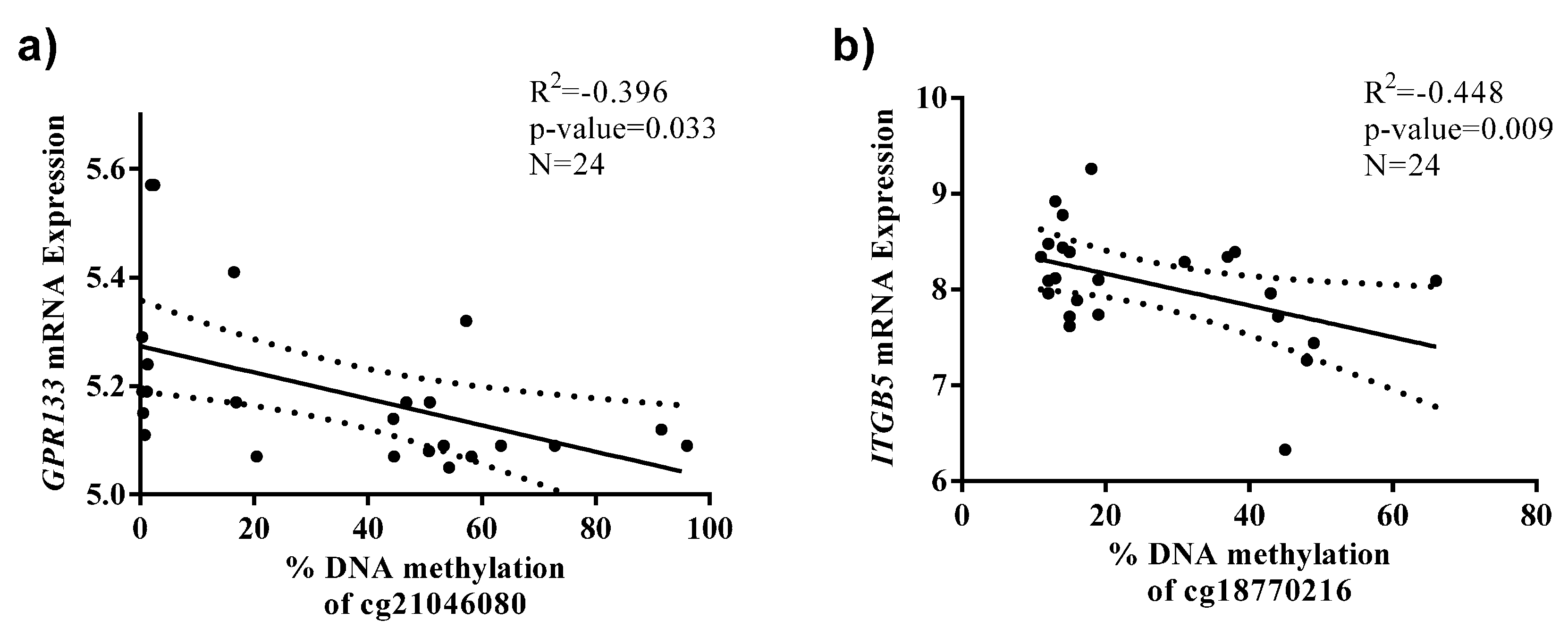{kind=link}
| Variables | Pooled Population | RESMENA Study | OBEPALIP Study | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TP | LR | HR | TP | LR | HR | TP | LR | HR | |
| n | 73 | 40 | 33 | 48 | 18 | 30 | 25 | 22 | 3 |
| Age (year) | 45 (10) | 41 (10) | 49 (10) + | 48 (10) | 47 (10) | 49 (10) | 37 (7) * | 37 (8) | 42 (2) |
| Sex (M/F) | 26/47 | 9/31 | 17/16 + | 26/22 | 9/9 | 17/13 | 0/25 | 0/22 | 0/3 |
| Weight (Kg) | 95.5 (17.8) | 85.8 (10.4) | 107.0 (18.0) + | 102.4 (17.9) | 90.5 (11.6) | 109.1 (17.4) + | 82.7 (7.6) * | 82.3 (7.9) | 86.0 (4.0) |
| BMI (kg/m2) | 34.6 (4.1) | 31.5 (2.3) | 38.1 (2.7) + | 36.2 (3.8) | 32.4 (2.7) | 38.2 (3.8) + | 31.5 (2.7) * | 30.9 (2.3) | 36.0 (0.6) + |
| WC (cm) | 106.0 (13.4) | 98.1 (8.8) | 115.3 (11.9) + | 112.0 (12.5) | 102.5 (10.2) | 117.3 (10.5) + | 94.7 (5.7) * | 94.6 (5.8) | 95.3 (5.7) |
| Metabolic Syndrome 1 | 39 | 12 | 27 | 39 | 12 | 27 | 0 * | 0 | 0 |
| Smoking | 12 | 7 | 5 | 8 | 3 | 5 | 4 | 4 | 0 |
2.1. Identification of CpG Sites Differentially Methylated with Age in WBC

2.2. Identification of CpG Sites Differentially Methylated in Relation to the Health Risks of Obesity (HRO)
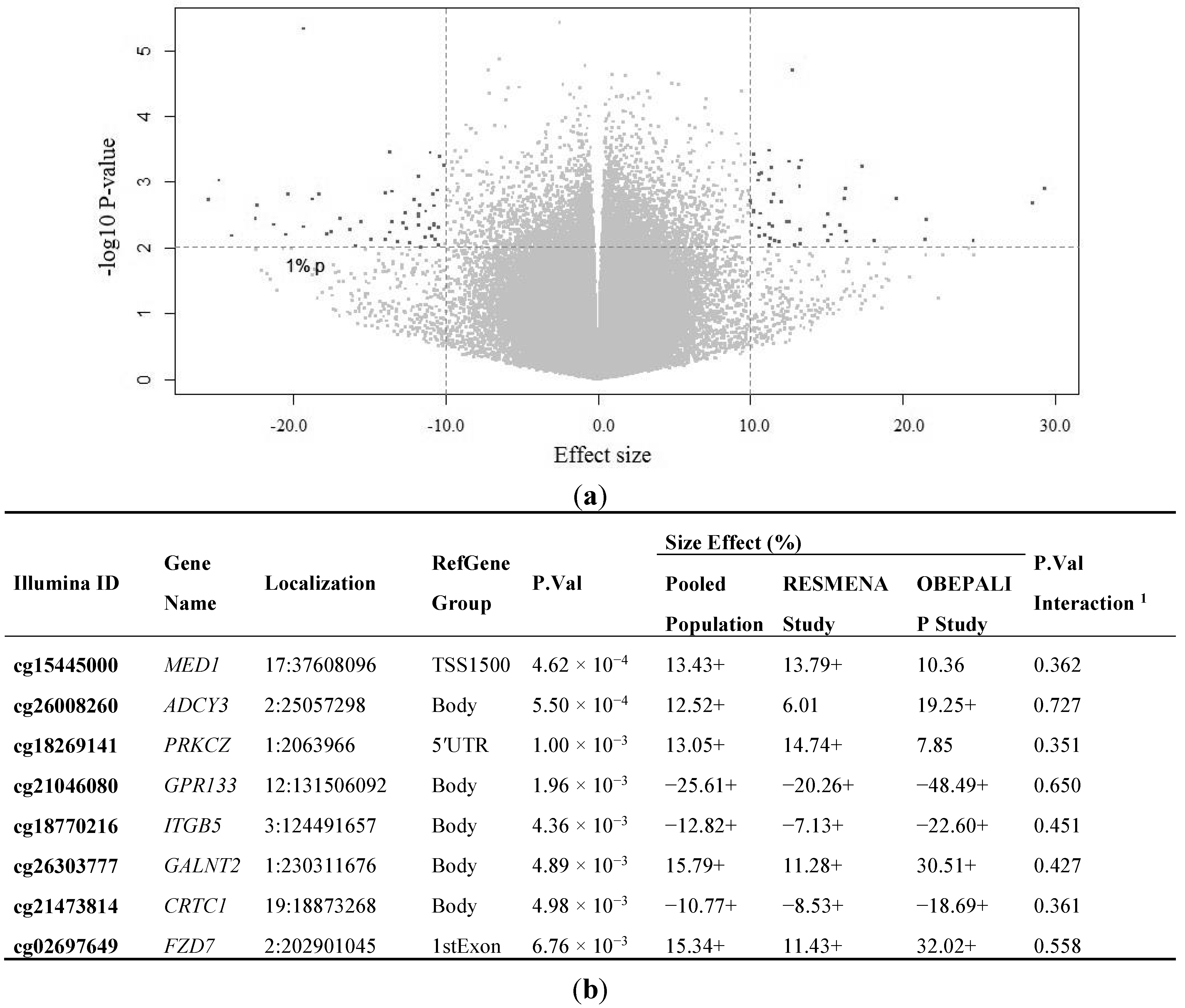
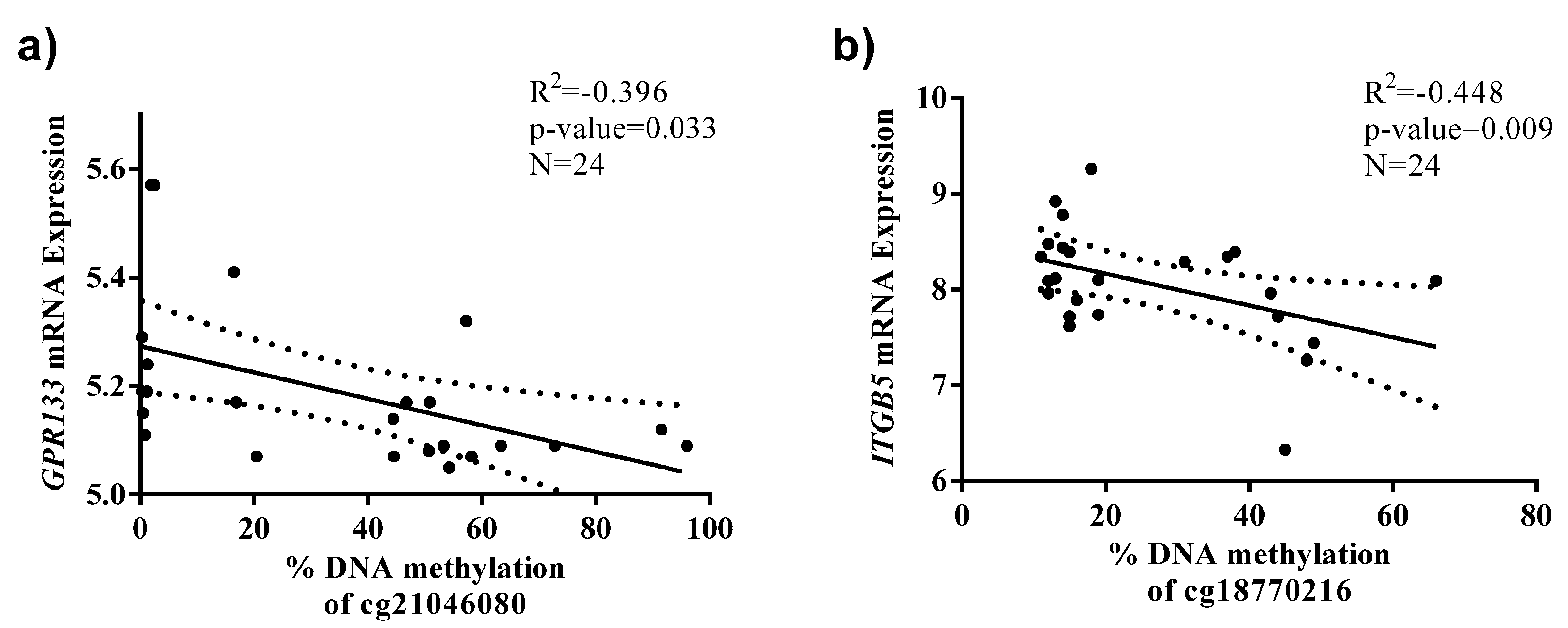
| BMI as Dependent | Standardized Beta Coefficient | Unstandardized B Coefficient (95% CI) | p-Value a | R2 |
|---|---|---|---|---|
| cg15445000_MED1 | −0.249 | −6.61 (−11.78; −1.43) | 0.013 | 0.335 |
| cg26008260_ADCY3 | −0.332 | −103.66 (−168.75; −38.6) | 0.002 | 0.349 |
| cg18269141_PRKCZ | −0.356 | −8.73 (−13.68; −3.78) | 0.003 | 0.397 |
| cg21046080_GPR133 | 0.278 | 5.11 (1.68; 10.01) | 0.017 | 0.292 |
| cg18770216_ITGB5 | 0.211 | 5.26 (0.21; 10.31) | 0.041 | 0.299 |
| cg26303777_GALNT2 | 0.122 | 2.61 (−1.91; 7.11) | 0.252 | 0.360 |
| cg21473814_CRTC1 | −0.039 | −1.13 (−7.28; 5.20) | 0.771 | 0.270 |
| cg02697649_FZD7 | −0.103 | −2.08 (−6.21; 2.05) | 0.318 | 0.279 |
3. Experimental Section
3.1. Subjects and Study Protocol
3.2. Procedures
3.3. DNA Isolation and DNA Methylation Analysis
3.4. Gene Ontology (GO) Analysis
3.5. Gene Expression Analysis
3.6. Statistical Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alberti, K.G.; Zimmet, P.; Shaw, J. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Marti, A.; Martinez-Gonzalez, M.A.; Martinez, J.A. Interaction between genes and lifestyle factors on obesity. Proc. Nutr. Soc. 2008, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, C.M.; Heid, I.M.; Randall, J.C.; Lamina, C.; Steinthorsdottir, V.; Qi, L.; Speliotes, E.K.; Thorleifsson, G.; Willer, C.J.; Herrera, B.M.; et al. Genome-wide association scan meta-analysis identifies three loci influencing adiposity and fat distribution. PLoS Genet. 2009, 5, e1000508. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, L.; Rabasa-Lhoret, R.; Faraj, M.; Lavoie, M.E.; Mill, J.; Perusse, L.; Vohl, M.C. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am. J. Clin. Nutr. 2010, 91, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Campion, J.; Milagro, F.I.; Martinez, J.A. Individuality and epigenetics in obesity. Obes. Rev. 2009, 10, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Claycombe, K.J.; Martinez, J.A.; Friso, S.; Schalinske, K.L. Nutritional epigenomics: A portal to disease prevention. Adv. Nutr. 2013, 4, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Milagro, F.I.; Mansego, M.L.; de Miguel, C.; Martinez, J.A. Dietary factors, epigenetic modifications and obesity outcomes: Progresses and perspectives. Mol. Asp. Med. 2013, 34, 782–812. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Pausova, Z. Cigarette smoking and DNA methylation. Front. Genet. 2013, 4, 132. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, S.; Peat, J.R.; Hore, T.A.; Santos, F.; Dean, W.; Reik, W. Reprogramming DNA methylation in the mammalian life cycle: Building and breaking epigenetic barriers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20110330. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A. Transgenerational epigenetic inheritance: Resolving uncertainty and evolving biology. Biomol. Concepts 2015, 6, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Sharp, G.C.; Lawlor, D.A.; Richmond, R.C.; Fraser, A.; Simpkin, A.; Suderman, M.; Shihab, H.A.; Lyttleton, O.; McArdle, W.; Ring, S.M.; et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: Findings from the AVON longitudinal study of parents and children. Int. J. Epidemiol. 2015. [Google Scholar] [CrossRef]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.F.; Atzmon, G.; Gheorghe, C.; Liang, H.Q.; Lowes, C.; Greally, J.M.; Barzilai, N. Tissue-specific dysregulation of DNA methylation in aging. Aging Cell 2010, 9, 506–518. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Obesity: Preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ. Tech. Rep. Ser. 2000, 894, 1–253. [Google Scholar]
- National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults—The evidence report. Obes. Res. 1998, 6 Suppl. 2, 51S–209S. [Google Scholar] [PubMed]
- Martinez, J.A.; Cordero, P.; Campion, J.; Milagro, F.I. Interplay of early-life nutritional programming on obesity, inflammation and epigenetic outcomes. Proc. Nutr. Soc. 2012, 71, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Wlodek, M.E.; Connelly, J.J.; Yan, Z. Epigenetic origins of metabolic disease: The impact of the maternal condition to the offspring epigenome and later health consequences. Food Sci. Hum. Wellness 2013, 2, 1–11. [Google Scholar] [CrossRef]
- Denis, G.V.; Obin, M.S. “Metabolically healthy obesity”: Origins and implications. Mol. Asp. Med. 2013, 34, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Roberson, L.L.; Aneni, E.C.; Maziak, W.; Agatston, A.; Feldman, T.; Rouseff, M.; Tran, T.; Blaha, M.J.; Santos, R.D.; Sposito, A.; et al. Beyond BMI: The “metabolically healthy obese” phenotype & its association with clinical/subclinical cardiovascular disease and all-cause mortality—A systematic review. BMC Public Health 2014, 14, 14. [Google Scholar] [PubMed]
- Badoud, F.; Perreault, M.; Zulyniak, M.A.; Mutch, D.M. Molecular insights into the role of white adipose tissue in metabolically unhealthy normal weight and metabolically healthy obese individuals. FASEB J. 2015, 29, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S. Epigenetic alterations in aging. J. Appl. Physiol. 2010, 109, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Carless, M.A.; Kulkarni, H.; Kos, M.Z.; Charlesworth, J.; Peralta, J.M.; Goring, H.H.; Curran, J.E.; Almasy, L.; Dyer, T.D.; Comuzzie, A.G.; et al. Genetic effects on DNA methylation and its potential relevance for obesity in Mexican Americans. PLoS ONE 2013, 8, e73950. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.E.; Kelder, B.; Bobik, E.G.; Chuang, L.T.; Lewis, C.J.; Kopchick, J.J.; Mukerji, P.; Huang, Y.S. Identification and expression of mammalian long-chain PUFA elongation enzymes. Lipids 2002, 37, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.; Vinuela, A.; Davies, M.N.; Ramasamy, A.; Parts, L.; Knowles, D.; Brown, A.A.; Hedman, A.K.; Small, K.S.; Buil, A.; et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013, 14, R75. [Google Scholar] [CrossRef] [PubMed]
- Acampora, D.; D’Esposito, M.; Faiella, A.; Pannese, M.; Migliaccio, E.; Morelli, F.; Stornaiuolo, A.; Nigro, V.; Simeone, A.; Boncinelli, E. The human HOX gene family. Nucleic Acids Res. 1989, 17, 10385–10402. [Google Scholar] [CrossRef] [PubMed]
- Stelnicki, E.J.; Komuves, L.G.; Kwong, A.O.; Holmes, D.; Klein, P.; Rozenfeld, S.; Lawrence, H.J.; Adzick, N.S.; Harrison, M.; Largman, C. HOX homeobox genes exhibit spatial and temporal changes in expression during human skin development. J. Investig. Dermatol. 1998, 110, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, K.; Futerman, A.H. Do longevity assurance genes containing HOX domains regulate cell development via ceramide synthesis? FEBS Lett. 2002, 528, 3–4. [Google Scholar] [CrossRef]
- Qi, S.; Song, Y.; Peng, Y.; Wang, H.; Long, H.; Yu, X.; Li, Z.; Fang, L.; Wu, A.; Luo, W.; et al. ZEB2 mediates multiple pathways regulating cell proliferation, migration, invasion, and apoptosis in glioma. PLoS ONE 2012, 7, e38842. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Burd, C.G. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011, 21, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Bacalini, M.G.; Boattini, A.; Gentilini, D.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Remondini, D.; Capri, M.; del Rio, A.; et al. A meta-analysis on age-associated changes in blood DNA methylation: Results from an original analysis pipeline for Infinium 450K data. Aging 2015, 7, 97–109. [Google Scholar] [PubMed]
- Chan, Y.F.; Jones, F.C.; McConnell, E.; Bryk, J.; Bunger, L.; Tautz, D. Parallel selection mapping using artificially selected mice reveals body weight control loci. Curr. Biol. 2012, 22, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Benton, M.C.; Johnstone, A.; Eccles, D.; Harmon, B.; Hayes, M.T.; Lea, R.A.; Griffiths, L.; Hoffman, E.P.; Stubbs, R.S.; Macartney-Coxson, D. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 2015, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, C.M.; Gomez-Llorente, C.; Tofe, I.; Gil-Campos, M.; Canete, R.; Gil, A. Genome-wide expression in visceral adipose tissue from obese prepubertal children. Int. J. Mol. Sci. 2015, 16, 7723–7737. [Google Scholar] [CrossRef] [PubMed]
- Marucci, A.; di Mauro, L.; Menzaghi, C.; Prudente, S.; Mangiacotti, D.; Fini, G.; Lotti, G.; Trischitta, V.; di Paola, R. GALNT2 expression is reduced in patients with type 2 diabetes: Possible role of hyperglycemia. PLoS ONE 2013, 8, e70159. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.J.; Lim, H.W.; Ho, Y.; Shapira, S.N.; Ishibashi, J.; Rajakumari, S.; Steger, D.J.; Lazar, M.A.; Won, K.J.; Seale, P. PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev. 2015, 29, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Howe, L.D.; Paternoster, L.; Kaakinen, M.; Herrala, S.; Huikari, V.; Wu, Y.Y.; Kemp, J.P.; Timpson, N.J.; Pourcain, B.S.; et al. A genome-wide association study of body mass index across early life and childhood. Int. J. Epidemiol. 2015. [Google Scholar] [CrossRef]
- Choong, E.; Quteineh, L.; Cardinaux, J.R.; Gholam-Rezaee, M.; Vandenberghe, F.; Dobrinas, M.; Bondolfi, G.; Etter, M.; Holzer, L.; Magistretti, P.; et al. Influence of CRTC1 polymorphisms on body mass index and fat mass in psychiatric patients and the general adult population. JAMA Psychiatry 2013, 70, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zheng, D.; Fan, C.; Liu, Z.; Dong, H.; Lu, Y.; Qi, K. Genome-wide screen of DNA methylation identifies novel markers in childhood obesity. Gene 2015, 566, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Yan, S.; Guan, X.; Pan, Y.; Qu, X. Hypermethylation of the PRKCZ gene in type 2 diabetes mellitus. J. Diabetes Res. 2013, 2013, 721493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.F.; Santella, R.M.; Wolff, M.; Kappil, M.A.; Markowitz, S.B.; Morabia, A. White blood cell global methylation and IL-6 promoter methylation in association with diet and lifestyle risk factors in a cancer-free population. Epigenetics 2012, 7, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, S.; Barnes, V.A.; de Miguel, C.; Pollock, J.; Ownby, D.; Shi, H.; Zhu, H.; Snieder, H.; Wang, X. A genome-wide methylation study on obesity: Differential variability and differential methylation. Epigenetics 2013, 8, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, H.; Snieder, H.; Su, S.; Munn, D.; Harshfield, G.; Maria, B.L.; Dong, Y.; Treiber, F.; Gutin, B.; et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Campion, J.; Milagro, F.I.; Goyenechea, E.; Steemburgo, T.; Javierre, B.M.; Martinez, J.A. Leptin and TNF-α promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J. Physiol. Biochem. 2011, 67, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Milagro, F.I.; Campion, J.; Cordero, P.; Goyenechea, E.; Gomez-Uriz, A.M.; Abete, I.; Zulet, M.A.; Martinez, J.A. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J. 2011, 25, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Moleres, A.; Campion, J.; Milagro, F.I.; Marcos, A.; Campoy, C.; Garagorri, J.M.; Gomez-Martinez, S.; Martinez, J.A.; Azcona-Sanjulian, M.C.; Marti, A. Differential DNA methylation patterns between high and low responders to a weight loss intervention in overweight or obese adolescents: The EVASYON study. FASEB J. 2013, 27, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, M.; Ismail, K.; Gervin, K.; Kyllonen, A.; Hakkarainen, A.; Lundbom, J.; Jarvinen, E.A.; Harris, J.R.; Lundbom, N.; Rissanen, A.; et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin. Epigenet. 2015, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccarelli, A.A.; Byun, H.M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenet. 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008, 9, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Uriz, A.M.; Milagro, F.I.; Mansego, M.L.; Cordero, P.; Abete, I.; de Arce, A.; Goyenechea, E.; Blazquez, V.; Martinez-Zabaleta, M.; Martinez, J.A.; et al. Obesity and ischemic stroke modulate the methylation levels of KCNQ1 in white blood cells. Hum. Mol. Genet. 2015, 24, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Udali, S.; Guarini, P.; Pellegrini, C.; Pattini, P.; Moruzzi, S.; Girelli, D.; Pizzolo, F.; Martinelli, N.; Corrocher, R.; et al. Global DNA hypomethylation in peripheral blood mononuclear cells as a biomarker of cancer risk. Cancer Epidemiol. Biomark. Prev. 2013, 22, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Zulet, M.A.; Bondia-Pons, I.; Abete, I.; de la Iglesia, R.; Lopez-Legarrea, P.; Forga, L.; Navas-Carretero, S.; Martinez, J.A. The reduction of the metabolyc syndrome in Navarra-Spain (RESMENA-S) study: A multidisciplinary strategy based on chrononutrition and nutritional education, together with dietetic and psychological control. Nutr. Hosp. 2011, 26, 16–26. [Google Scholar] [PubMed]
- Huerta, A.E.; Navas-Carretero, S.; Prieto-Hontoria, P.L.; Martinez, J.A.; Moreno-Aliaga, M.J. Effects of α-lipoic acid and eicosapentaenoic acid in overweight and obese women during weight loss. Obesity 2015, 23, 313–321. [Google Scholar] [CrossRef] [PubMed]
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) final report. Circulation 2002, 106, 3143–3421. [Google Scholar]
- Seidell, J.C.; Flegal, K.M. Assessing obesity: Classification and epidemiology. Br. Med. Bull. 1997, 53, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Touleimat, N.; Tost, J. Complete pipeline for Infinium® Human Methylation 450K Beadchip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012, 4, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Naeem, H.; Wong, N.C.; Chatterton, Z.; Hong, M.K.; Pedersen, J.S.; Corcoran, N.M.; Hovens, C.M.; Macintyre, G. Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the HumanMethylation450 array. BMC Genomics 2014, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Wettenhall, J.M.; Smyth, G.K. Limmagui: A graphical user interface for linear modeling of microarray data. Bioinformatics 2004, 20, 3705–3706. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansego, M.L.; Milagro, F.I.; Zulet, M.Á.; Moreno-Aliaga, M.J.; Martínez, J.A. Differential DNA Methylation in Relation to Age and Health Risks of Obesity. Int. J. Mol. Sci. 2015, 16, 16816-16832. https://doi.org/10.3390/ijms160816816
Mansego ML, Milagro FI, Zulet MÁ, Moreno-Aliaga MJ, Martínez JA. Differential DNA Methylation in Relation to Age and Health Risks of Obesity. International Journal of Molecular Sciences. 2015; 16(8):16816-16832. https://doi.org/10.3390/ijms160816816
Chicago/Turabian StyleMansego, María Luisa, Fermín I. Milagro, María Ángeles Zulet, María J. Moreno-Aliaga, and José Alfredo Martínez. 2015. "Differential DNA Methylation in Relation to Age and Health Risks of Obesity" International Journal of Molecular Sciences 16, no. 8: 16816-16832. https://doi.org/10.3390/ijms160816816