
Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage

{kind=link}
{kind=link}
Abstract
:1. Introduction
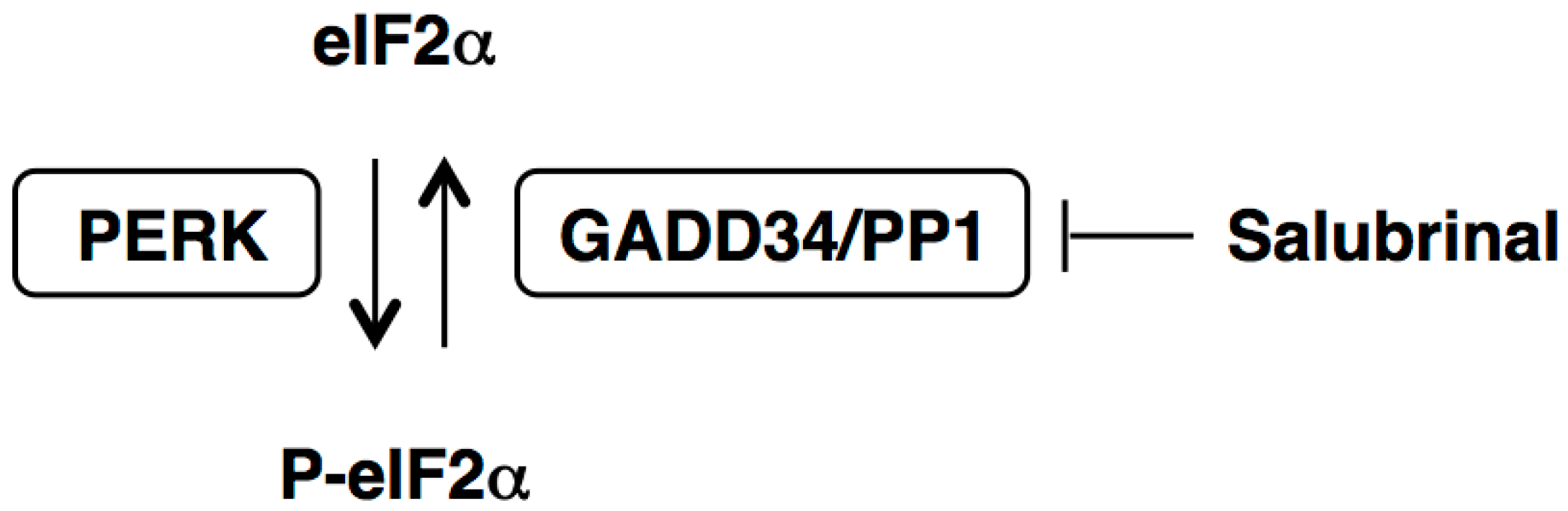
2. Effects of Salubrinal on Xenotoxicant-Induced Cellular Damage
2.1. Cadmium
2.2. Arsenic
2.3. Paraquat
2.4. Rotenone
2.5. Benzo[a]pyrene-7,8-diol-9,10-epoxide
2.6. 2,3,7,8-Tetrachlorodibenzo-p-dioxin
2.7. Cigarette Smoke
2.8. Cisplatin
2.9. Cyclosporine
3. Possible Mechanisms of Protection by Salubrinal from Xenotoxicant-Induced Cellular Damage
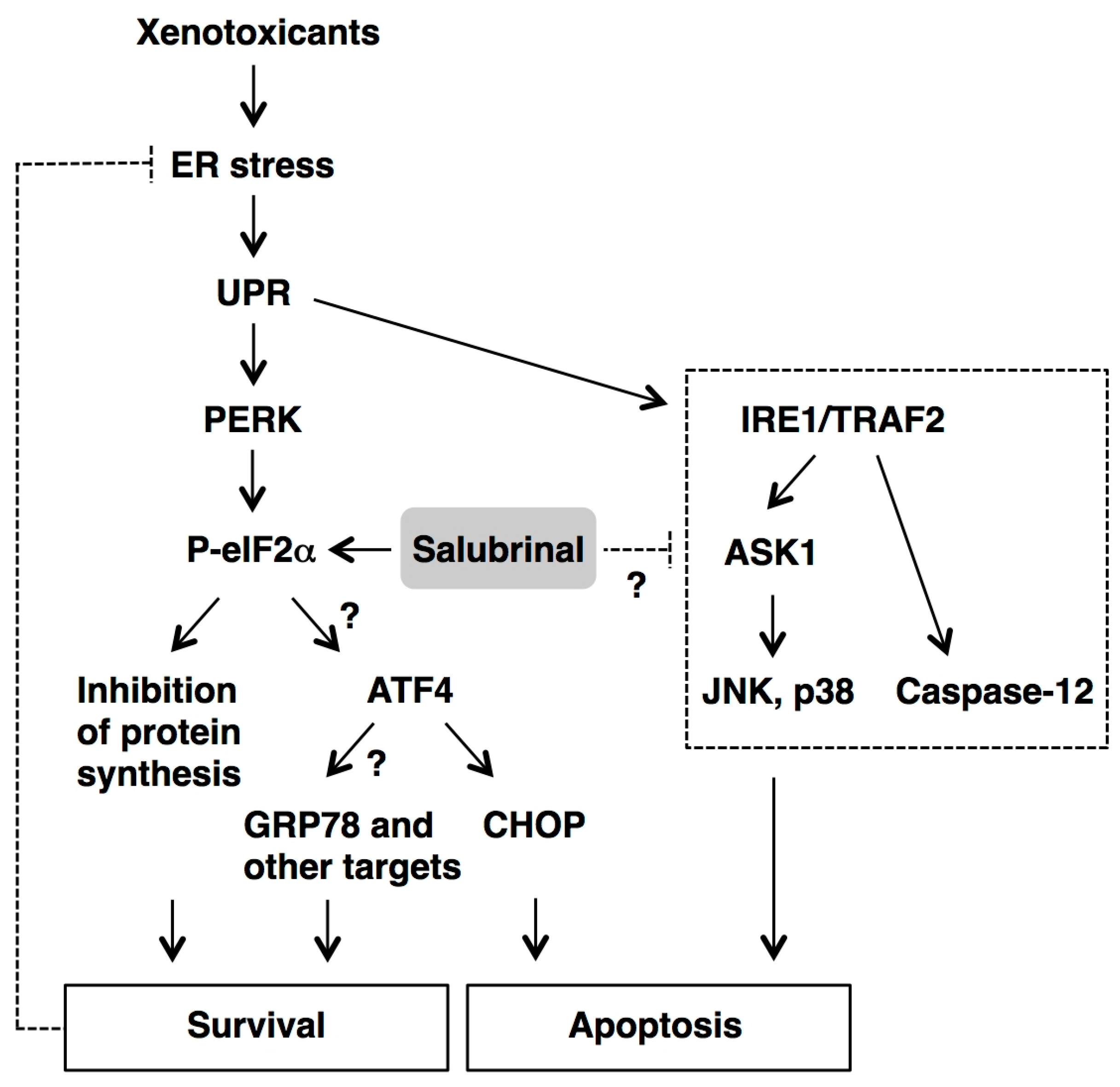
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brostrom, M.A.; Brostrom, C.O. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: Implications for cell growth and adaptability. Cell Calcium 2003, 34, 345–363. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Endoplasmic reticulum stress as a progression factor for kidney injury. Curr. Opin. Pharmacol. 2010, 10, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M. The unfolded protein response triggered by environmental factors. Semin. Immunopathol. 2013, 35, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Long, K.; Boyce, M.; Lin, H.; Yuan, J.; Ma, D. Structure-activity relationship studies of salubrinal lead to its active biotinylated derivative. Bioorg. Med. Chem. Lett. 2005, 15, 3849–3852. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Komoike, Y.; Inamura, H.; Matsuoka, M. Effects of salubrinal on cadmium-induced apoptosis in HK-2 human renal proximal tubular cells. Arch. Toxicol. 2012, 86, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Inageda, K.; Nishitai, G.; Matsuoka, M. Cadmium induces the expression of Grp78, an endoplasmic reticulum molecular chaperone, in LLC-PK1 renal epithelial cells. Environ. Health Perspect. 2006, 114, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Yokouchi, M.; Hiramatsu, N.; Hayakawa, K.; Kasai, A.; Takano, Y.; Yao, J.; Kitamura, M. Atypical, bidirectional regulation of cadmium-induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ. 2007, 14, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Hiramatsu, N. The oxidative stress: Endoplasmic reticulum stress axis in cadmium toxicity. Biometals 2010, 23, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of Grp78/BiP by translational block: Activation of the Grp78 promoter by ATF4 through an upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.H.; Chao, P.L.; Tsai, M.J.; Cheng, H.H.; Chen, K.B.; Yang, D.M.; Yang, C.H.; Lin, A.M.Y. Arsenite-induced cytotoxicity in dorsal root ganglion explants. Free Radic. Biol. Med. 2008, 44, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-Y.; Chiou, S.-Y.; Wang, L.; Kou, M.-C.; Wang, Y.-J.; Wu, M.-J. Arsenic trioxide induces unfolded protein response in vascular endothelial cells. Arch. Toxicol. 2014, 88, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jiang, C.; Sun, X. Therapeutical effects and mechanism of salubrinal combined with ulinastatin on treating paraquat poisoning. Cell Biochem. Biophys. 2014, 70, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tiffany-Castiglioni, E.; Koh, H.C.; Son, I.H. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxicol. Lett. 2009, 191, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Bravo-San Pedro, J.M.; Gómez-Sánchez, R.; Climent, V.; Soler, G.; Fuentes, J.M.; González-Polo, R.A. ASK1 overexpression accelerates paraquat-induced autophagy via endoplasmic reticulum stress. Toxicol. Sci. 2011, 119, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Luo, N.; Zhao, H.-R.; Gao, Q.; Lu, J.; Pan, Y.; Shi, J.-P.; Tian, Y.-Y.; Zhang, Y.-D. Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 2014, 1549, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Gupta, S.; Biswas, J.; Joshi, N.; Swarnkar, S.; Nath, C.; Singh, S. Endoplasmic reticulum stress plays a key role in rotenone-induced apoptotic death of neurons. Mol. Neurobiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jiang, H.; Fan, Y.; Huang, X.; Shen, J.; Qi, H.; Li, Q.; Lu, X.; Shao, J. Phosphorylation of the α-subunit of the eukaryotic initiation factor-2 (eIF2α) alleviates benzo[a]pyrene-7,8-diol-9,10-epoxide induced cell cycle arrest and apoptosis in human cells. Environ. Toxicol. Pharmacol. 2011, 31, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zhao, J.; Fan, X.; Tang, C.; Liang, L.; Nie, X.; Liu, J.; Wu, Q.; Xu, G. The PERK-eIF2α signaling pathway is involved in TCDD-induced ER stress in PC12 cells. Neurotoxicology 2014, 44, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Luo, B.-L.; Wei, T.-H.; Zhang, L.; He, B.-M.; Niu, R.-C. Salubrinal protects against cigarette smoke extract-induced HBEpC apoptosis likely via regulating the activity of PERK-eIF2α signaling pathway. Arch. Med. Res. 2012, 43, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Min, T.; Bodas, M.; Mazur, S.; Vij, N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 2011, 89, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-I.; Kang, K.-L.; Shin, S.-I.; Herr, Y.; Lee, Y.-M.; Kim, E.-C. Endoplasmic reticulum stress modulates nicotine-induced extracellular matrix degradation in human periodontal ligament cells. J. Periodontal Res. 2012, 47, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Sheu, M.L.; Tsai, K.S.; Weng, T.I.; Chiang, C.K.; Liu, S.H. The role of endoplasmic reticulum stress-related unfolded protein response in the radiocontrast medium-induced renal tubular cell injury. Toxicol. Sci. 2010, 114, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Sheu, M.L.; Tsai, K.S.; Chiang, C.K.; Liu, S.H. Salubrinal, an eIF2α dephosphorylation inhibitor, enhances cisplatin-induced oxidative stress and nephrotoxicity in a mouse model. Free Radic. Biol. Med. 2011, 51, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Bouvier, N.; Bendjallabah, A.; Rabant, M.; Flinois, J.P.; Hertig, A.; Legendre, C.; Beaune, P.; Thervet, E.; Anglicheau, D. Cyclosporine-induced endoplasmic reticulum stress triggers tubular phenotypic changes and death. Am. J. Transplant. 2008, 8, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Bouvier, N.; Legendre, C.; Gilleron, J.; Codogno, P.; Beaune, P.; Thervet, E.; Anglicheau, D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 2008, 4, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, N.; Flinois, J.P.; Gilleron, J.; Sauvage, F.-L.; Legendre, C.; Beaune, P.; Thervet, E.; Anglicheau, D.; Pallet, N. Cyclosporine triggers endoplasmic reticulum stress in endothelial cells: A role for endothelial phenotypic changes and death. Am. J. Physiol. Ren. Physiol. 2009, 296, F160–F169. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Brewer, J.W. Regulatory crosstalk within the mammalian unfolded protein response. Cell. Mol. Life Sci. 2014, 71, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, Y.; Zhang, H.; Ma, Q.; Zhang, Y.-W.; Xu, H. Salubrinal attenuates β-amyloid-induced neuronal death and microglial activation by inhibition of the NF-κB pathway. Neurobiol. Aging 2012, 33, 1007.e9–1007.e17. [Google Scholar] [CrossRef] [PubMed]
- Hamamura, K.; Lin, C.-C.; Yokota, H. Salubrinal reduces expression and activity of MMP13 in chondrocytes. Osteoarthr. Cartil. 2013, 21, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D. Protection of Bcl-2 by salubrinal. Biochem. Biophys. Res. Commun. 2006, 346, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Basal levels of eIF2α phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J. Biol. Chem. 2009, 284, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, K.-S.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Jung, H.-Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.; Lee, W.; Chung, H.; Jung, Y.-K. Amyloid β-induced FOXRED2 mediates neuronal cell death via inhibition of proteasome activity. Cell. Mol. Life Sci. 2011, 68, 2115–2127. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Reijonen, S.; Putkonen, N.; Nørremølle, A.; Lindholm, D.; Korhonen, L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res. 2008, 314, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.K.; Shin, K.S.; Yuan, J.; Kang, S.J. Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J. Neurochem. 2008, 104, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Wang, Q.; Lin, Z.; Chen, M.-L.; Sun, G.-Z. Endoplasmic reticulum (ER) stress inhibitor salubrinal protects against ceramide-induced SH-SY5Y cell death. Biochem. Biophys. Res. Commun. 2012, 427, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Sokka, A.-L.; Putkonen, N.; Mudo, G.; Pryazhnikov, E.; Reijonen, S.; Khiroug, L.; Belluardo, N.; Lindholm, D.; Korhonen, L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J. Neurosci. 2007, 27, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Heo, R.W.; Kim, H.; Yi, C.-O.; Shin, H.J.; Han, J.W.; Roh, G.S. Salubrinal, ER stress inhibitor, attenuates kainic acid-induced hippocampal cell death. J. Neural Transm. 2014, 121, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuoka, M.; Komoike, Y. Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. Int. J. Mol. Sci. 2015, 16, 16275-16287. https://doi.org/10.3390/ijms160716275
Matsuoka M, Komoike Y. Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. International Journal of Molecular Sciences. 2015; 16(7):16275-16287. https://doi.org/10.3390/ijms160716275
Chicago/Turabian StyleMatsuoka, Masato, and Yuta Komoike. 2015. "Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage" International Journal of Molecular Sciences 16, no. 7: 16275-16287. https://doi.org/10.3390/ijms160716275