
A Method for Systematic Assessment of Intrinsically Disordered Protein Regions by NMR


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
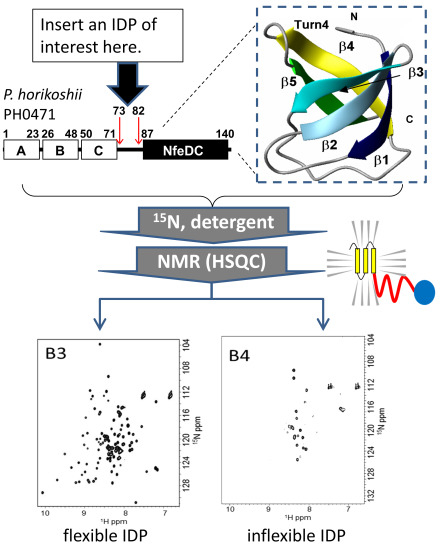
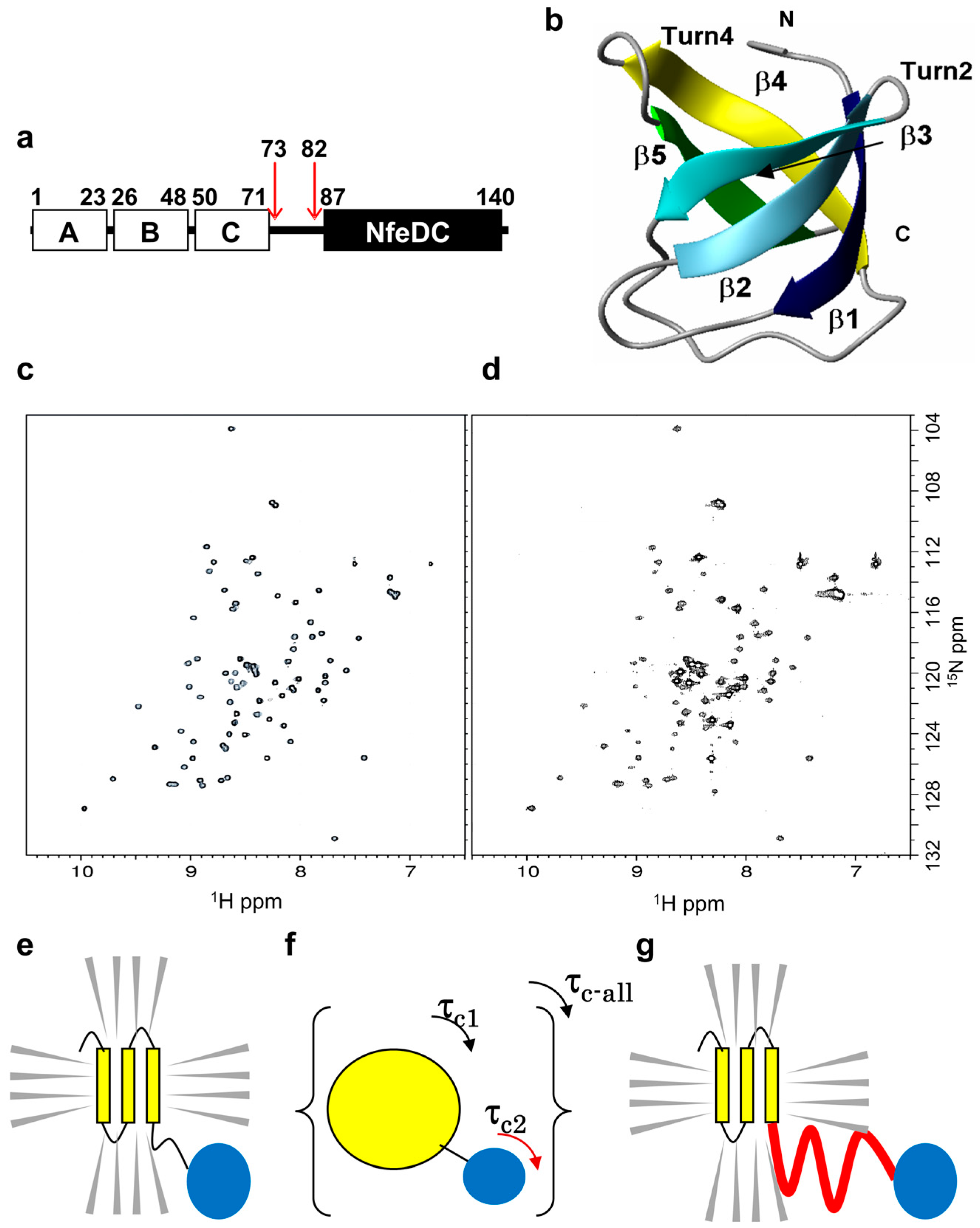
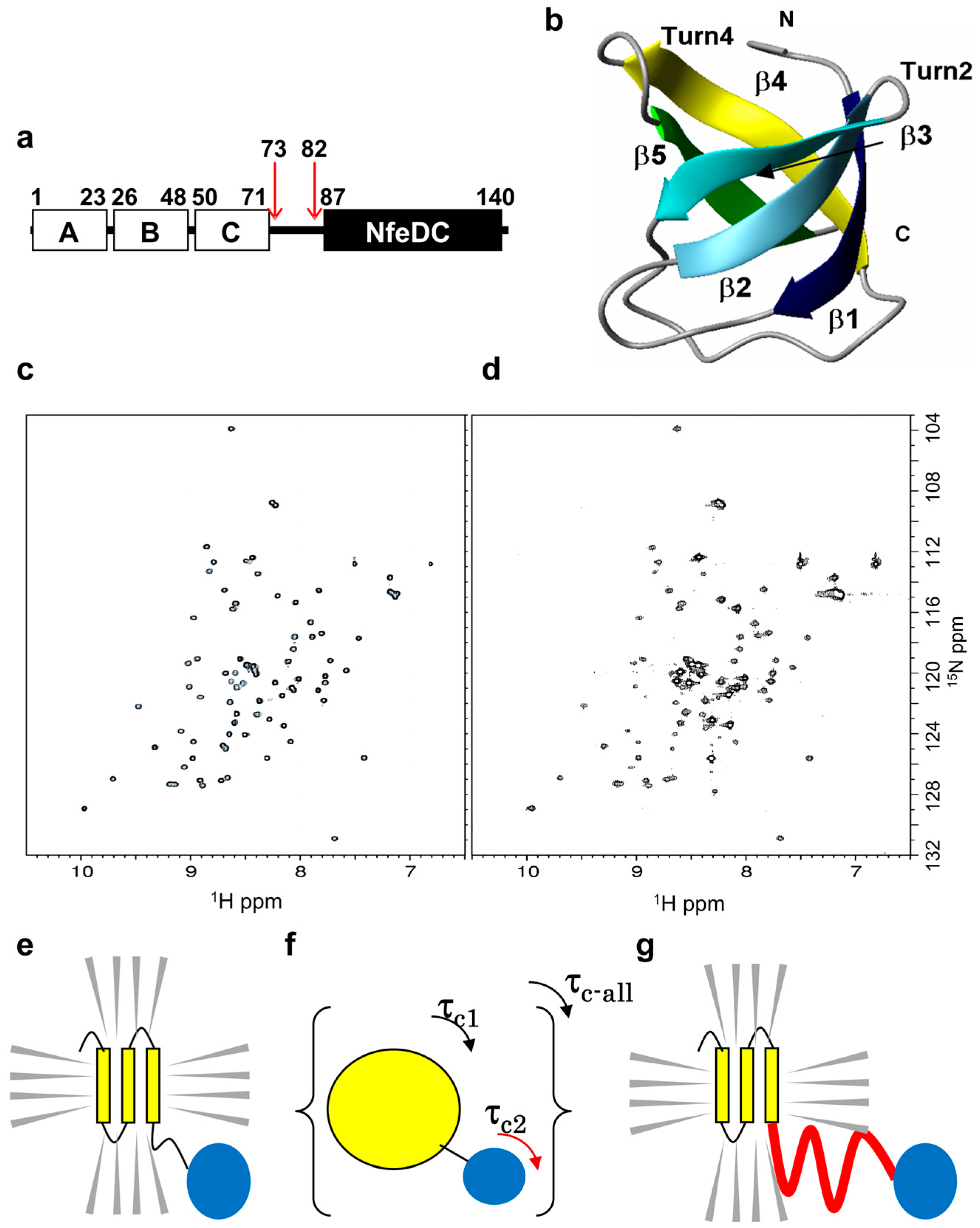
2.1. Theoretical Background
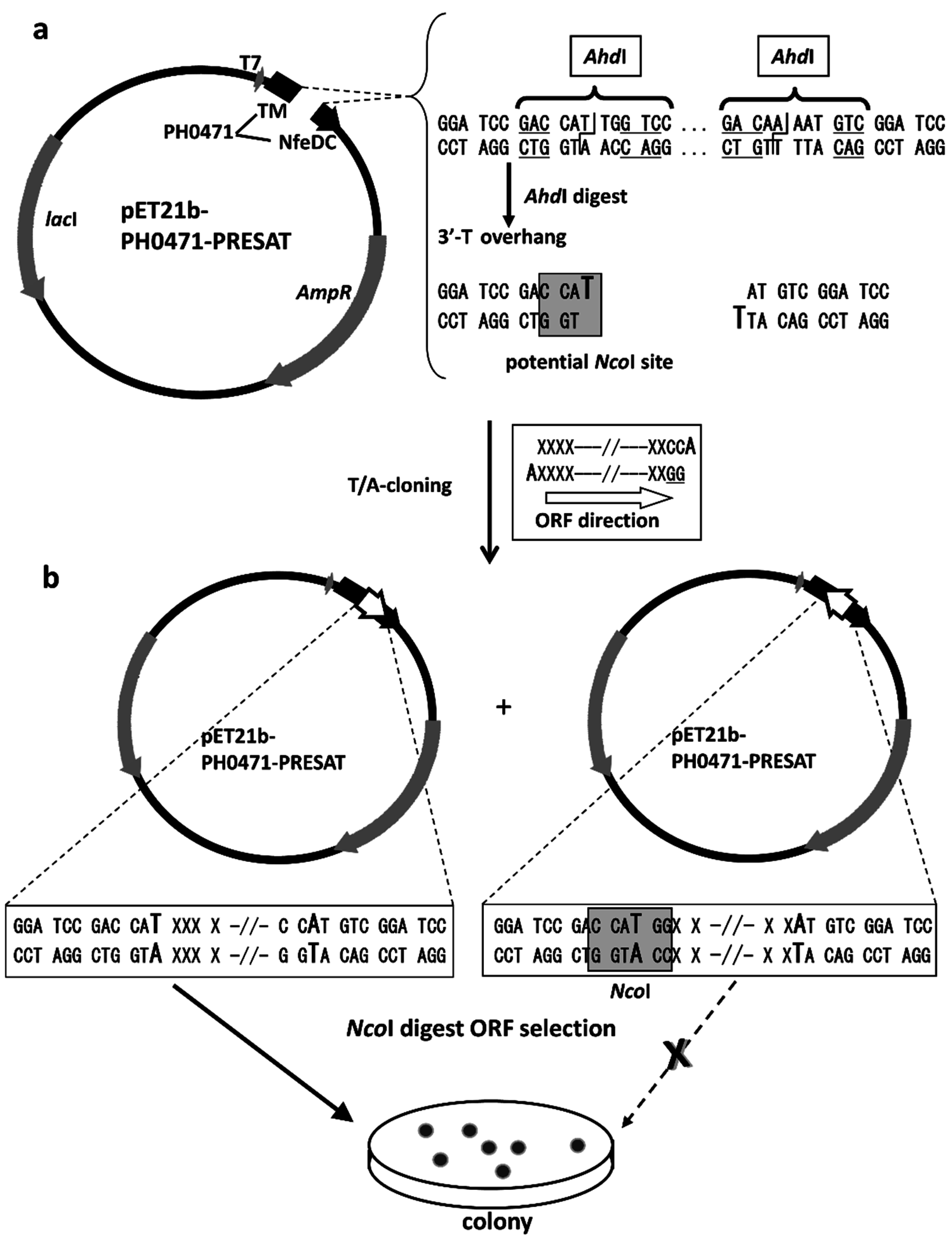
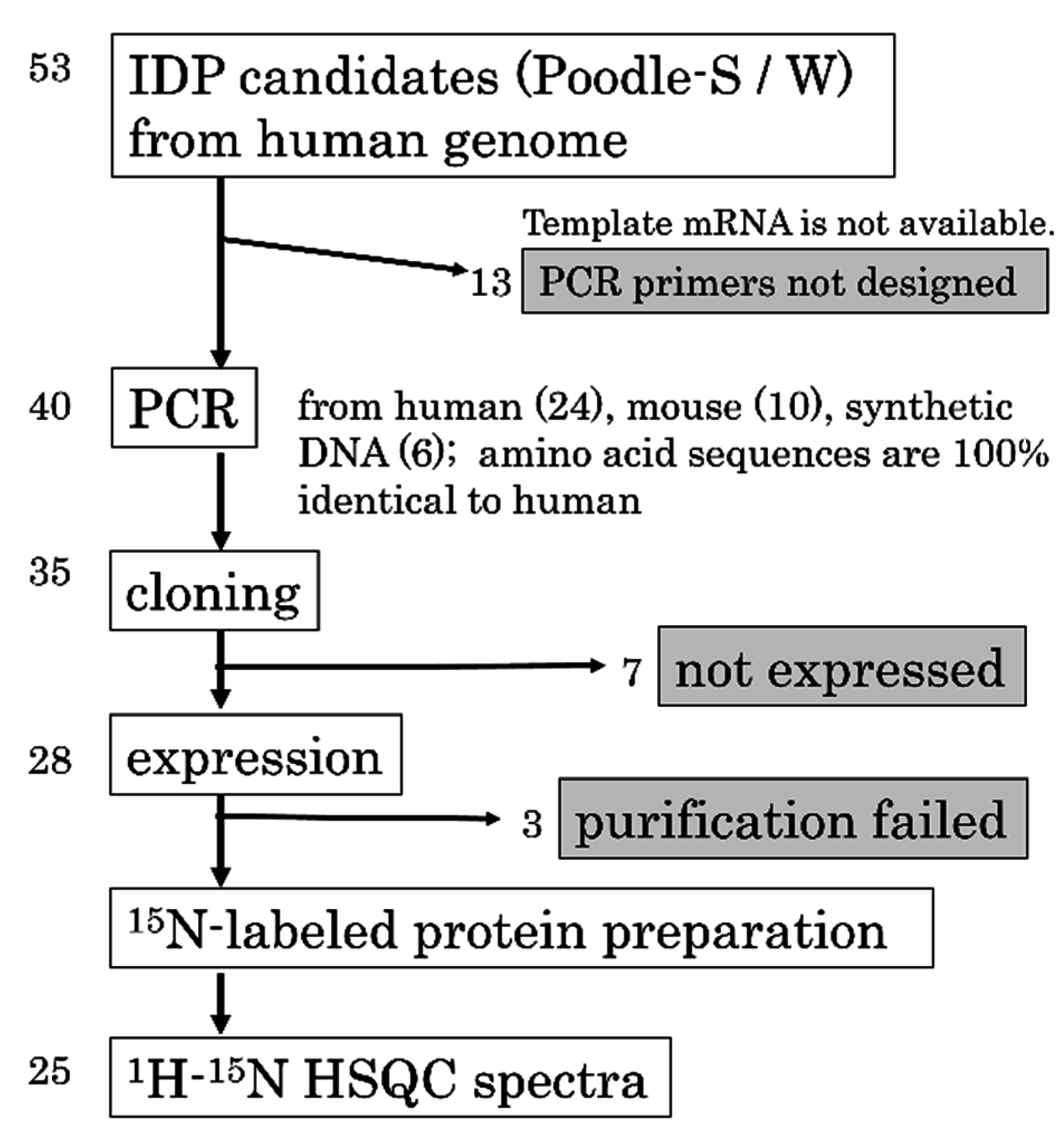
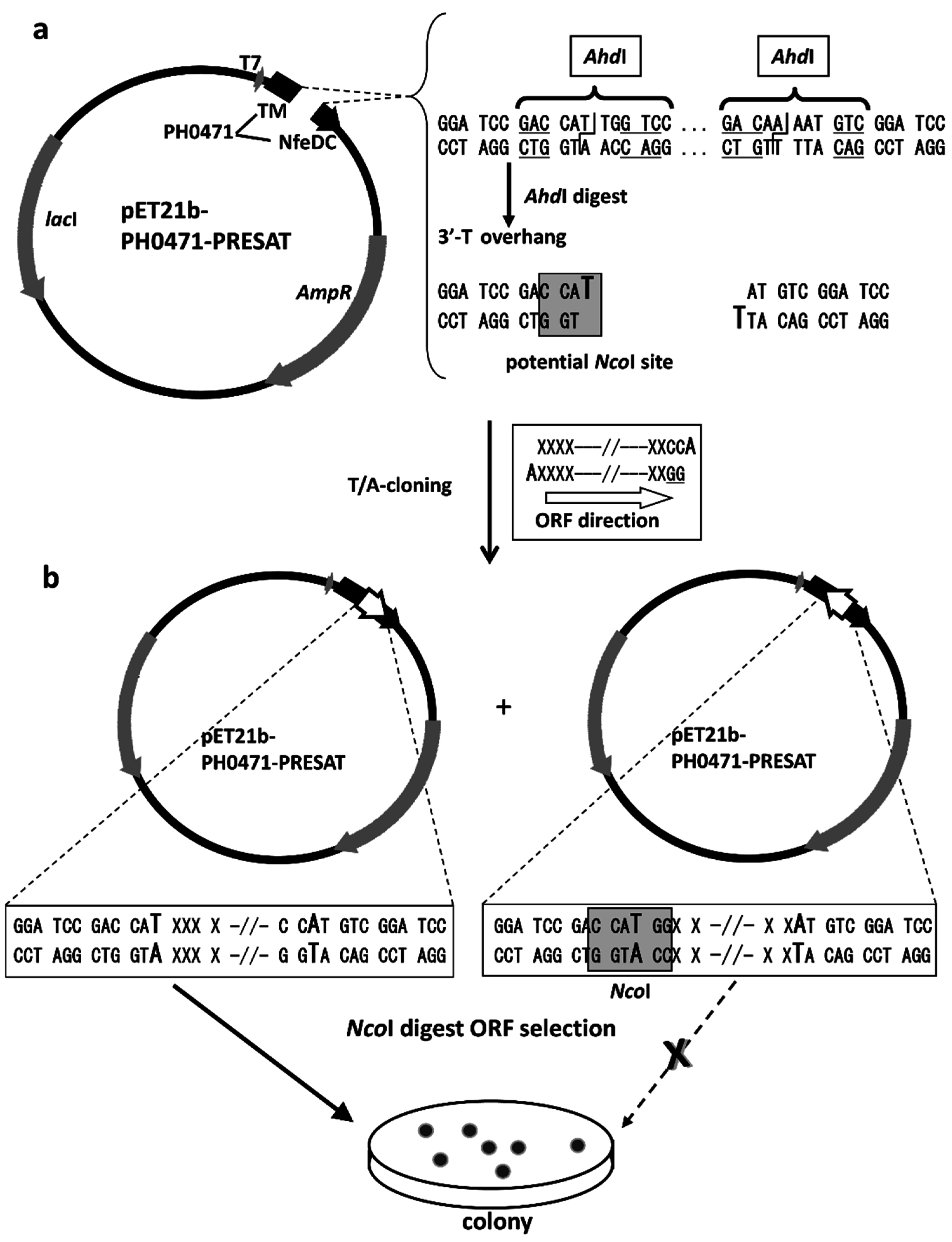
2.2. Design of the PH0471-Based PRESAT Vector
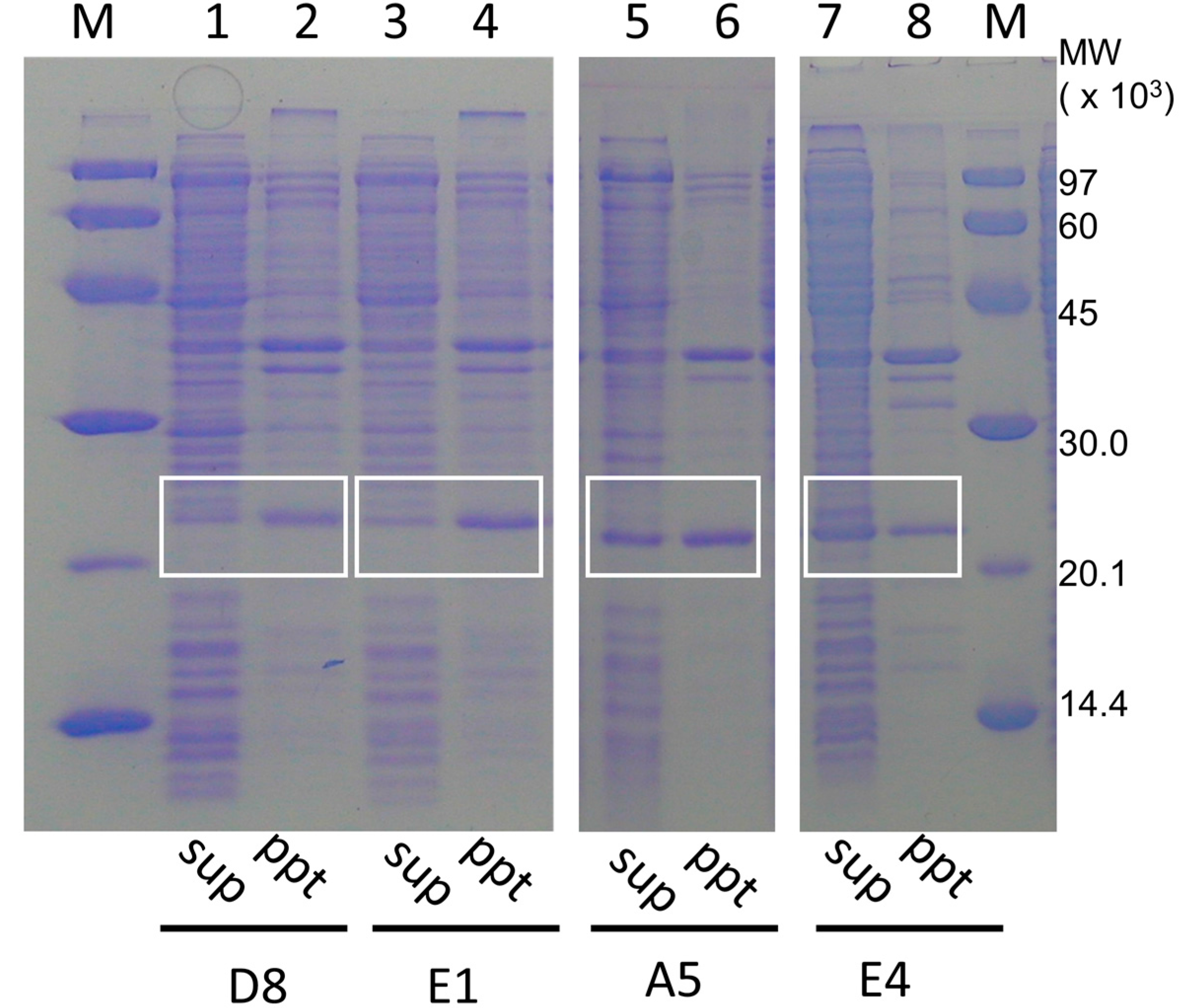
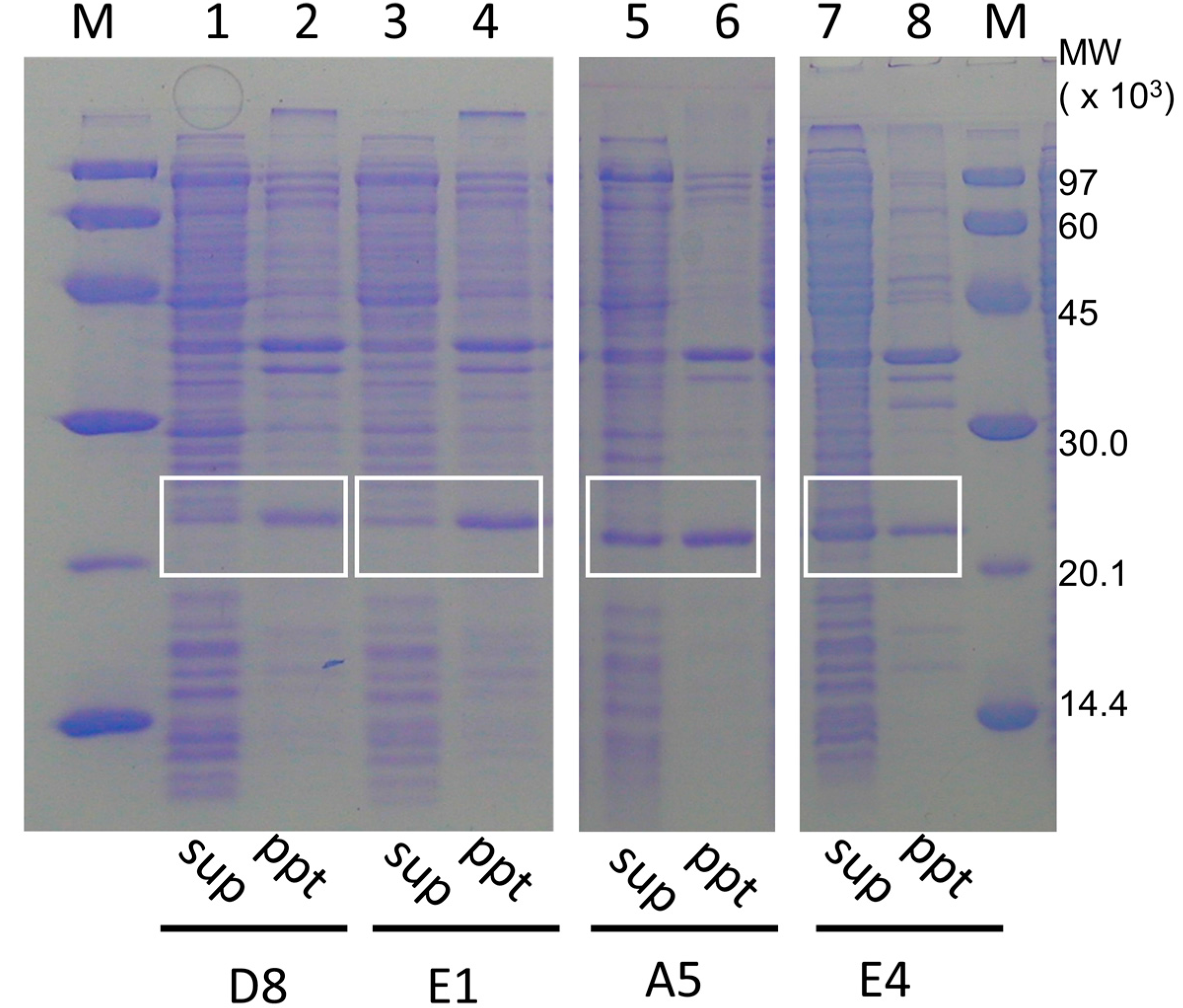
2.3. Proof of Method
2.4. Strategy and Workflow of the Systematic Assessment of IDPs by NMR

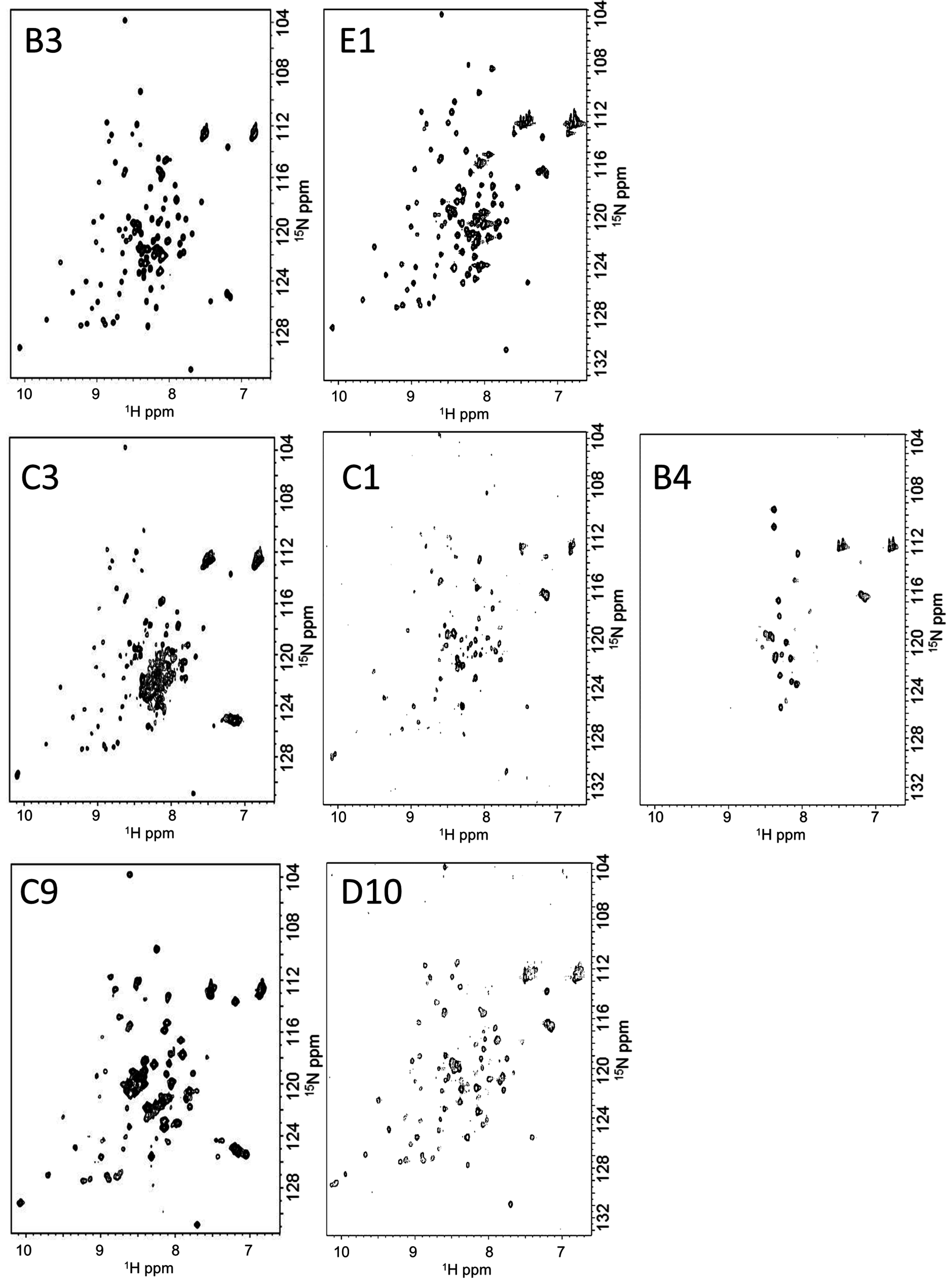
2.5. IDPs Have Different Flexibility in Solution

3. Discussion
4. Experimental Section
4.1. Materials
4.2. Vector Construction
4.3. Protein Techniques
4.4. NMR Spectroscopy
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tompa, P. Structure and Function of Intrinsically Disordered Proteins; Chapman and Hall/CRC: New York, NY, USA, 2009. [Google Scholar]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Ota, M.; Koike, R.; Amemiya, T.; Tenno, T.; Romero, P.R.; Hiroaki, H.; Dunker, A.K.; Fukuchi, S. An assignment of intrinsically disordered regions of proteins based on NMR structures. J. Struct. Biol. 2013, 181, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 2000, 11, 161–171. [Google Scholar] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Minezaki, Y.; Homma, K.; Kinjo, A.R.; Nishikawa, K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006, 359, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Toh, H. Interaction between intrinsically disordered proteins frequently occurs in a human protein-protein interaction network. J. Mol. Biol. 2009, 392, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Cannon, P.; Romero, P.; Obradovic, Z.; Dunker, A. Predicting disordered regions from amino acid sequence: Common themes despite differing structural characterization. Genome Inform. Ser. Workshop Genome Inform. 1998, 9, 201–213. [Google Scholar] [PubMed]
- Jones, D.T.; Ward, J.J. Prediction of disordered regions in proteins from position specific score matrices. Proteins 2003, 53 (Suppl. 6), 573–578. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.; Obradovic, Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Work Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K. Predicting intrinsic disorder from amino acid sequence. Proteins 2003, 53 (Suppl. 6), 566–572. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Dunker, A.K. Order, disorder, and flexibility: Prediction from protein sequence. Structure 2003, 11, 1316–1317. [Google Scholar] [CrossRef] [PubMed]
- Hecker, J.; Yang, J.Y.; Cheng, J. Protein disorder prediction at multiple levels of sensitivity and specificity. BMC Genomics 2008, 9 (Suppl. 1), S9. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Homma, K.; Minezaki, Y.; Gojobori, T.; Nishikawa, K. Development of an accurate classification system of proteins into structured and unstructured regions that uncovers novel structural domains: Its application to human transcription factors. BMC Struct. Biol. 2009, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Hirose, S.; Noguchi, T. POODLE-S: Web application for predicting protein disorder by using physicochemical features and reduced amino acid set of a position-specific scoring matrix. Bioinformatics 2007, 23, 2337–2338. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Muraoka, Y.; Hirose, S.; Tomii, K.; Noguchi, T. Predicting mostly disordered proteins by using structure-unknown protein data. BMC Bioinform. 2007, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Shimizu, K.; Kanai, S.; Kuroda, Y.; Noguchi, T. POODLE-L: A two-level SVM prediction system for reliably predicting long disordered regions. Bioinformatics 2007, 23, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Weathers, E.A.; Paulaitis, M.E.; Woolf, T.B.; Hoh, J.H. Reduced amino acid alphabet is sufficient to accurately recognize intrinsically disordered protein. FEBS Lett. 2004, 576, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Vucetic, S.; Obradovic, Z.; Vacic, V.; Radivojac, P.; Peng, K.; Iakoucheva, L.M.; Cortese, M.S.; Lawson, J.D.; Brown, C.J.; Sikes, J.G.; et al. DisProt: A database of protein disorder. Bioinformatics 2005, 21, 137–140. [Google Scholar]
- Fukuchi, S.; Sakamoto, S.; Nobe, Y.; Murakami, S.D.; Amemiya, T.; Hosoda, K.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL: Intrinsically disordered proteins with extensive annotations and literature. Nucleic Acids Res. 2012, 40, D507–D511. [Google Scholar] [CrossRef] [PubMed]
- Potenza, E.; di Domenico, T.; Walsh, I.; Tosatto, S.C.E. MobiDB 2.0: An improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2014, 43, D315–D320. [Google Scholar]
- Varadi, M.; Kosol, S.; Lebrun, P.; Valentini, E.; Blackledge, M.; Dunker, A.K.; Felli, I.C.; Forman-Kay, J.D.; Kriwacki, R.W.; Pierattelli, R.; et al. pE-DB: A database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucleic Acids Res. 2014, 42, D326–D335. [Google Scholar]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wüthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J. Biomol. NMR 1998, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Robien, M.A.; Gronenborn, A.M. Exploring the limits of precision and accuracy of protein structures determined by nuclear magnetic resonance spectroscopy. J. Mol. Biol. 1993, 231, 82–102. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, A.M.; Brünger, A.T. Do NOE distances contain enough information to assess the relative populations of multi-conformer structures? J. Biomol. NMR 1996, 7, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Nilges, M.; Habeck, M.; O’Donoghue, S.I.; Rieping, W. Error distribution derived NOE distance restraints. Proteins 2006, 64, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Goda, N.; Tenno, T.; Takasu, H.; Hiroaki, H.; Shirakawa, M. The PRESAT-vector: Asymmetric T-vector for high-throughput screening of soluble protein domains for structural proteomics. Protein Sci. 2004, 13, 652–628. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Ohno, A.; Morii, T.; Yokoyama, H.; Matsui, I.; Tochio, H.; Shirakawa, M.; Hiroaki, H. The solution structure of the C-terminal domain of NfeD reveals a novel membrane-anchored OB-fold. Protein Sci. 2008, 17, 1915–1924. [Google Scholar] [CrossRef] [PubMed]
- Tenno, T.; Goda, N.; Tateishi, Y.; Tochio, H.; Mishima, M.; Hayashi, H.; Shirakawa, M.; Hiroaki, H. High-throughput construction method for expression vector of peptides for NMR study suited for isotopic labeling. Protein Eng. Des. Sel. 2004, 17, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Goda, N.; Tenno, T.; Inomata, K.; Iwaya, N.; Sasaki, Y.; Shirakawa, M.; Hiroaki, H. LBT/PTD dual tagged vector for purification, cellular protein delivery and visualization in living cells. Biochim. Biophys. Acta 2007, 1773, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.R.; Suresh, M.; Kumaran, K.; Kannabiran, N.; Suresh, S.; Bala, P.; Shivakumar, K.; Anuradha, N.; Reddy, R.; Raghavan, T.M.; et al. Human protein reference database—2006 Update. Nucleic Acids Res. 2006, 34, D411–D414. [Google Scholar]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Tsai, C.-J.; Kumar, S.; Zanuy, D.; Nussinov, R. Extended disordered proteins: Targeting function with less scaffold. Trends Biochem. Sci. 2003, 28, 81–85. [Google Scholar] [CrossRef]
- Dafforn, T.R.; Smith, C.J.I. Natively unfolded domains in endocytosis: Hooks, lines and linkers. EMBO Rep. 2004, 5, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Owen, D.J. Endocytosis and vesicle trafficking. Curr. Opin. Struct. Biol. 2002, 12, 814–821. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N.; Wolynes, P.G. Fly-casting in protein-DNA binding: Frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 2007, 129, 738–739. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Dötsch, V. In-cell NMR spectroscopy. Biochemistry 2001, 40, 14317–14323. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Tochio, H.; Tenno, T.; Ito, Y.; Kokubo, T.; Hiroaki, H.; Shirakawa, M. In-cell NMR spectroscopy of proteins inside Xenopus laevis oocytes. J. Biomol. NMR 2006, 36, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Selenko, P.; Hänsel, R.; Reckel, S.; Löhr, F.; Ferrell, J.E.; Wagner, G.; Dötsch, V. Investigating macromolecules inside cultured and injected cells by in-cell NMR spectroscopy. Nat. Protoc. 2006, 1, 2701–2709. [Google Scholar] [CrossRef] [PubMed]
- Inomata, K.; Ohno, A.; Tochio, H.; Isogai, S.; Tenno, T.; Nakase, I.; Takeuchi, T.; Futaki, S.; Ito, Y.; Hiroaki, H.; et al. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458, 106–109. [Google Scholar]
- Livernois, A.M.; Hnatchuk, D.J.; Findlater, E.E.; Graether, S.P. Obtaining highly purified intrinsically disordered protein by boiling lysis and single step ion exchange. Anal. Biochem. 2009, 392, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Csokova, N.; Skrabana, R.; Liebig, H.-D.; Mederlyova, A.; Kontsek, P.; Novak, M. Rapid purification of truncated tau proteins: Model approach to purification of functionally active fragments of disordered proteins, implication for neurodegenerative diseases. Protein Expr. Purif. 2004, 35, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Moncoq, K.; Broutin, I.; Larue, V.; Perdereau, D.; Cailliau, K.; Browaeys-Poly, E.; Burnol, A.-F.; Ducruix, A. The PIR domain of Grb14 is an intrinsically unstructured protein: Implication in insulin signaling. FEBS Lett. 2003, 554, 240–246. [Google Scholar] [CrossRef]
- Uegaki, K.; Nemoto, N.; Shimizu, M.; Wada, T.; Kyogoku, Y.; Kobayashi, Y. 15N labeling method of peptides using a thioredoxin gene fusion expression system: An application to ACTH-(1–24). FEBS Lett. 1996, 379, 47–50. [Google Scholar] [CrossRef]
- Achmüller, C.; Kaar, W.; Ahrer, K.; Wechner, P.; Hahn, R.; Werther, F.; Schmidinger, H.; Cserjan-Puschmann, M.; Clementschitsch, F.; Striedner, G.; et al. N(pro) fusion technology to produce proteins with authentic N termini in E. coli. Nat. Methods 2007, 4, 1037–1043. [Google Scholar]
- Goda, N.; Matsuo, N.; Tenno, T.; Ishino, S.; Ishino, Y.; Fukuchi, S.; Ota, M.; Hiroaki, H. An optimized Npro-based method for the expression and purification of intrinsically disordered proteins for an NMR study. Intrinsically Disord. Proteins 2015, 3, 1–6. [Google Scholar] [CrossRef]
- Santner, A.A.; Croy, C.H.; Vasanwala, F.H.; Uversky, V.N.; Van, Y.-Y.J.; Dunker, A.K. Sweeping away protein aggregation with entropic bristles: Intrinsically disordered protein fusions enhance soluble expression. Biochemistry 2012, 51, 7250–7262. [Google Scholar] [CrossRef] [PubMed]
- Reumers, J.; Maurer-Stroh, S.; Schymkowitz, J.; Rousseau, F. Protein sequences encode safeguards against aggregation. Hum. Mutat. 2009, 30, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Ishino, S.; Yamagami, T.; Kitamura, M.; Kodera, N.; Mori, T.; Sugiyama, S.; Ando, T.; Goda, N.; Tenno, T.; Hiroaki, H.; et al. Multiple interactions of the intrinsically disordered region between the helicase and nuclease domains of the archaeal Hef protein. J. Biol. Chem. 2014, 289, 21627–21639. [Google Scholar]
- Mori, S.; Abeygunawardana, C.; Johnson, M.O.; Vanzijl, P.C.M. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. Ser. B 1995, 108, 94–98. [Google Scholar] [CrossRef]
- Kay, L.; Keifer, P.; Saarinen, T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992, 114, 10663–10665. [Google Scholar] [CrossRef]
- Piotto, M.; Saudek, V.; Sklenár, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. Sparky 3; NMR Assignment and Integration Software; University of California: San Francisco, CA, USA, 2004. [Google Scholar]
- Edanz. Available online: http://www.edanzediting.co.jp/ (accessed on 25 May 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goda, N.; Shimizu, K.; Kuwahara, Y.; Tenno, T.; Noguchi, T.; Ikegami, T.; Ota, M.; Hiroaki, H. A Method for Systematic Assessment of Intrinsically Disordered Protein Regions by NMR. Int. J. Mol. Sci. 2015, 16, 15743-15760. https://doi.org/10.3390/ijms160715743
Goda N, Shimizu K, Kuwahara Y, Tenno T, Noguchi T, Ikegami T, Ota M, Hiroaki H. A Method for Systematic Assessment of Intrinsically Disordered Protein Regions by NMR. International Journal of Molecular Sciences. 2015; 16(7):15743-15760. https://doi.org/10.3390/ijms160715743
Chicago/Turabian StyleGoda, Natsuko, Kana Shimizu, Yohta Kuwahara, Takeshi Tenno, Tamotsu Noguchi, Takahisa Ikegami, Motonori Ota, and Hidekazu Hiroaki. 2015. "A Method for Systematic Assessment of Intrinsically Disordered Protein Regions by NMR" International Journal of Molecular Sciences 16, no. 7: 15743-15760. https://doi.org/10.3390/ijms160715743