
Global Reprogramming of Transcription in Chinese Fir (Cunninghamia lanceolata) during Progressive Drought Stress and after Rewatering

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. High-Throughput Transcriptome Sequencing and Read Assembly
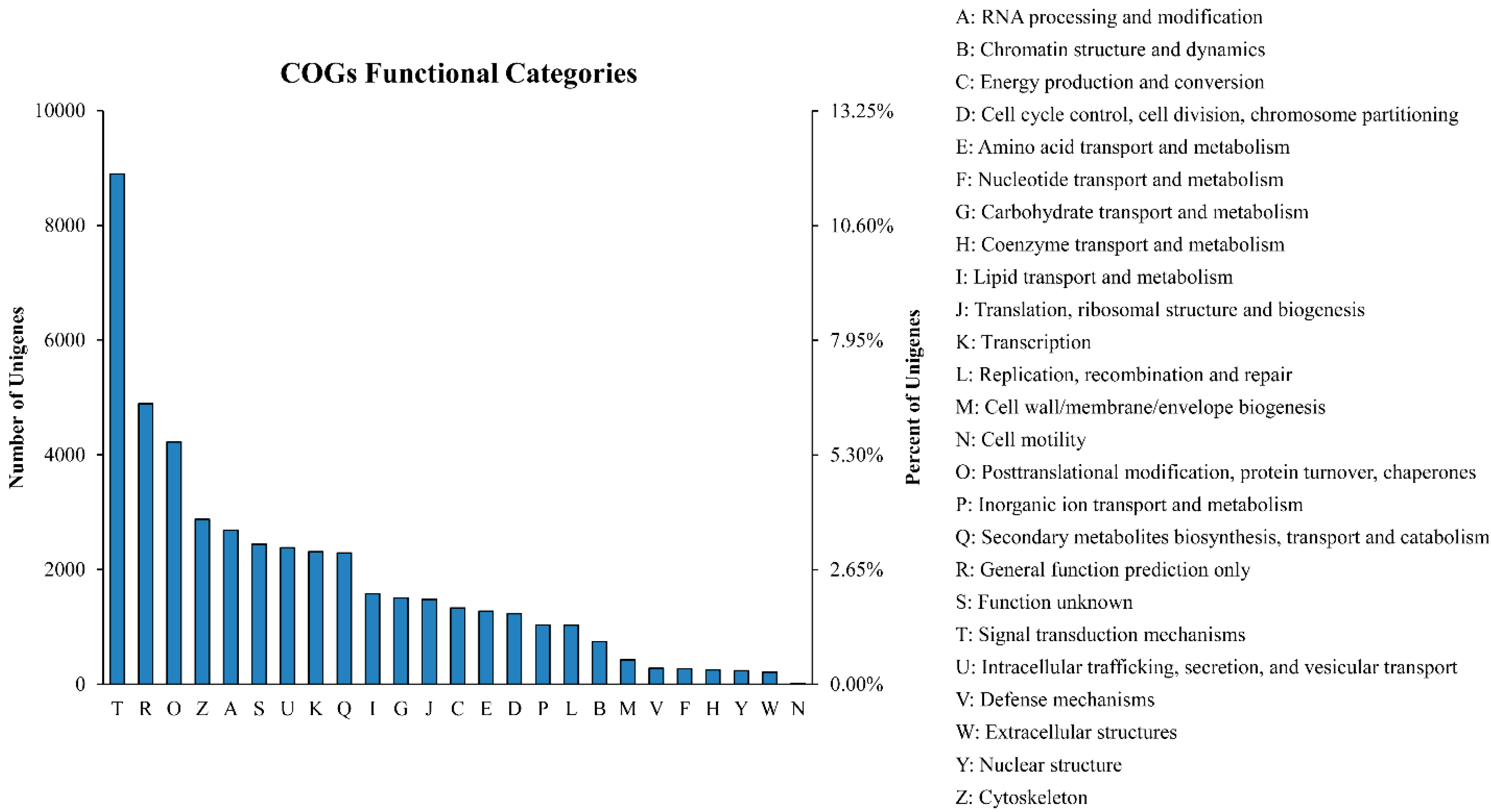
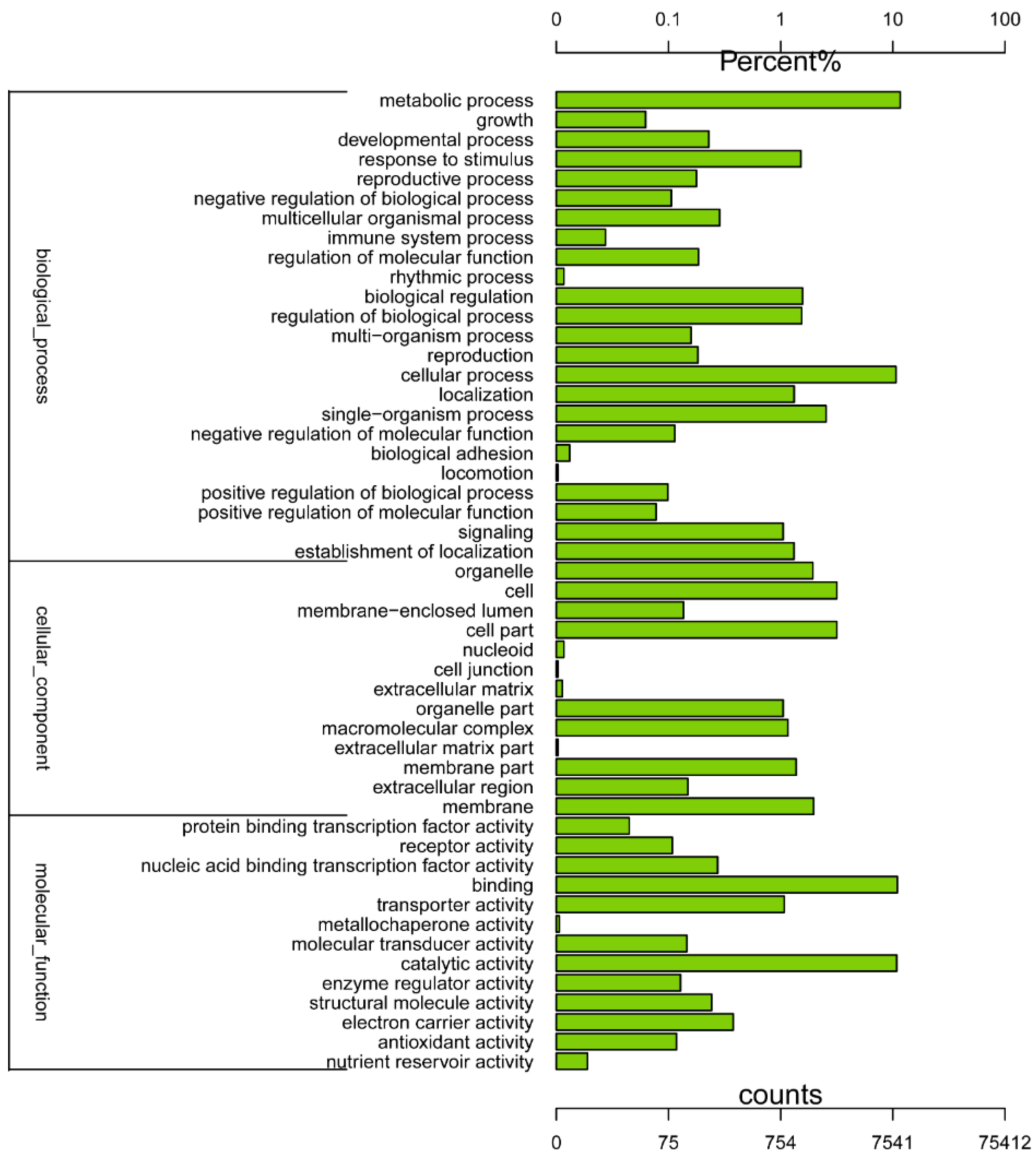
2.2. Gene Annotation and Functional Classification
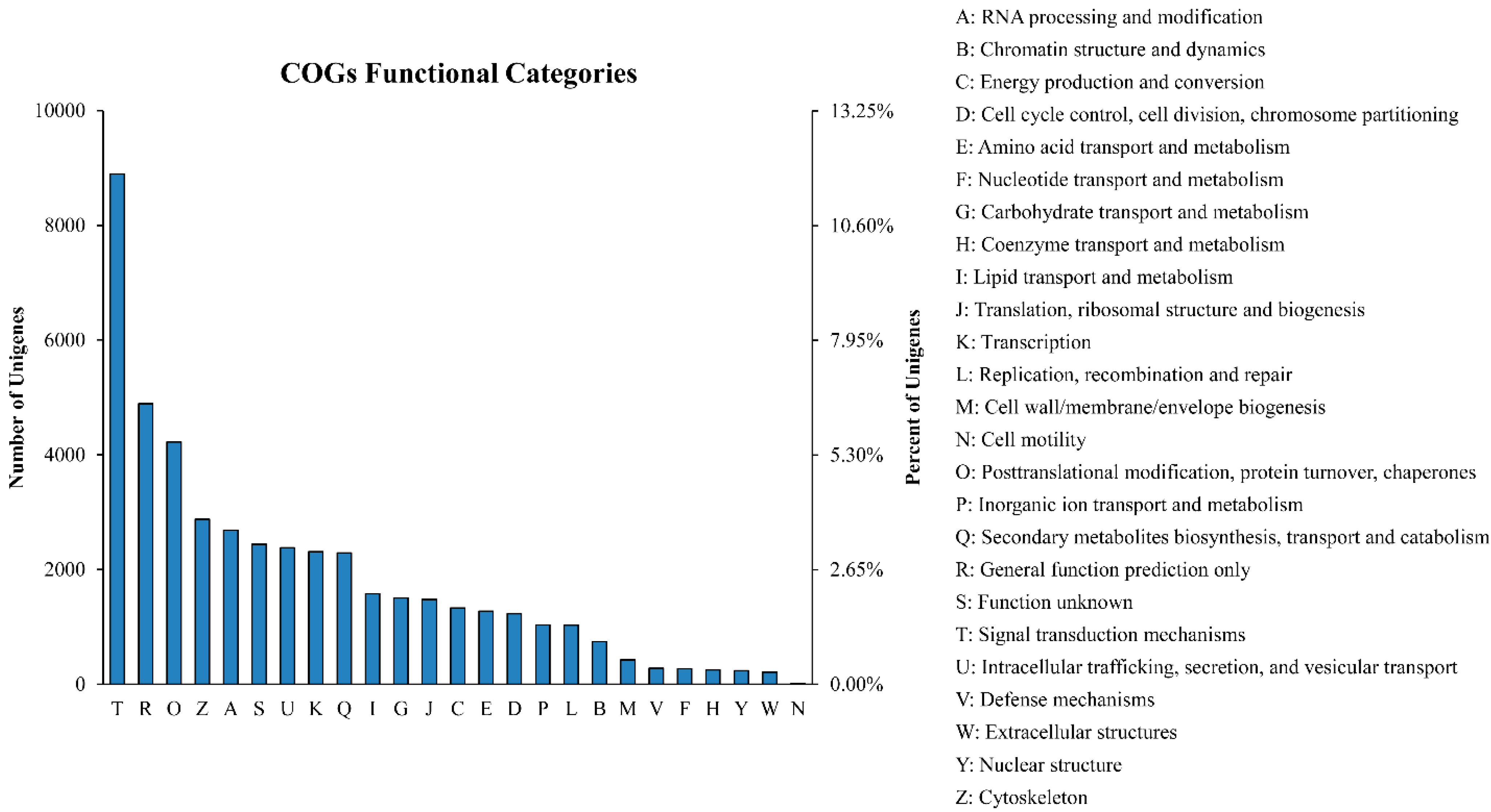
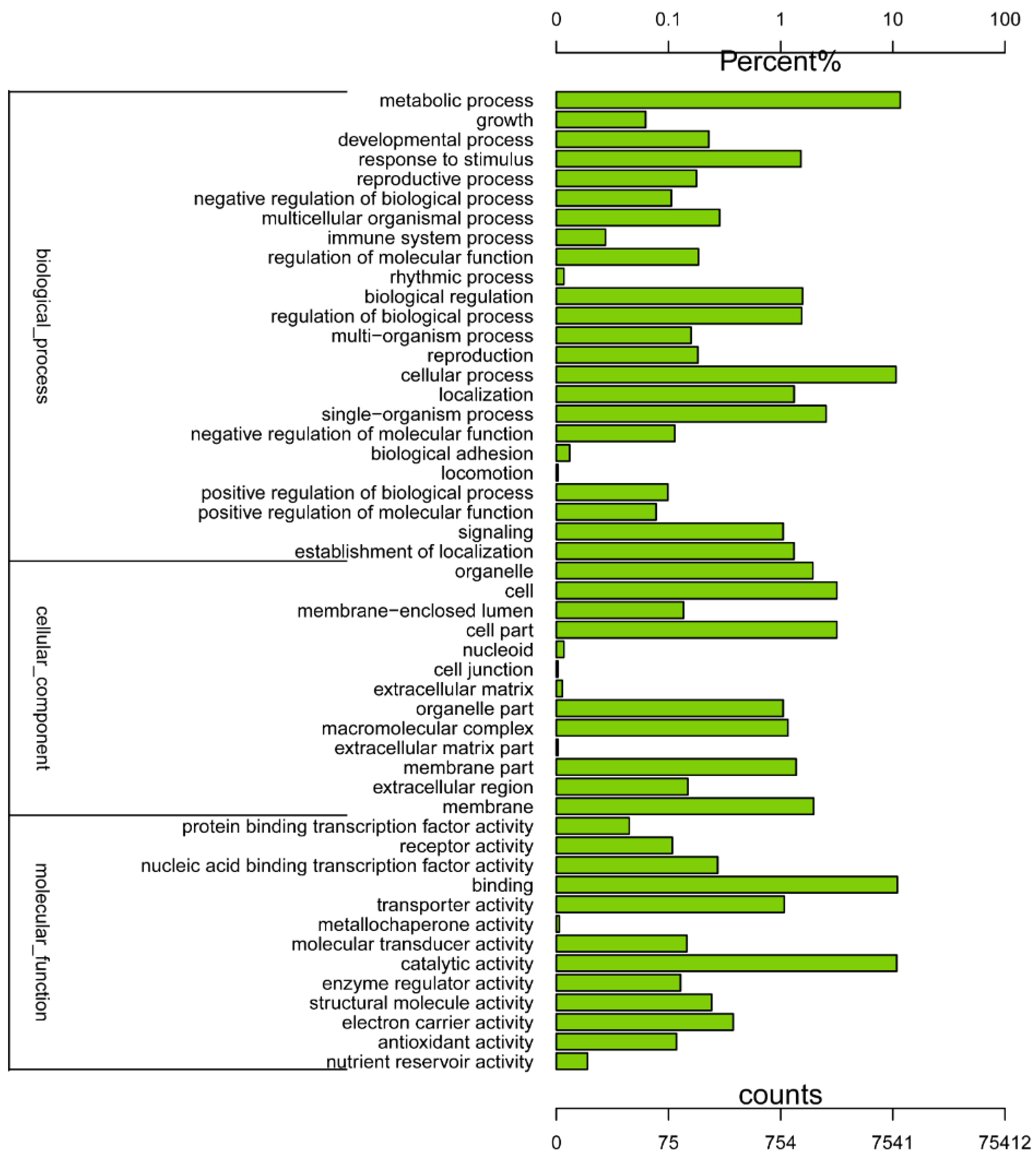
2.3. Differently Expressed Genes during the Drought-Stress Treatments

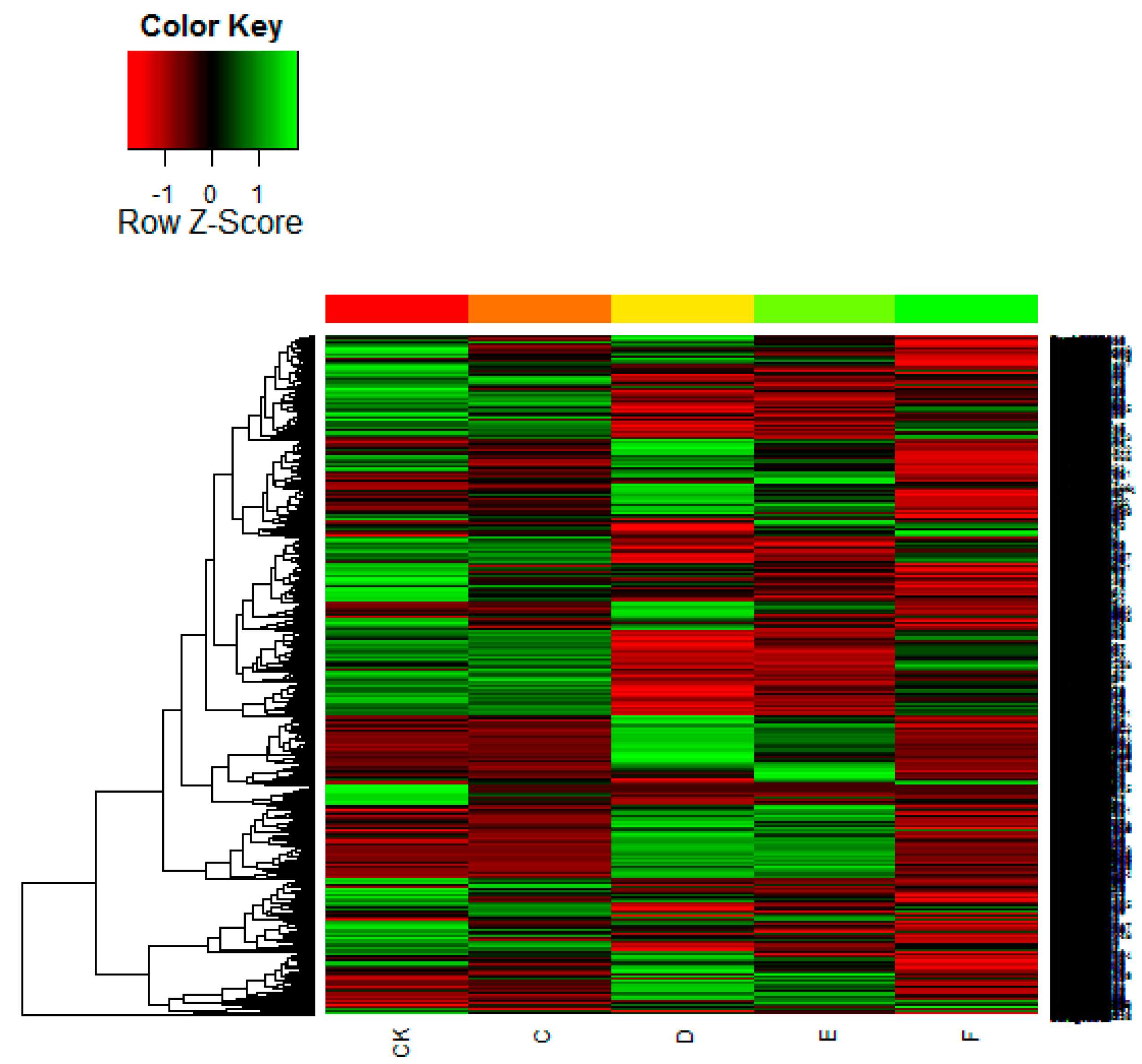
2.4. SOM Cluster Analysis of Gene Expression Data
2.5. Gene Enrichment Analysis for DEGs among Five Libraries
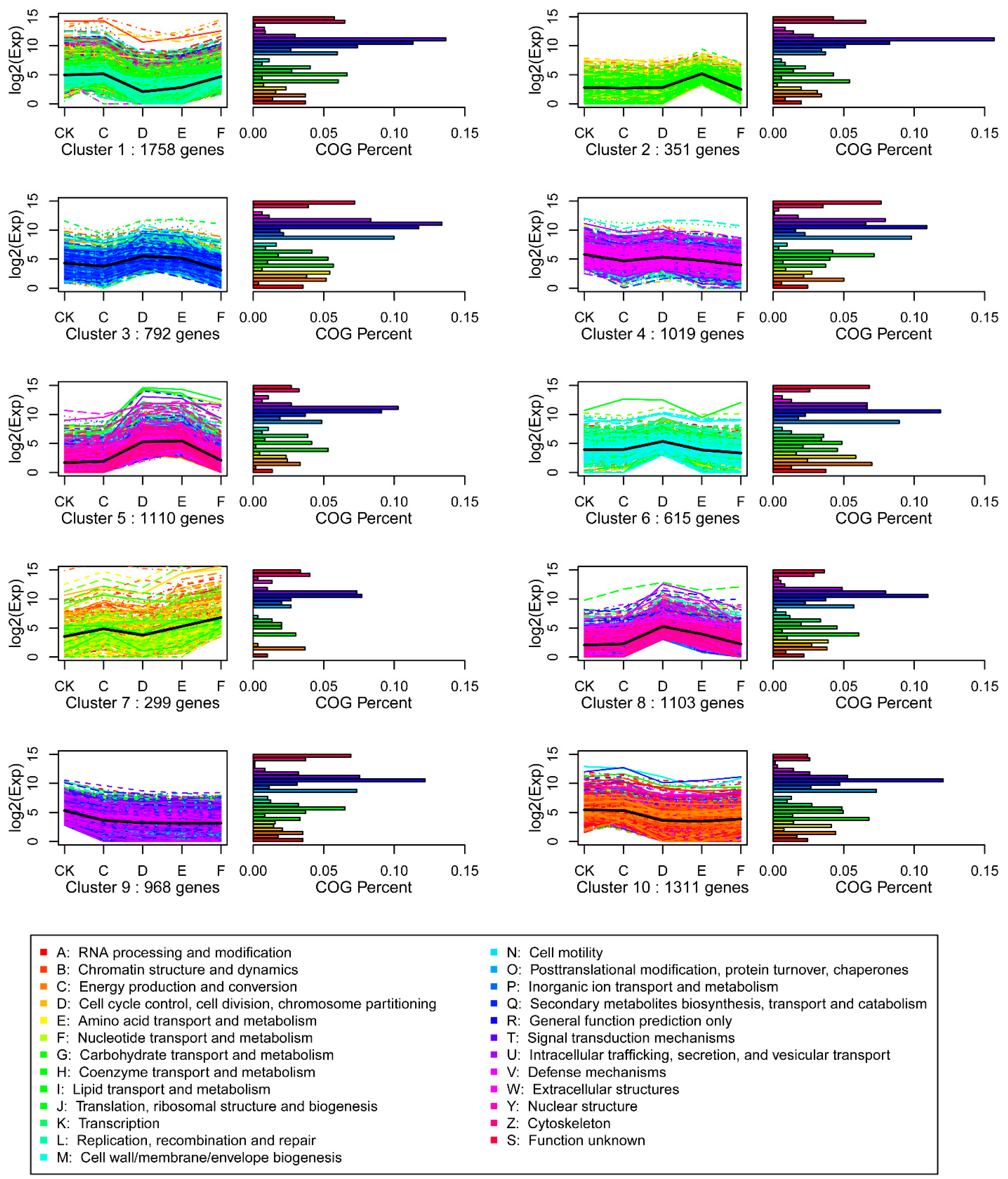
2.6. The Changes of Number of DEGs Enriched in GO Oxidation-Reduction Process and KEGG Metabolic Pathways
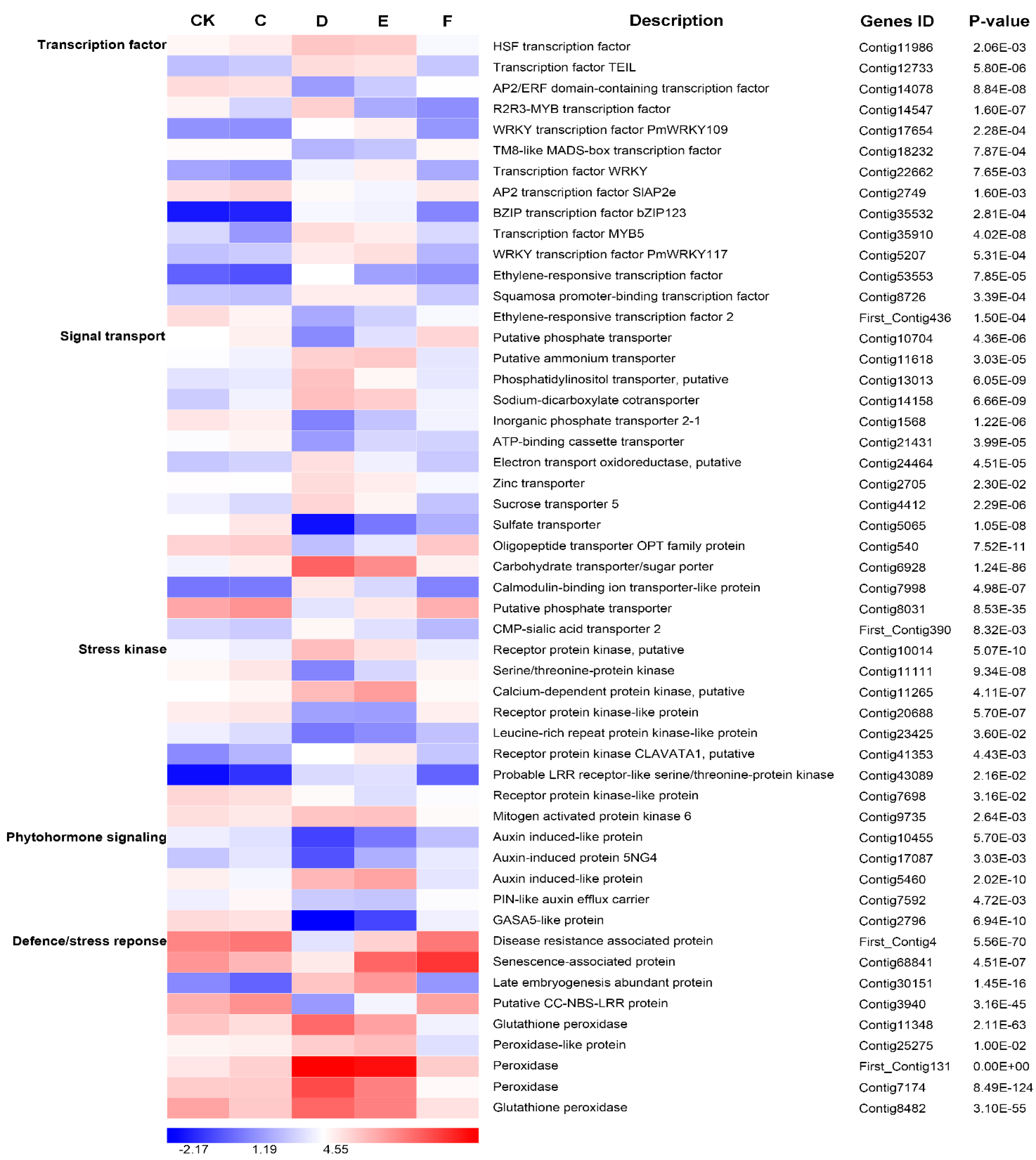
2.7. Responses of Important Differentially Expressed Genes to Drought Stress
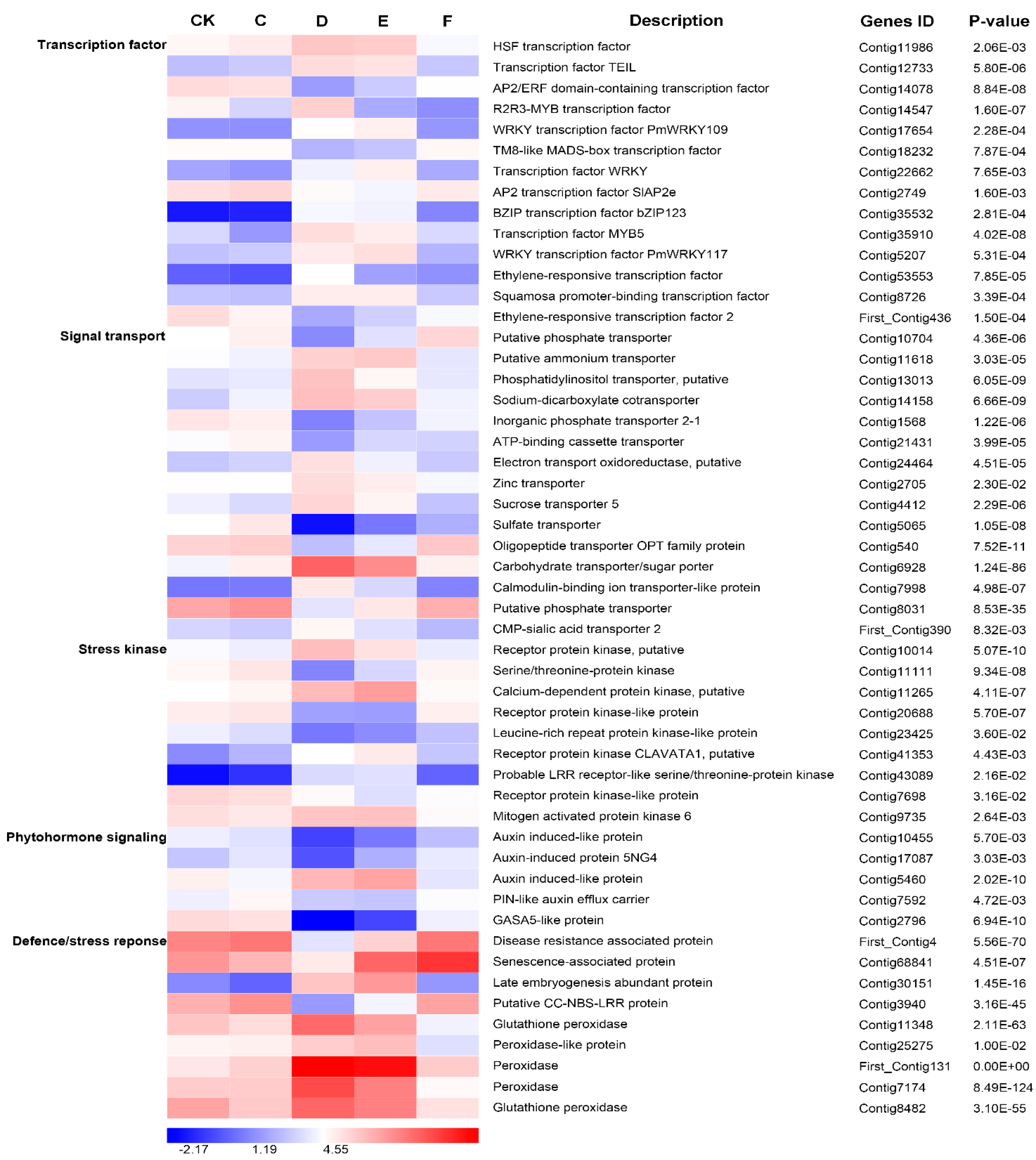
3. Discussion
3.1. Transcriptome Sequencing and Functional Annotation
3.2. The Most Abundant Categories of DEGs Enriched in GO and KEGG Pathways
3.3. Important TF Genes Involved in Drought Stress and Re-Watering
3.4. Differentially Expressed Genes Related to Phytohormone Signaling under Drought Stress and Re-Watering
3.5. The Other Candidate Functional and Regulatory Genes Involved in Drought Stress and Re-Watering
4. Experimental Section
4.1. Ethics Statement
4.2. Plant Growth, Drought Stress, and Sampling
4.3. cDNA Library Preparation and Transcriptome Sequencing
4.4. Analysis of Illumina Transcriptome Sequencing Results
4.5. Bioinformatics for Functional Annotation of Differentially Expressed Genes
4.6. Gene Ontology Functional and KEGG Pathway Enrichment Analysis for DEGs
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Allen, C.D.; Macalady, A.K.; Chenchouni, H.; Bachelet, D.; McDowell, N.; Vennetier, M.; Kitzberger, T.; Rigling, A.; Breshears, D.D.; Hogg, E.H.; et al. A global overview of drought and heat-induced tree mortality reveals emerging climate change risks for forests. For. Ecol. Manag. 2010, 259, 660–684. [Google Scholar] [CrossRef]
- Choat, B.; Jansen, S.; Brodribb, T.J.; Cochard, H.; Delzon, S.; Bhaskar, R.; Bucci, S.J.; Feild, T.S.; Gleason, S.M.; Hacke, U.G.; et al. Global convergence in the vulnerability of forests to drought. Nature 2012, 491, 752–755. [Google Scholar] [PubMed]
- Davidson, E.A.; de Araujo, A.C.; Artaxo, P.; Balch, J.K.; Brown, I.F.; Bustamante, M.M.C.; Coe, M.T.; DeFries, R.S.; Keller, M.; Longo, M.; et al. The Amazon basin in transition. Nature 2012, 481, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Peng, C.; Li, Y.; Liu, S.; Zhang, Q.; Tang, X.; Liu, J.; Yan, J.; Zhang, D.; Chu, G. A climate change-induced threat to the ecological resilience of a subtropical monsoon evergreen broad-leaved forest in Southern China. Glob. Chang. Biol. 2013, 19, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Fitzgerald, J.B.; Zimmermann, N.E.; Reyer, C.; Delzon, S.; van der Maaten, E.; Schelhaas, M.J.; Lasch, P.; Eggers, J.; van der Maaten-Theunissen, M.; et al. Climate change and European forests: What do we know, what are the uncertainties, and what are the implications for forest management? J. Environ. Manag. 2014, 146C, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.; Sawant, K. Drought stress adaptation: metabolic adjustment and regulation of gene expression. Plant Breed. 2013, 132, 21–32. [Google Scholar] [CrossRef]
- Mittler, R. Abiotic stress, the field environment and stress combination. Trends Plant Sci. 2006, 11, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, I.; de Vos, R.C.; Bones, A.M.; Hall, R.D. Plant molecular stress responses face climate change. Trends Plant Sci. 2010, 15, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, E.T.; Campbell, M.M. Genome-wide responses to drought in forest trees. Forestry 2011, 84, 273–283. [Google Scholar] [CrossRef]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Chaves, M.M. Photosynthesis and drought: Can we make metabolic connections from available data? J. Exp. Bot. 2011, 62, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Cabello, J.V.; Lodeyro, A.F.; Zurbriggen, M.D. Novel perspectives for the engineering of abiotic stress tolerance in plants. Curr. Opin. Biotechnol. 2014, 26C, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ng, C.K.Y.; Fan, L. MYB transcription factors, active players in abiotic stress signaling. Environ. Exp. Bot. 2015, 114, 80–91. [Google Scholar] [CrossRef]
- Liu, X.; Zhai, S.; Zhao, Y.; Sun, B.; Liu, C.; Yang, A.; Zhang, J. Overexpression of the phosphatidylinositol synthase gene (ZmPIS) conferring drought stress tolerance by altering membrane lipid composition and increasing ABA synthesis in maize. Plant Cell Environ. 2013, 36, 1037–1055. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Improving plant drought, salt and freezing tolerance by gene transfer of a single stress-inducible transcription factor. Novartis Found. Symp. 2001, 236, 176–186. [Google Scholar] [PubMed]
- Wang, W.; Vinocur, B.; Altman, A. Plant responses to drought, salinity and extreme temperatures: Towards genetic engineering for stress tolerance. Planta 2003, 218, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Achievements and challenges in understanding plant abiotic stress responses and tolerance. Plant Cell Physiol. 2011, 52, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Cominelli, E.; Conti, L.; Tonelli, C.; Galbiati, M. Challenges and perspectives to improve crop drought and salinity tolerance. N. Biotechnol. 2013, 30, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Imelfort, M.; Duran, C.; Batley, J.; Edwards, D. Discovering genetic polymorphisms in next-generation sequencing data. Plant Biotechnol. J. 2009, 7, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Nayak, S.N.; May, G.D.; Jackson, S.A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2009, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Habash, D.Z.; Baudo, M.; Hindle, M.; Powers, S.J.; Defoin-Platel, M.; Mitchell, R.; Saqi, M.; Rawlings, C.; Latiri, K.; Araus, J.L.; et al. Systems responses to progressive water stress in durum wheat. PLoS ONE 2014, 9, e108431. [Google Scholar] [CrossRef] [PubMed]
- Cossu, R.M.; Giordani, T.; Cavallini, A.; Natali, L. High-throughput analysis of transcriptome variation during water deficit in a poplar hybrid: A general overview. Tree Genet. Genomes 2014, 10, 53–66. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, M.; Li, X.; Cao, B.; Ma, X. Identification of differentially expressed genes in leaf of Reaumuria soongorica under PEG-induced drought stress by digital gene expression profiling. PLoS ONE 2014, 9, e94277. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.H. Physiological and biotechnological implications of transcript-level variation under abiotic stress. Plant. Biol. (Stuttg.) 2013, 15, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Harfouche, A.; Meilan, R.; Altman, A. Molecular and physiological responses to abiotic stress in forest trees and their relevance to tree improvement. Tree Physiol. 2014, 34, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Li, Y.; Jiang, Y.; Jiang, Z.; Hao, W.; Yuan, Y. Transcriptome profile analysis of maize seedlings in response to high-salinity, drought and cold stresses by deep sequencing. Plant Mol. Biol. Rep. 2013, 31, 1485–1491. [Google Scholar] [CrossRef]
- Swamy, B.P.; Kumar, A. Genomics-based precision breeding approaches to improve drought tolerance in rice. Biotechnol. Adv. 2013, 31, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Fleury, D.; Jefferies, S.; Kuchel, H.; Langridge, P. Genetic and genomic tools to improve drought tolerance in wheat. J. Exp. Bot. 2010, 61, 3211–3222. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Zhang, J.; Li, H.; Yang, C.; Zhang, C.; Zhang, X.; Khurram, Z.; Zhang, Y.; Wang, T.; Fei, Z.; et al. Transcriptional profiles of drought-responsive genes in modulating transcription signal transduction, and biochemical pathways in tomato. J. Exp. Bot. 2010, 61, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, O.; Waldron, L.; Nahal, H.; Provart, N.J.; Campbell, M.M. Genotype and time of day shape the Populus drought response. Plant J. 2009, 60, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Baum, M.; Grando, S.; Ceccarelli, S.; Bai, G.; Li, R.; von Korff, M.; Varshney, R.K.; Graner, A.; Valkoun, J. Differentially expressed genes between drought-tolerant and drought-sensitive barley genotypes in response to drought stress during the reproductive stage. J. Exp. Bot. 2009, 60, 3531–3544. [Google Scholar] [CrossRef] [PubMed]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2011, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Zhao-Hua, L.; Gang, W.; Xin, M.; Guang-Xin, B. Current situation of Chinese forestry tactics and strategy of sustainable development. J. For. Res. 2002, 13, 319–322. [Google Scholar] [CrossRef]
- Liu, A.; Ma, X.; Feng, L. A study on physiological responses of different Chinese fir clones under water stress. J. Fujian Coll. For. 1997, 18, 28–31. [Google Scholar]
- Li, S.; Ding, G.; Cao, G.; Lin, S.; Guo, W. Drought resistance estimation of different Chinese fir clones under simulated water stress. J. Fujian Agric. For. Univ. 2012, 5, 10. [Google Scholar]
- Zhang, J.Y.; Cruz, D.C.M.; Torres-Jerez, I.; Kang, Y.; Allen, S.N.; Huhman, D.V.; Tang, Y.; Murray, J.; Sumner, L.W.; Udvardi, M.K. Global reprogramming of transcription and metabolism in Medicago truncatula during progressive drought and after re-watering. Plant Cell Environ. 2014, 37, 2553–2576. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Wan, L.; Chen, T.; Wan, Y.; He, X.; Lu, S.; Wang, Y.; Lin, J. The regulation of cambial activity in Chinese fir (Cunninghamia lanceolata) involves extensive transcriptome remodeling. New Phytol. 2013, 199, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Liu, W.; Luo, Z.; Wang, P.; Zhang, Y.; Zheng, R.; Shi, J. Transcriptome characteristics and six alternative expressed genes positively correlated with the phase transition of annual cambial activities in Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook). PLoS ONE 2013, 8, e71562. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, J.; Liu, S.; Liu, X.; Lin, Y.; Li, L. Identification of rapidly induced genes in the response of peanut (Arachis hypogaea) to water deficit and abscisic acid. BMC Biotechnol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, L.; Li, L.; Wang, S. De novo assembly of the common bean transcriptome using short reads for the discovery of drought-responsive genes. PLoS ONE 2014, 9, e109262. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Dong, Y.; Liang, D.; Zhang, Z.; Ye, C.; Shuai, P.; Han, X.; Zhao, Y.; Yin, W.; Xia, X. Analysis of the drought stress-responsive transcriptome of black cottonwood (Populus trichocarpa) using deep RNA sequencing. Plant Mol. Biol. Rep. 2014, 33, 1–15. [Google Scholar] [CrossRef]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Compatible solute, transporter protein, transcription factor, and hormone-related gene expression provides an indicator of drought stress in Paulownia fortunei. Funct. Integr. Genomic 2014, 14, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J. 2011, 9, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Li, S.; An, X.; Liu, X.; Qin, H.; Wang, D. Transgenic expression of MYB15 confers enhanced sensitivity to abscisic acid and improved drought tolerance in Arabidopsis thaliana. J. Genet. Genomics 2009, 36, 17–29. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, G.; Xia, C.; Jia, J.; Liu, X.; Kong, X. A wheat R2R3-MYB gene, TaMYB30-B, improves drought stress tolerance in transgenic Arabidopsis. J. Exp. Bot. 2012, 63, 5873–5885. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, M.; Tian, Y.; He, W.; Han, L.; Xia, G. Over-expression of TaMYB33 encoding a novel wheat MYB transcription factor increases salt and drought tolerance in Arabidopsis. Mol. Biol. Rep. 2012, 39, 7183–7192. [Google Scholar] [CrossRef] [PubMed]
- Rushton, D.L.; Tripathi, P.; Rabara, R.C.; Lin, J.; Ringler, P.; Boken, A.K.; Langum, T.J.; Smidt, L.; Boomsma, D.D.; Emme, N.J.; et al. WRKY transcription factors: Key components in abscisic acid signalling. Plant Biotechnol. J. 2012, 10, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, W.W.; Alba, R.; Yu, Y.S.; Bordeaux, J.M.; Simoes, M.; Dean, J.F. Microarray analysis and scale-free gene networks identify candidate regulators in drought-stressed roots of loblolly pine (P. taeda L.). BMC Genomics 2011, 12, 264. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Liu, Q.; Dubouzet, J.G.; Abe, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 2002, 290, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chen, M.; Li, L.; Xu, Z.; Chen, X.; Guo, J.; Ma, Y. Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. J. Exp. Bot. 2009, 60, 3781–3796. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tang, N.; Du, H.; Ye, H.; Xiong, L. Characterization of OsbZIP23 as a key player of the basic leucine zipper transcription factor family for conferring abscisic acid sensitivity and salinity and drought tolerance in rice. Plant Physiol. 2008, 148, 1938–1952. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Liu, G.; Liu, Y.; Zheng, L.; Nie, X.; Wang, Y. The bZIP protein from Tamarix hispida, ThbZIP1, is ACGT elements binding factor that enhances abiotic stress signaling in transgenic Arabidopsis. BMC Plant Biol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Zhang, D.F.; Fu, J.; Shi, Y.S.; Song, Y.C.; Wang, T.Y.; Li, Y. Cloning and characterization of a maize bZIP transcription factor, ZmbZIP72, confers drought and salt tolerance in transgenic Arabidopsis. Planta 2012, 235, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Vanstraelen, M.; Benkova, E. Hormonal interactions in the regulation of plant development. Annu. Rev. Cell Dev. Biol. 2012, 28, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Liu, H.; Xiong, L. Endogenous auxin and jasmonic acid levels are differentially modulated by abiotic stresses in rice. Front. Plant Sci. 2013, 4, 397. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, L.; Xiong, L. Comprehensive expression profiling analysis of OsIAA gene family in developmental processes and in response to phytohormone and stress treatments. Planta 2009, 229, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, D.K.; Choi, Y.D.; Kim, J.K. OsIAA6, a member of the rice Aux/IAA gene family, is involved in drought tolerance and tiller outgrowth. Plant Sci. 2015, 236, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Wisniewska, J.; Xu, J.; Seifertova, D.; Brewer, P.B.; Ruzicka, K.; Blilou, I.; Rouquie, D.; Benkova, E.; Scheres, B.; Friml, J. Polar PIN localization directs auxin flow in plants. Science 2006, 312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, J.; Zhang, W.; Yan, S.; Wang, R.; Zhao, J.; Li, Y.; Qi, Z.; Sun, Z.; Zhu, Z. The putative auxin efflux carrier OsPIN3t is involved in the drought stress response and drought tolerance. Plant J. 2012, 72, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.; Zhang, W.; Deng, F.; Zhao, J.; Wang, X. A bifurcating pathway directs abscisic acid effects on stomatal closure and opening in Arabidopsis. Science 2006, 312, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Nakashima, K.; Miyakawa, T.; Kuromori, T.; Tanokura, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular basis of the core regulatory network in ABA responses: sensing, signaling and transport. Plant Cell Physiol. 2010, 51, 1821–1839. [Google Scholar] [CrossRef] [PubMed]
- Rathinasabapathi, B. Metabolic engineering for stress tolerance: Installing osmoprotectant synthesis pathways. Ann. Bot. 2000, 86, 709–716. [Google Scholar] [CrossRef]
- Boursiac, Y.; Leran, S.; Corratge-Faillie, C.; Gojon, A.; Krouk, G.; Lacombe, B. ABA transport and transporters. Trends Plant Sci. 2013, 18, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. ABA control of plant macroelement membrane transport systems in response to water deficit and high salinity. New Phytol. 2014, 202, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, S.; Kobayashi, M.; Taji, T.; Naramoto, M.; Seki, M.; Kato, T.; Tabata, S.; Kakubari, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis. Plant J. 2001, 27, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zeevaart, J.A. Overexpression of a 9-cis-epoxycarotenoid dioxygenase gene in Nicotiana plumbaginifolia increases abscisic acid and phaseic acid levels and enhances drought tolerance. Plant Physiol. 2002, 128, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Speirs, J.; Binney, A.; Collins, M.; Edwards, E.; Loveys, B. Expression of ABA synthesis and metabolism genes under different irrigation strategies and atmospheric VPDs is associated with stomatal conductance in grapevine (Vitis vinifera L. cv Cabernet Sauvignon). J. Exp. Bot. 2013, 64, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Kuromori, T.; Miyaji, T.; Yabuuchi, H.; Shimizu, H.; Sugimoto, E.; Kamiya, A.; Moriyama, Y.; Shinozaki, K. ABC transporter AtABCG25 is involved in abscisic acid transport and responses. Proc. Natl. Acad. Sci. USA 2010, 107, 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Hwang, J.U.; Lee, M.; Kim, Y.Y.; Assmann, S.M.; Martinoia, E.; Lee, Y. PDR-type ABC transporter mediates cellular uptake of the phytohormone abscisic acid. Proc. Natl. Acad. Sci. USA 2010, 107, 2355–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danquah, A.; de Zelicourt, A.; Colcombet, J.; Hirt, H. The role of ABA and MAPK signaling pathways in plant abiotic stress responses. Biotechnol. Adv. 2014, 32, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Aalen, R.B.; Audenaert, D.; Beeckman, T.; Broadley, M.R.; Butenko, M.A.; Cano-Delgado, A.I.; de Vries, S.; Dresselhaus, T.; Felix, G.; et al. Tackling drought stress: Receptor-like kinases present new approaches. Plant Cell 2012, 24, 2262–2278. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.Q.; Liu, Y.F.; Liu, P.; Lei, G.; He, S.J.; Ma, B.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. Receptor-like kinase OsSIK1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J. 2010, 62, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Samajova, O.; Plihal, O.; Al-Yousif, M.; Hirt, H.; Samaj, J. Improvement of stress tolerance in plants by genetic manipulation of mitogen-activated protein kinases. Biotechnol. Adv. 2013, 31, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Campo, S.; Baldrich, P.; Messeguer, J.; Lalanne, E.; Coca, M.; San Segundo, B. Overexpression of a calcium-dependent protein kinase confers salt and drought tolerance in rice by preventing membrane lipid peroxidation. Plant Physiol. 2014, 165, 688–704. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Sensing the environment: Key roles of membrane-localized kinases in plant perception and response to abiotic stress. J. Exp. Bot. 2013, 64, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.M.; Gao, Q.; Xue, H.W.; Sui, Z.H.; Yue, G.D.; Yang, A.F.; Zhang, J.R. Overexpression of the phosphatidylinositol synthase gene from Zea mays in tobacco plants alters the membrane lipids composition and improves drought stress tolerance. Planta 2012, 235, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Arinaga, N.; Umezawa, T.; Katsura, S.; Nagamachi, K.; Tanaka, H.; Ohiraki, H.; Yamada, K.; Seo, S.U.; Abo, M.; et al. Osmotic stress responses and plant growth controlled by potassium transporters in Arabidopsis. Plant Cell 2013, 25, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, Y.; Zhang, H.; Zhang, D. Genome-wide analysis of gene expression in response to drought stress in Populus simonii. Plant Mol. Biol. Rep. 2013, 31, 946–962. [Google Scholar] [CrossRef]
- Iskandar, H.M.; Casu, R.E.; Fletcher, A.T.; Schmidt, S.; Xu, J.; Maclean, D.J.; Manners, J.M.; Bonnett, G.D. Identification of drought-response genes and a study of their expression during sucrose accumulation and water deficit in sugarcane culms. BMC Plant Biol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, M.; Hincha, D.K. LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, Y.; Jeong, J.C.; Kim, C.Y.; Lee, H.; Bang, J.; Kwak, S. Sweetpotato late embryogenesis abundant 14 (IbLEA14) gene influences lignification and increases osmotic- and salt stress-tolerance of transgenic calli. Planta 2011, 233, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Bräutigam, A.; Mullick, T.; Schliesky, S.; Weber, A.P. Critical assessment of assembly strategies for non-model species mRNA-Seq data and application of next-generation sequencing to the comparison of C3 and C4 species. J. Exp. Bot. 2011, 62, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Patel, R.K.; Tyagi, A.K.; Jain, M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011, 18, 53–63. [Google Scholar] [CrossRef] [PubMed]
- UniProtKB/Swiss-Prot. Available online: http://www.expasy.ch/sprot (accessed on 25 April 2013).
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Wu, J.; Zhang, Y.; Zhang, H.; Huang, H.; Folta, K.M.; Lu, J. Whole genome wide expression profiles of Vitis amurensis grape responding to downy mildew by using Solexa sequencing technology. BMC Plant Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, R.; Wu, B.; Zheng, H.; Hu, D.; Wang, X.; Duan, H.; Sun, Y.; Wang, J.; Zhang, Y.; Li, Y. Global Reprogramming of Transcription in Chinese Fir (Cunninghamia lanceolata) during Progressive Drought Stress and after Rewatering. Int. J. Mol. Sci. 2015, 16, 15194-15219. https://doi.org/10.3390/ijms160715194
Hu R, Wu B, Zheng H, Hu D, Wang X, Duan H, Sun Y, Wang J, Zhang Y, Li Y. Global Reprogramming of Transcription in Chinese Fir (Cunninghamia lanceolata) during Progressive Drought Stress and after Rewatering. International Journal of Molecular Sciences. 2015; 16(7):15194-15219. https://doi.org/10.3390/ijms160715194
Chicago/Turabian StyleHu, Ruiyang, Bo Wu, Huiquan Zheng, Dehuo Hu, Xinjie Wang, Hongjing Duan, Yuhan Sun, Jinxing Wang, Yue Zhang, and Yun Li. 2015. "Global Reprogramming of Transcription in Chinese Fir (Cunninghamia lanceolata) during Progressive Drought Stress and after Rewatering" International Journal of Molecular Sciences 16, no. 7: 15194-15219. https://doi.org/10.3390/ijms160715194