
Synthesis of Unsaturated Polyester Resins from Various Bio-Derived Platform Molecules



Abstract
:
1. Introduction
2. Results and Discussion
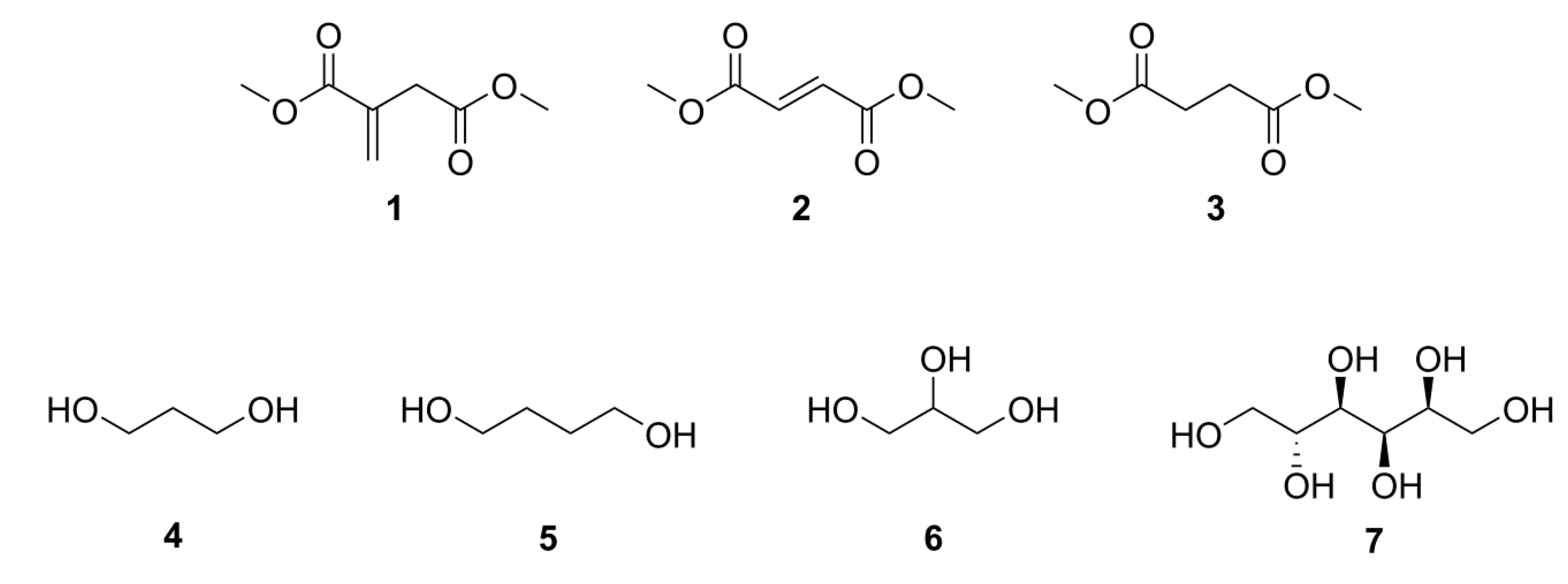
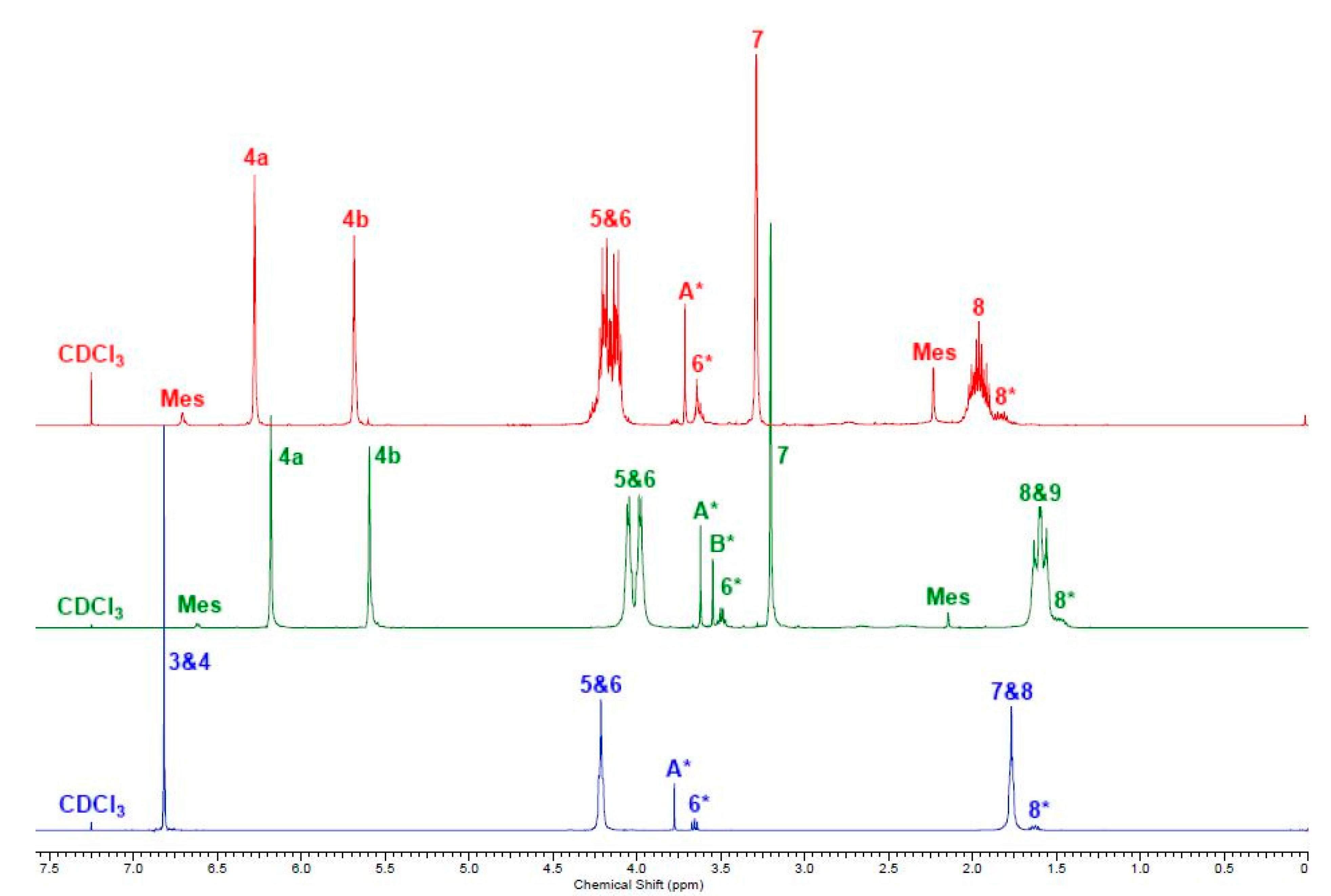
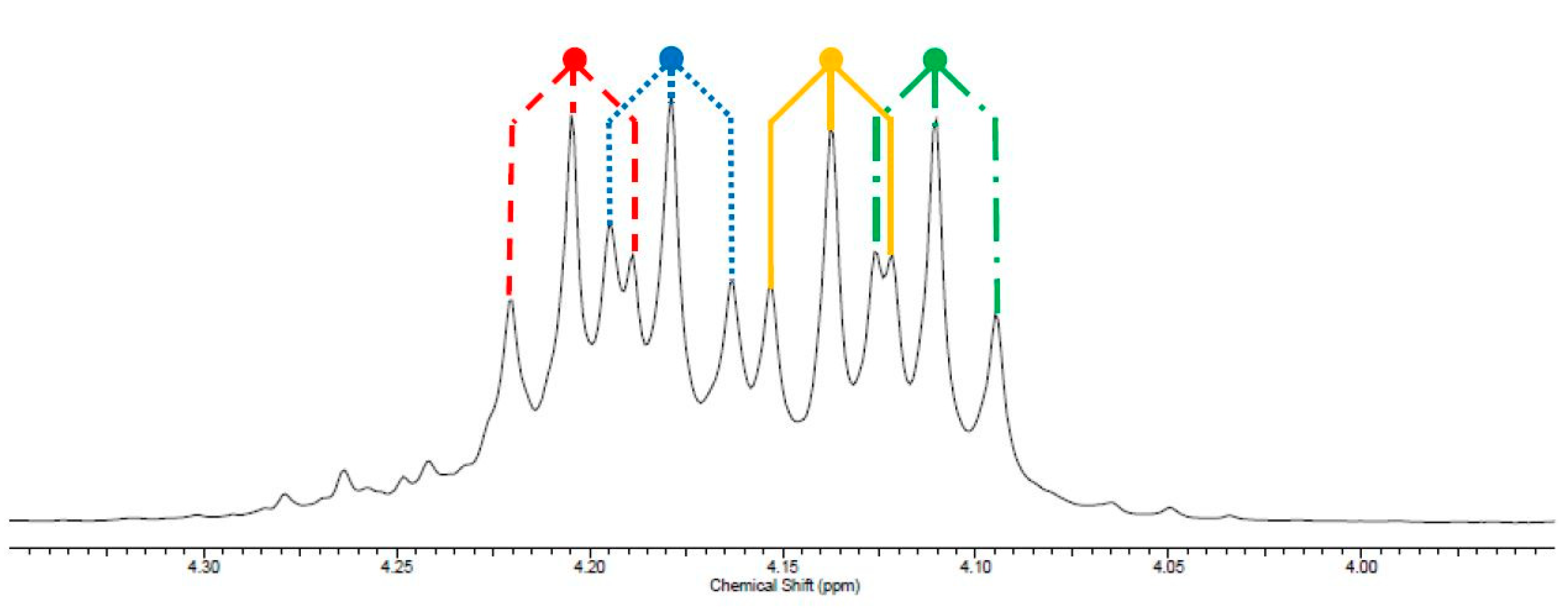
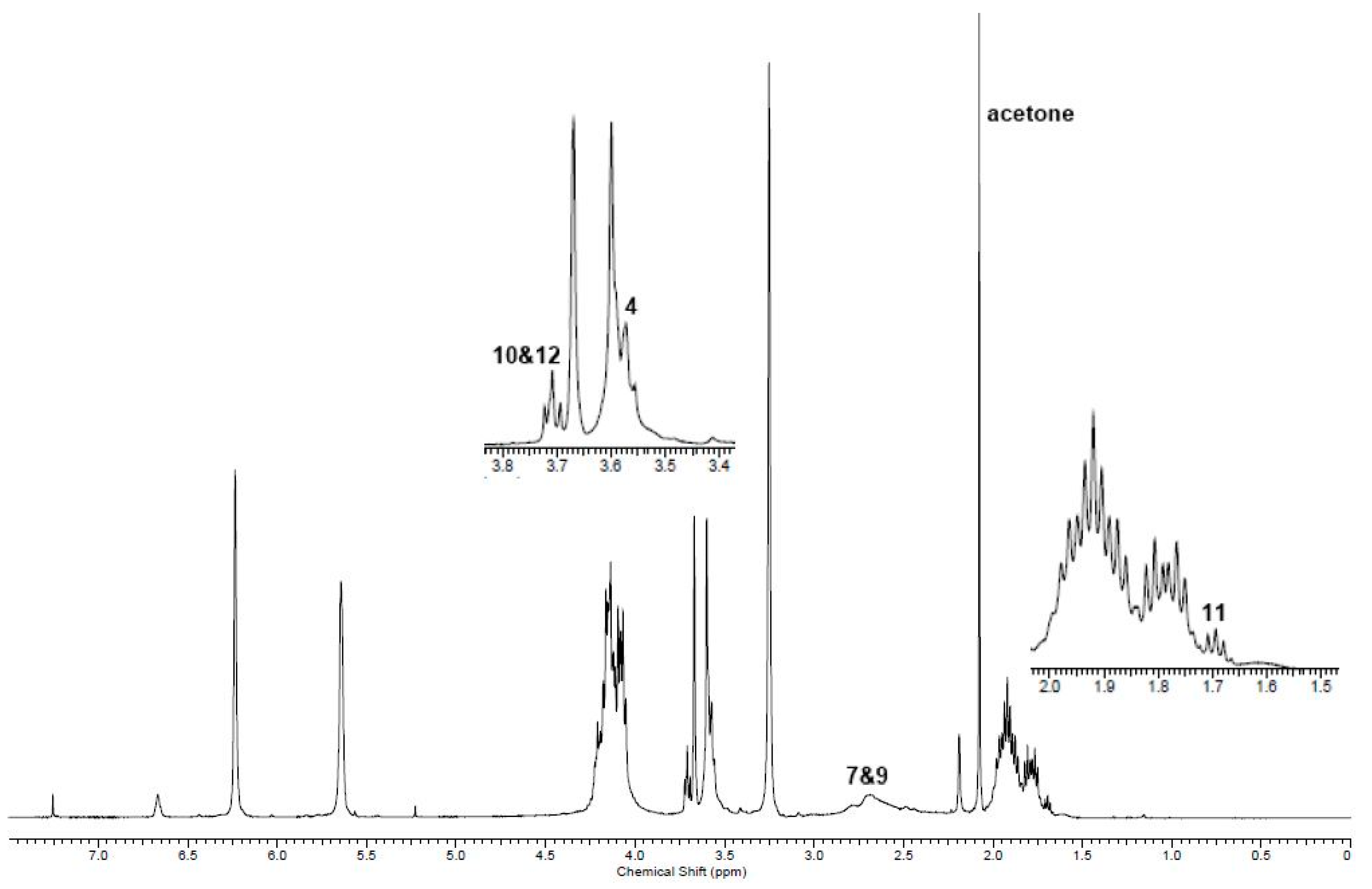
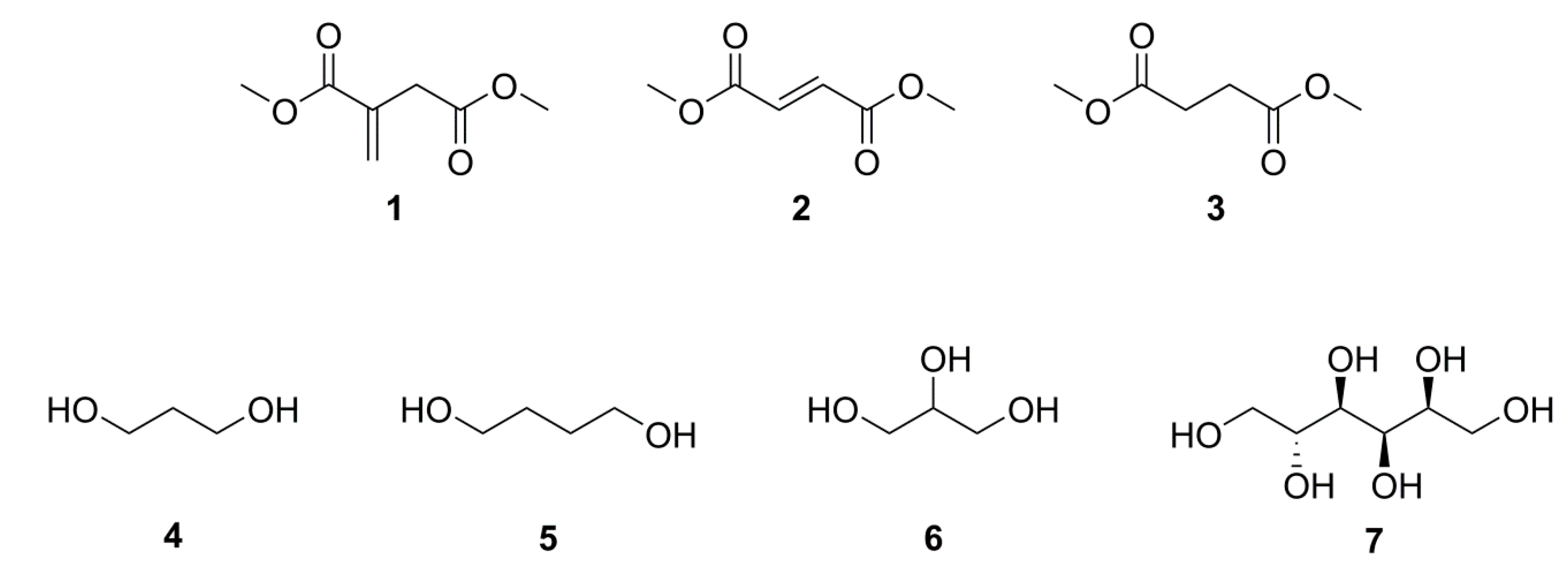
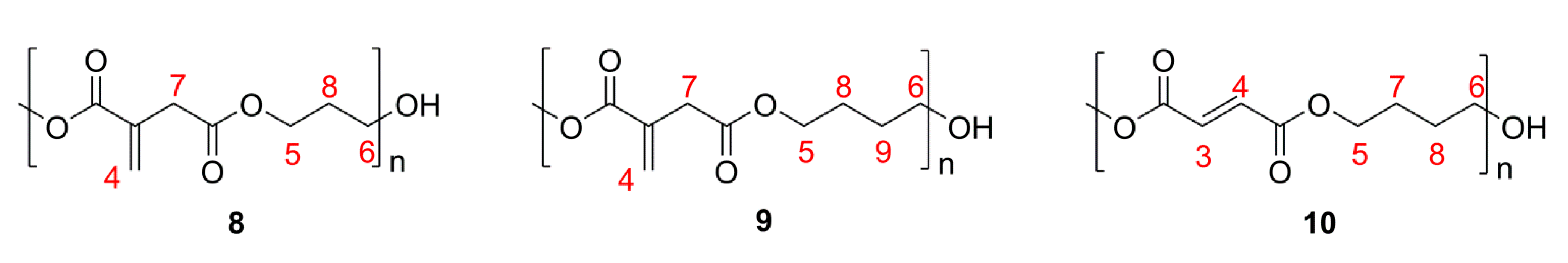
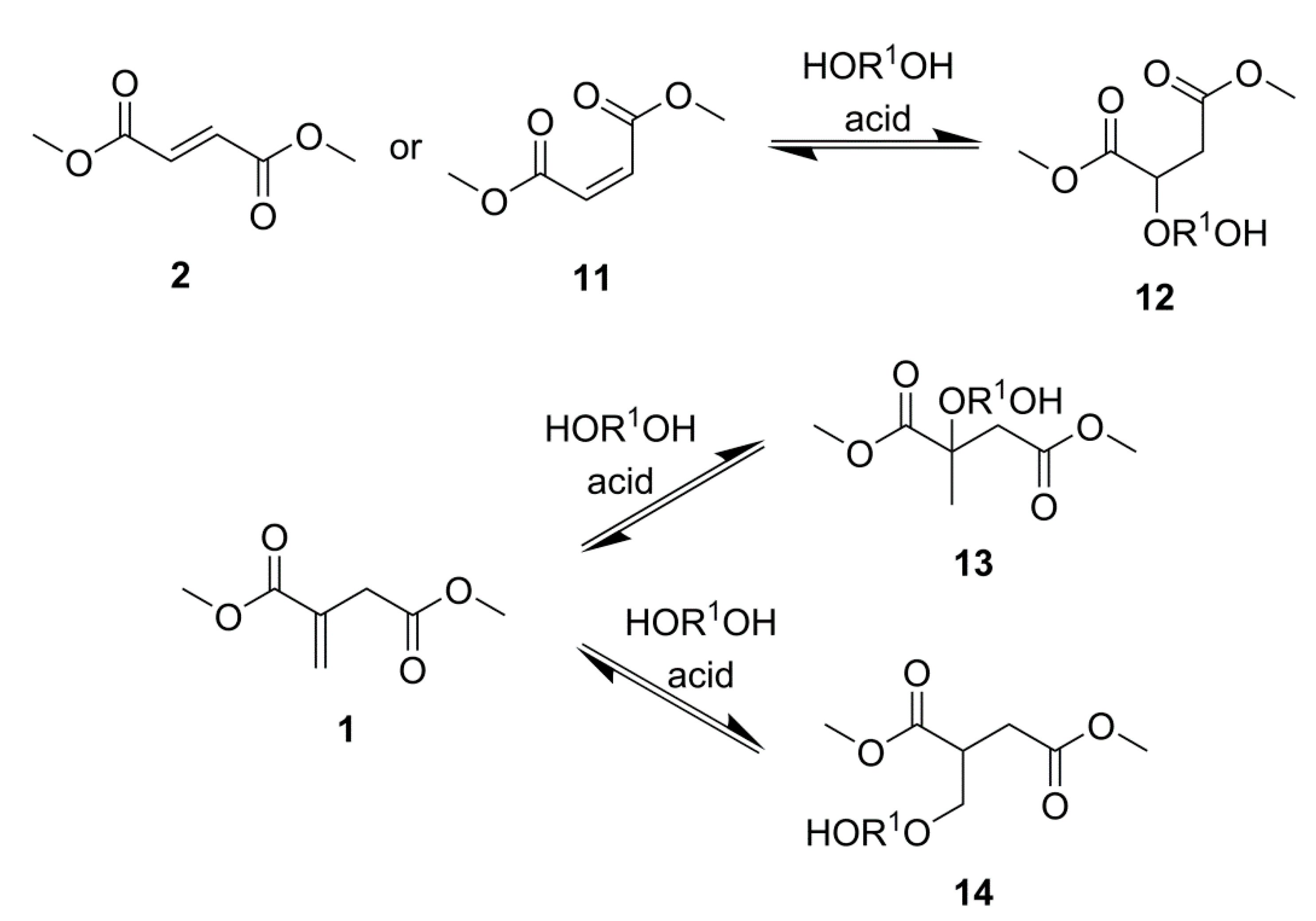
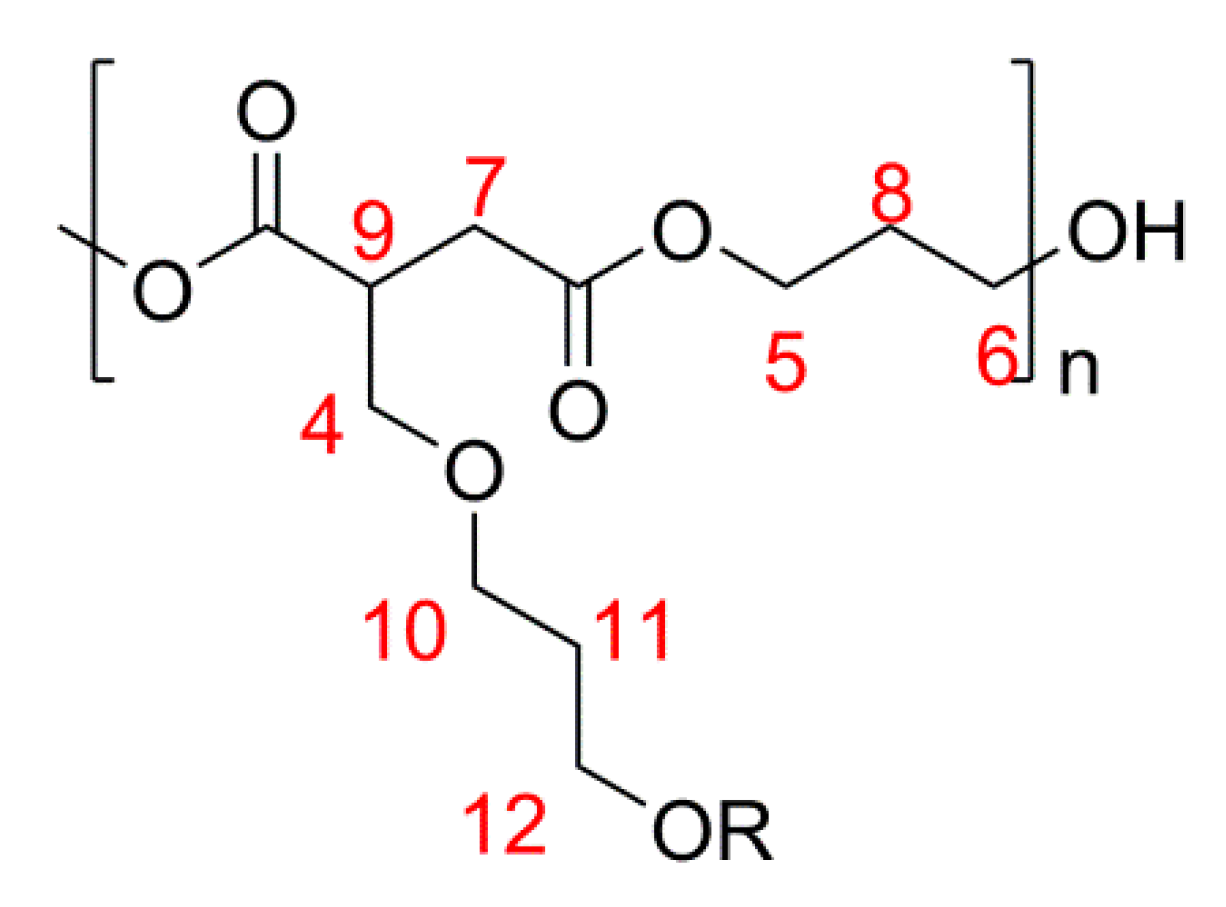
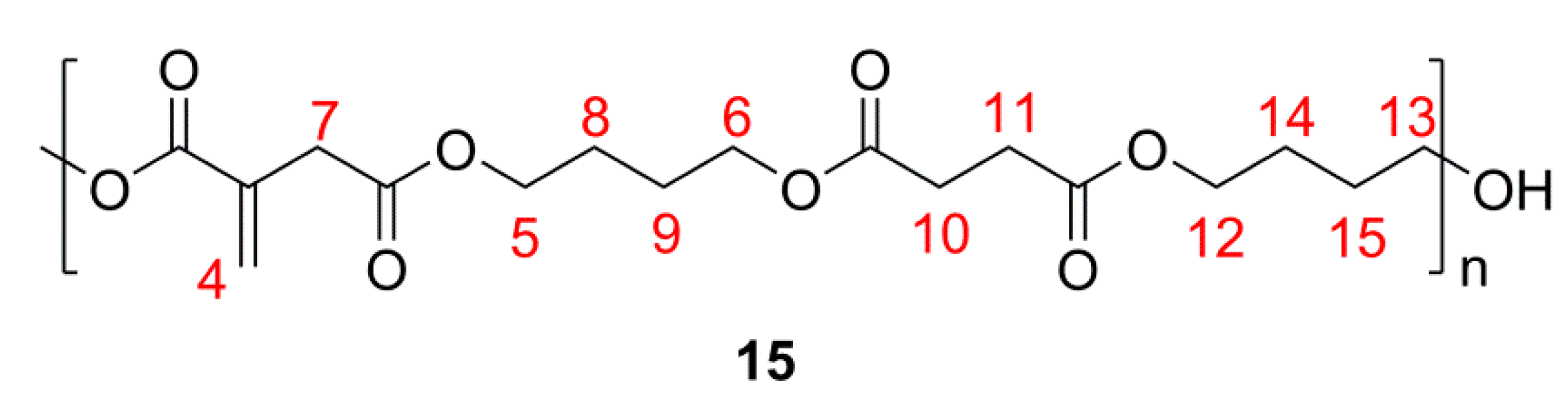
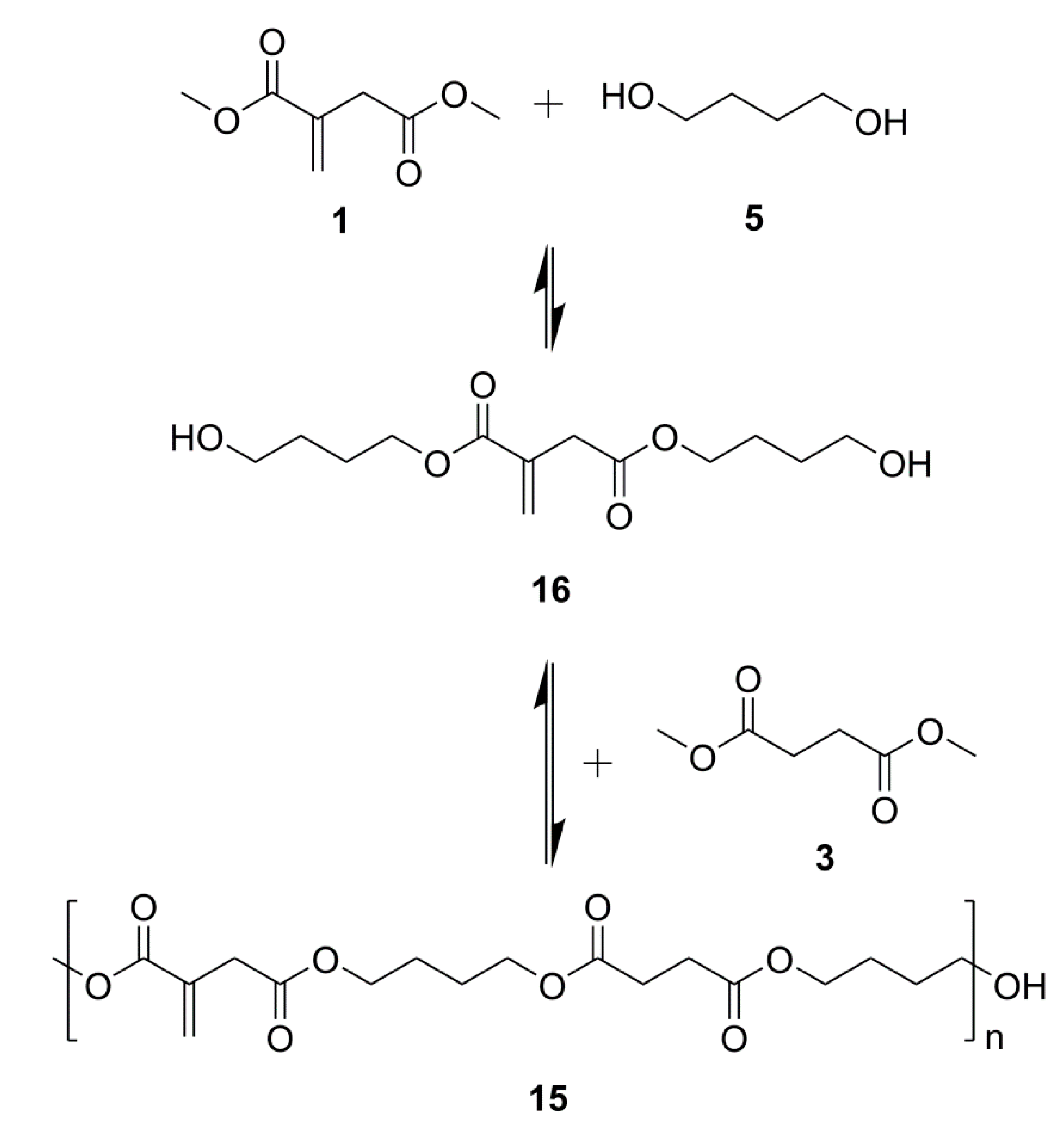
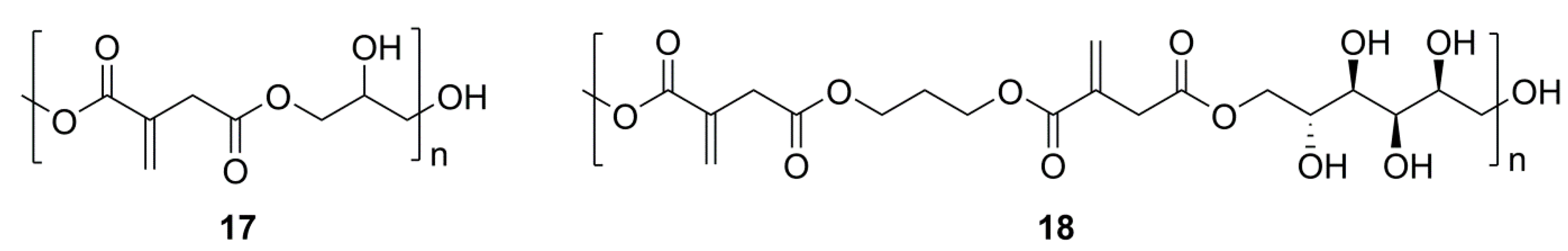
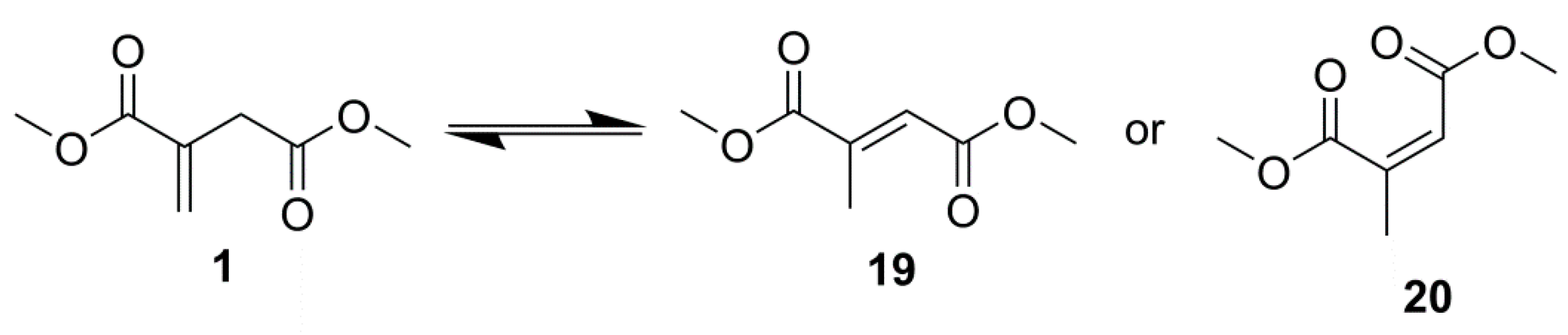
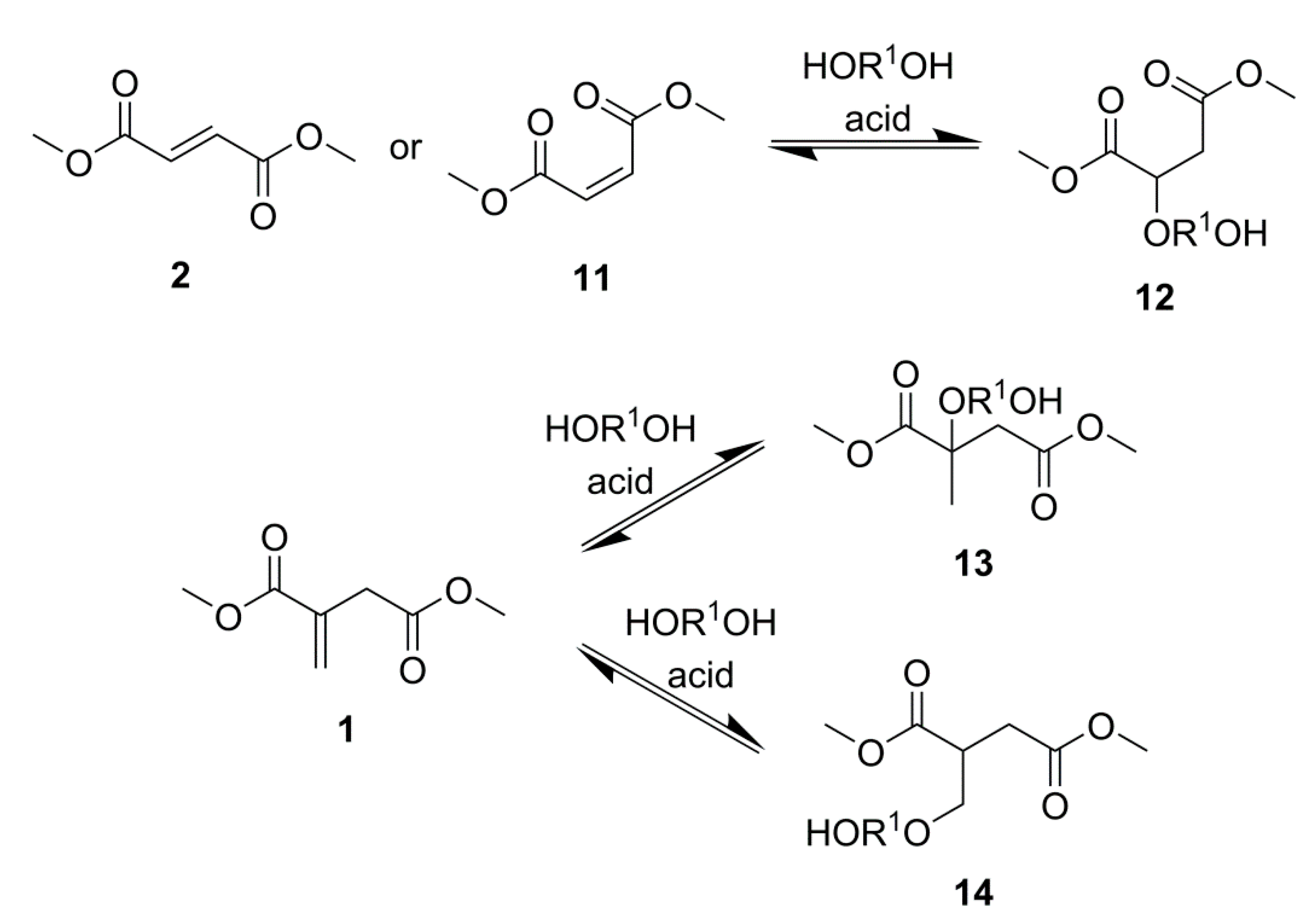
2.1. Simple UPEs
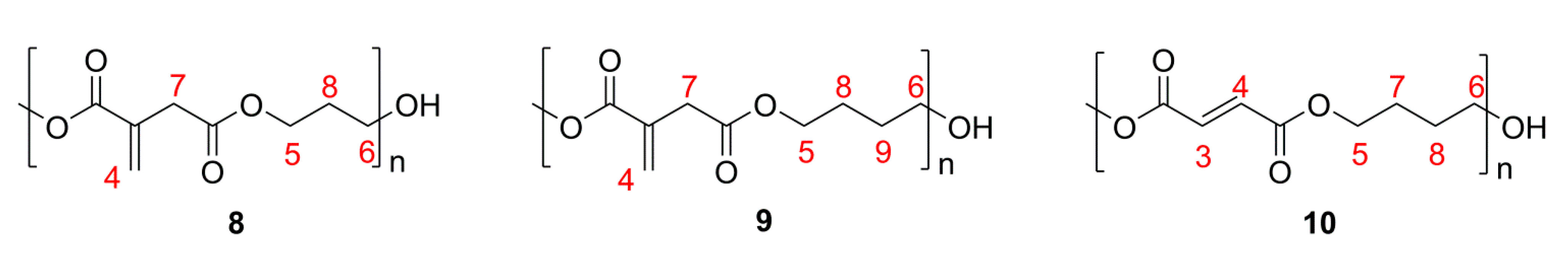


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPC Analysis | PPI | PBI | PBFu |
|---|---|---|---|
| MN | 3600 | 580 | 2400 |
| MW | 10,600 | 2600 | 4300 |
| Pdi | 2.9 | 3.4 |


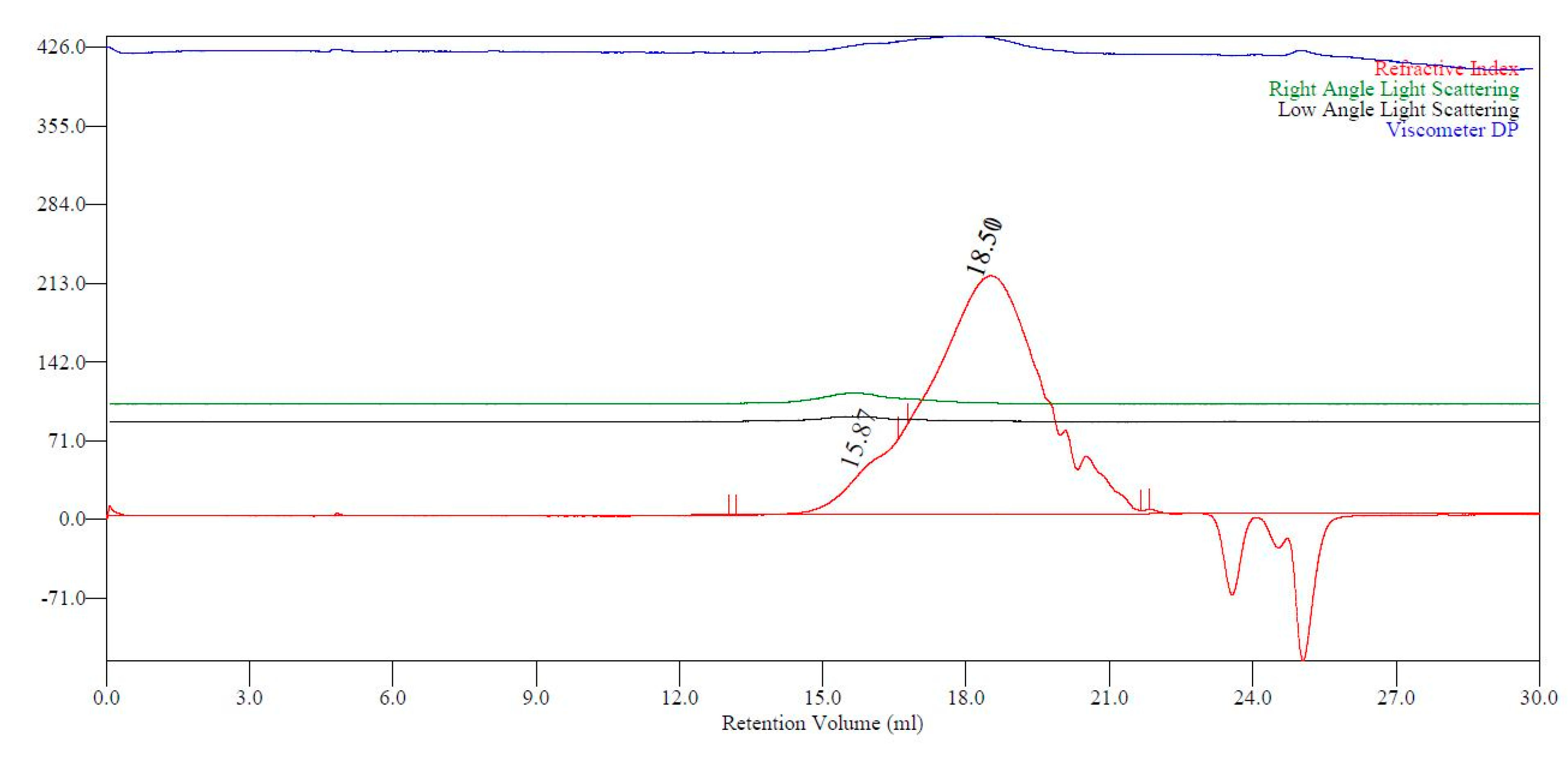

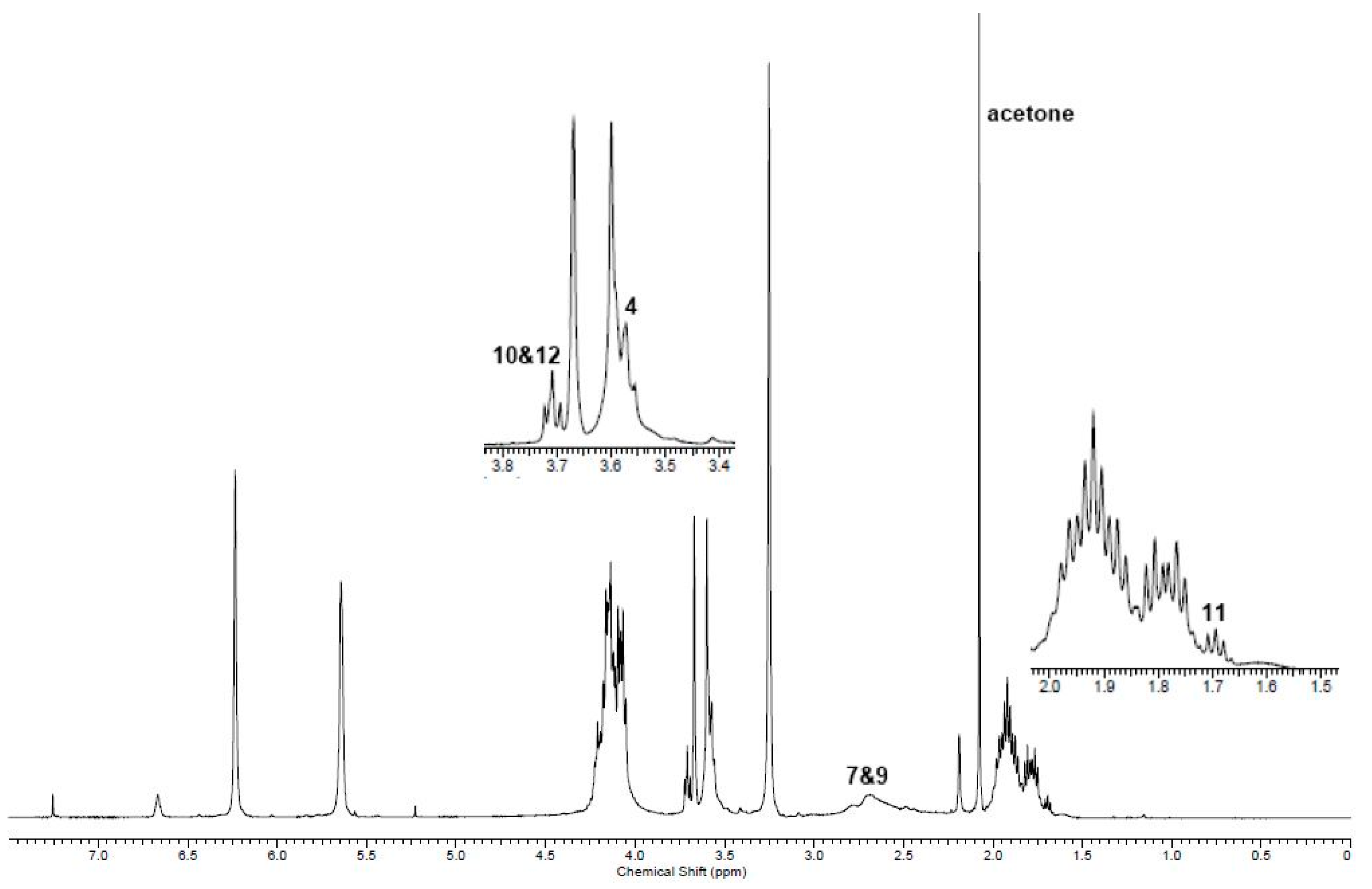
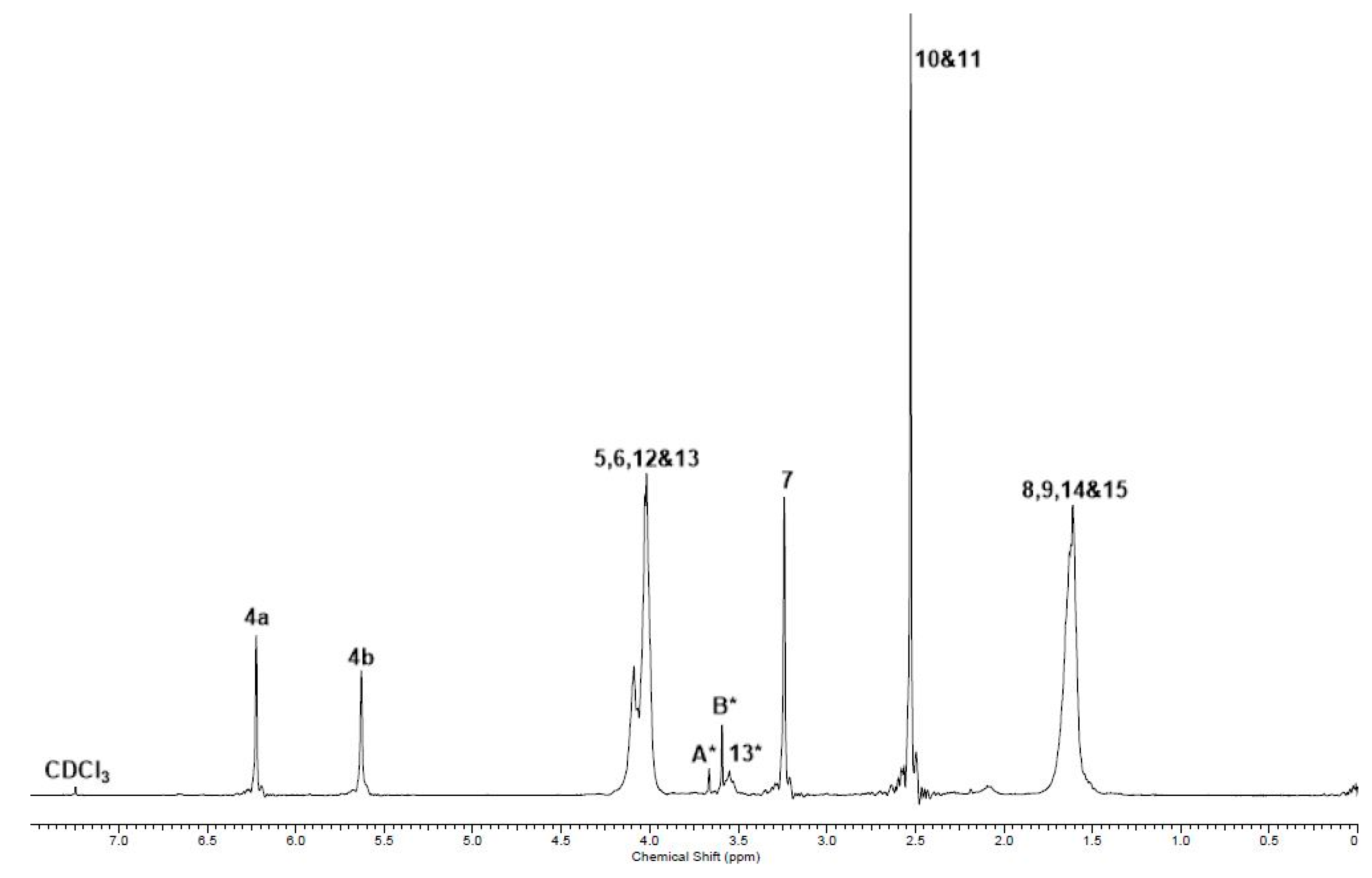
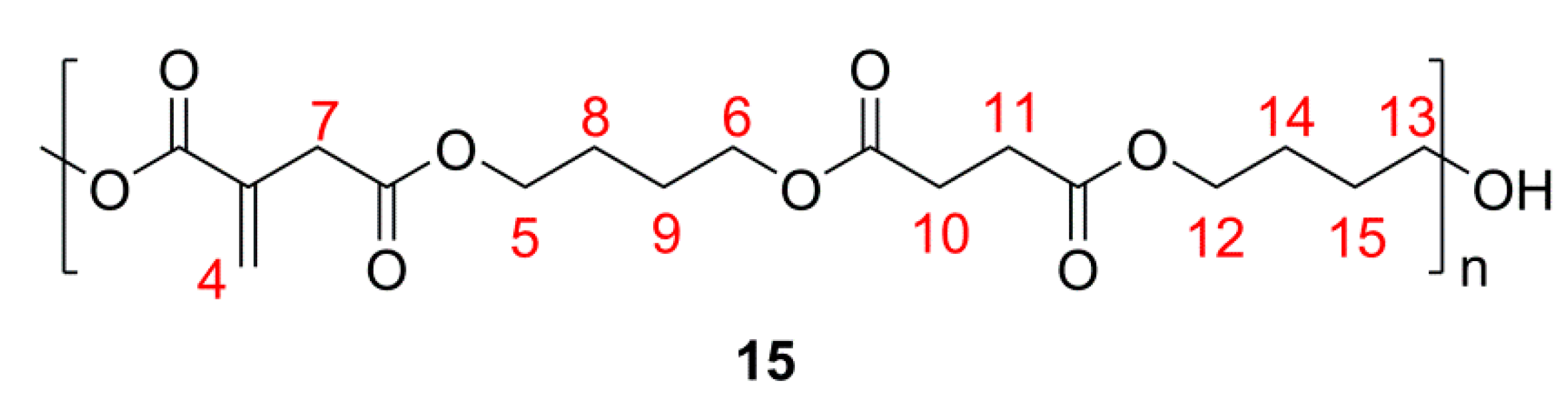
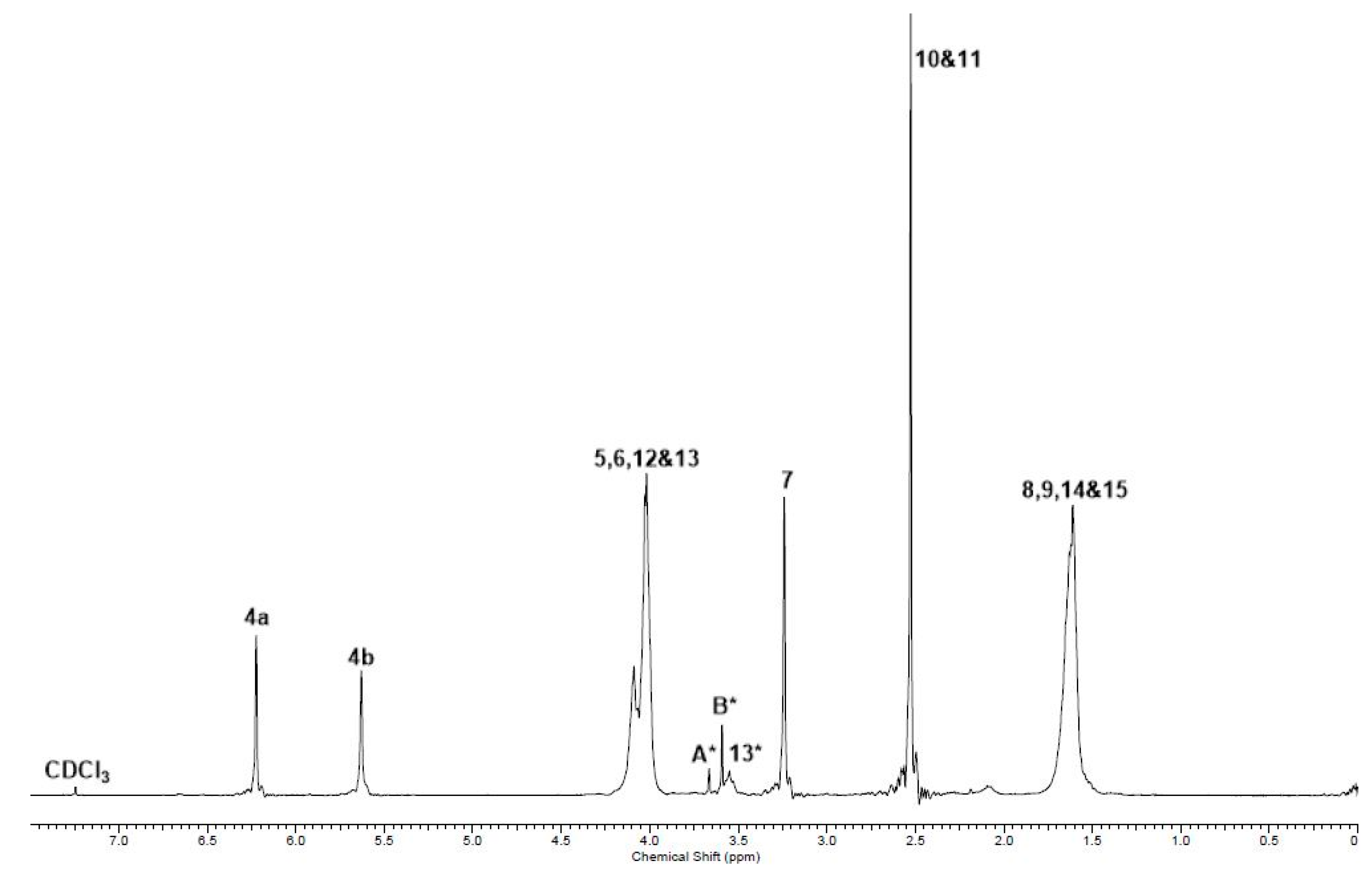
2.2. Co-UPEs

| GPC Analysis | Total Polymer (a) | High Conc./Low Mass (b) | Low Conc./High Mass (c) |
|---|---|---|---|
| MN | 780 | 900 | 130,000 |
| MW | 69,000 | 8300 | 350,000 |
| Pdi | 85 | 5.0 | 2.8 |
2.3. Glycerol or Sorbitol UPEs

| Polymer | % Itaconate | % Mesaconate | % Citraconate |
|---|---|---|---|
| PGI | 46 | 49 | 5 |
| PPISI | 57 | 39 | 4 |
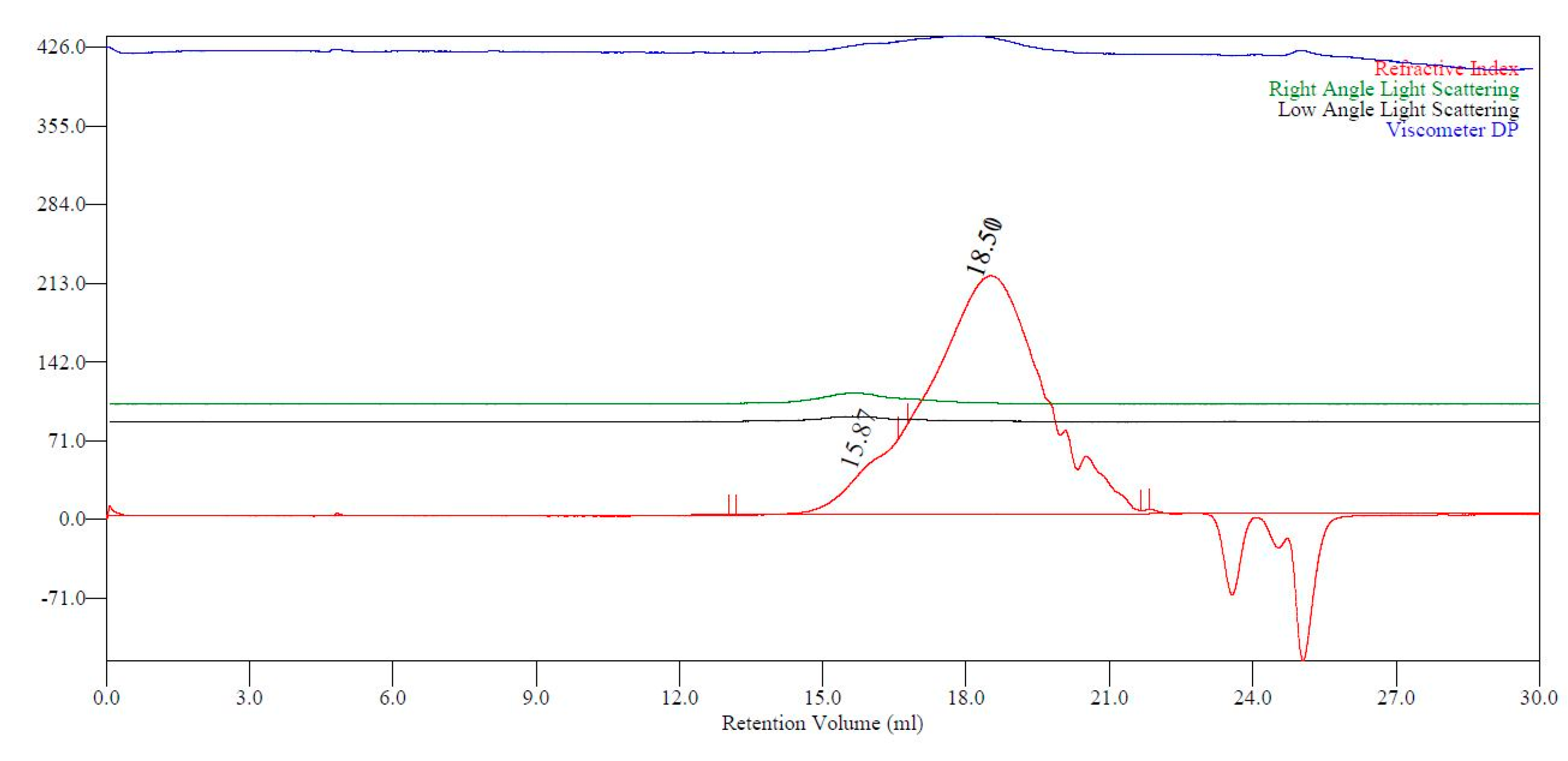
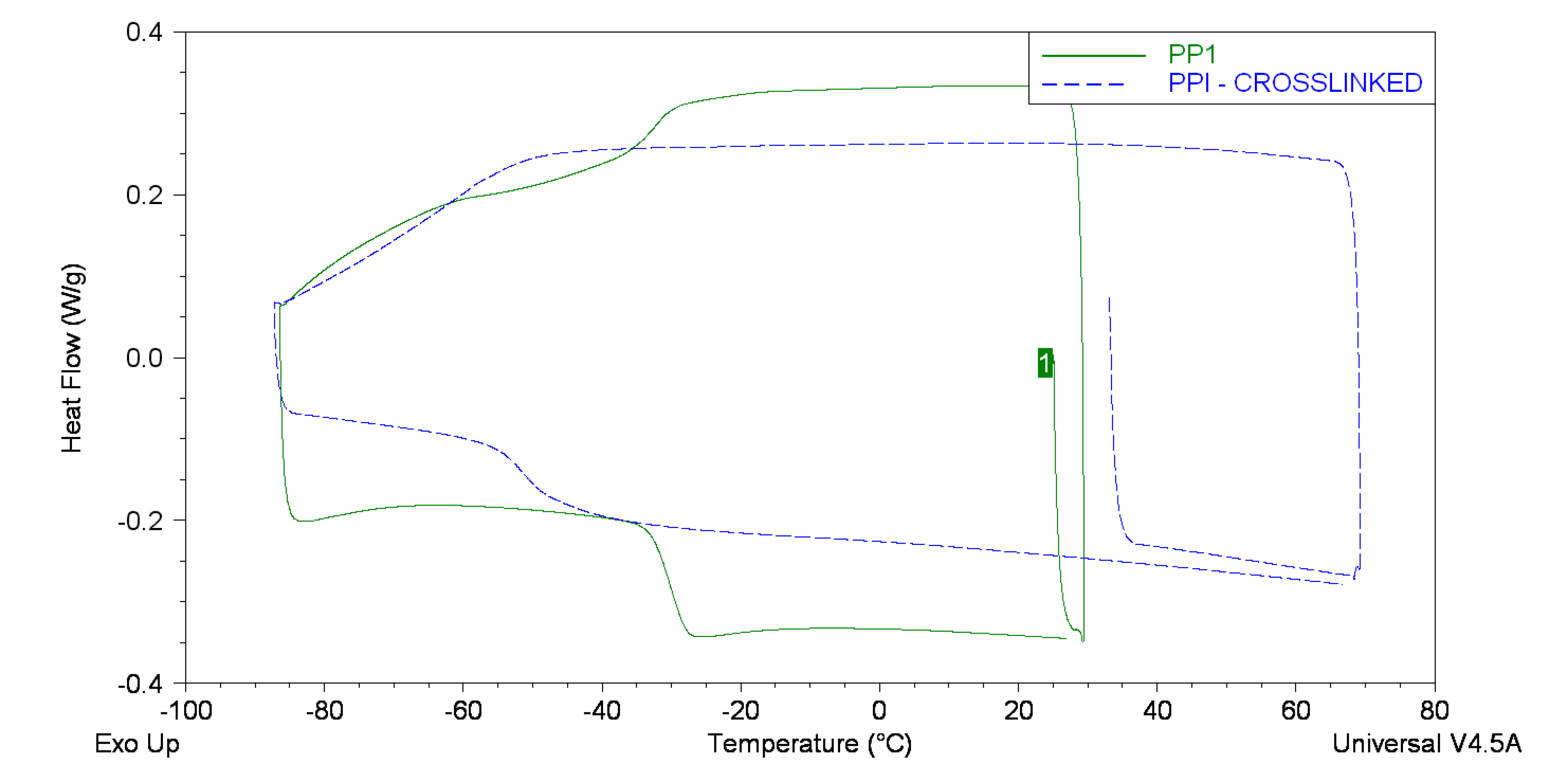
2.4. Advanced Analysis
| Backbone | 1st Decomposition (Minor) | 2nd Decomposition (Major) | Range of Decomposition |
|---|---|---|---|
| PPI | 345.1 | 407.2 | 291–437 |
| PBI | 395.6 | 351–426 | |
| PBFu | 258.7 | 381.8 | 337–425 |
| PBIBS | 304.5 | 399.3 | 333–413 |
| PGI | 280.3 | 336.8 | 273–446 |
| PPISI | 304.1 | 265–407 |
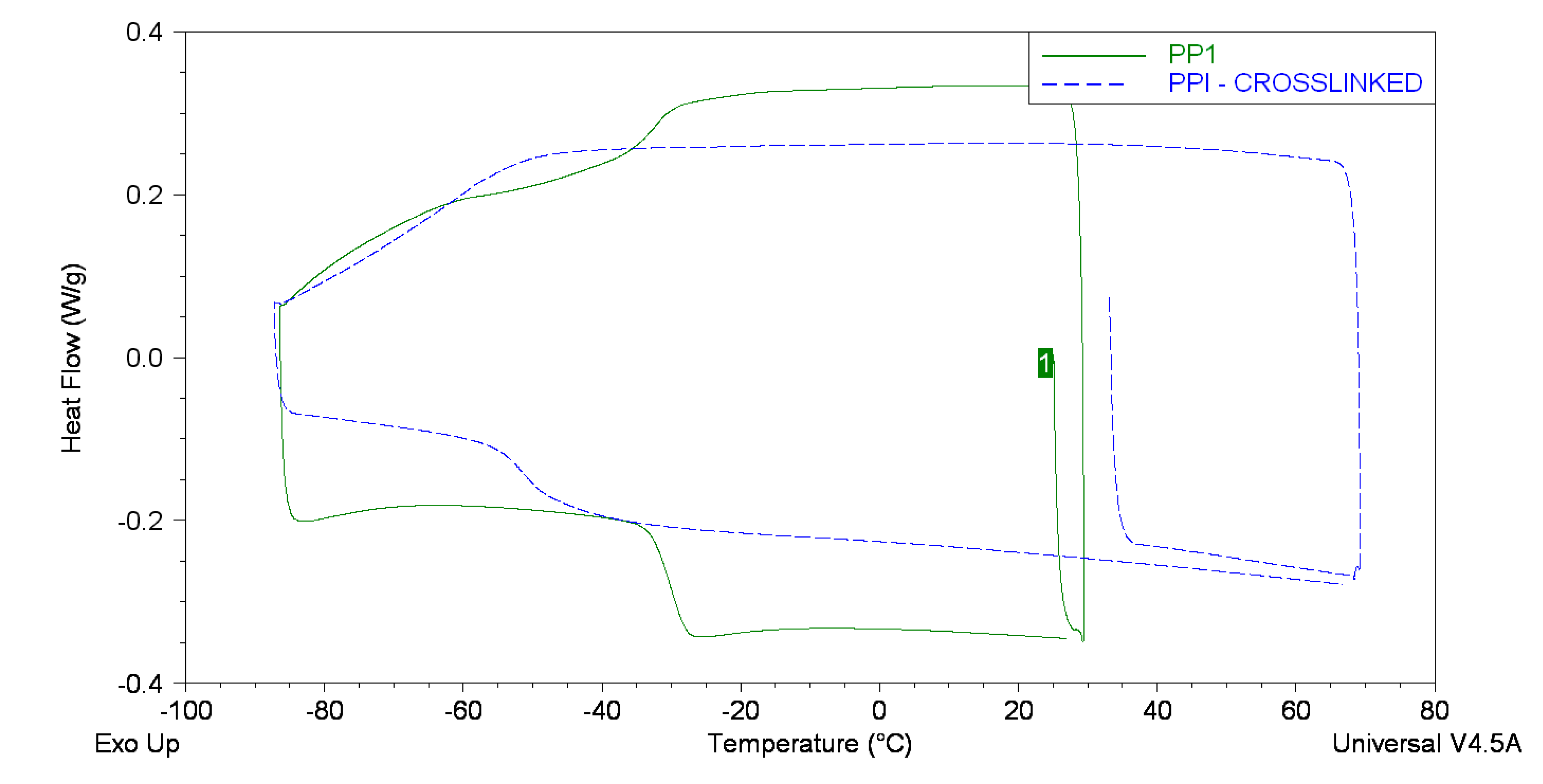
| Backbone | Tg/°C | Range/°C |
|---|---|---|
| PPI | −30.1 | −32.8 to −28.0 |
| PPI (crosslinked) | −51.2 | −54.3 to −48.9 |
| PBI | −41.7 | −44.8 to −39.6 |
| PBFu @ | 135.4 | 112 to 138 |
| PBIBS | −41.3 | −43.5 to −39.8 |
| PGI | −20.9 | −25.5 to −16.4 |
| PPISI | −16.6 | −21.0 to −11.8 |
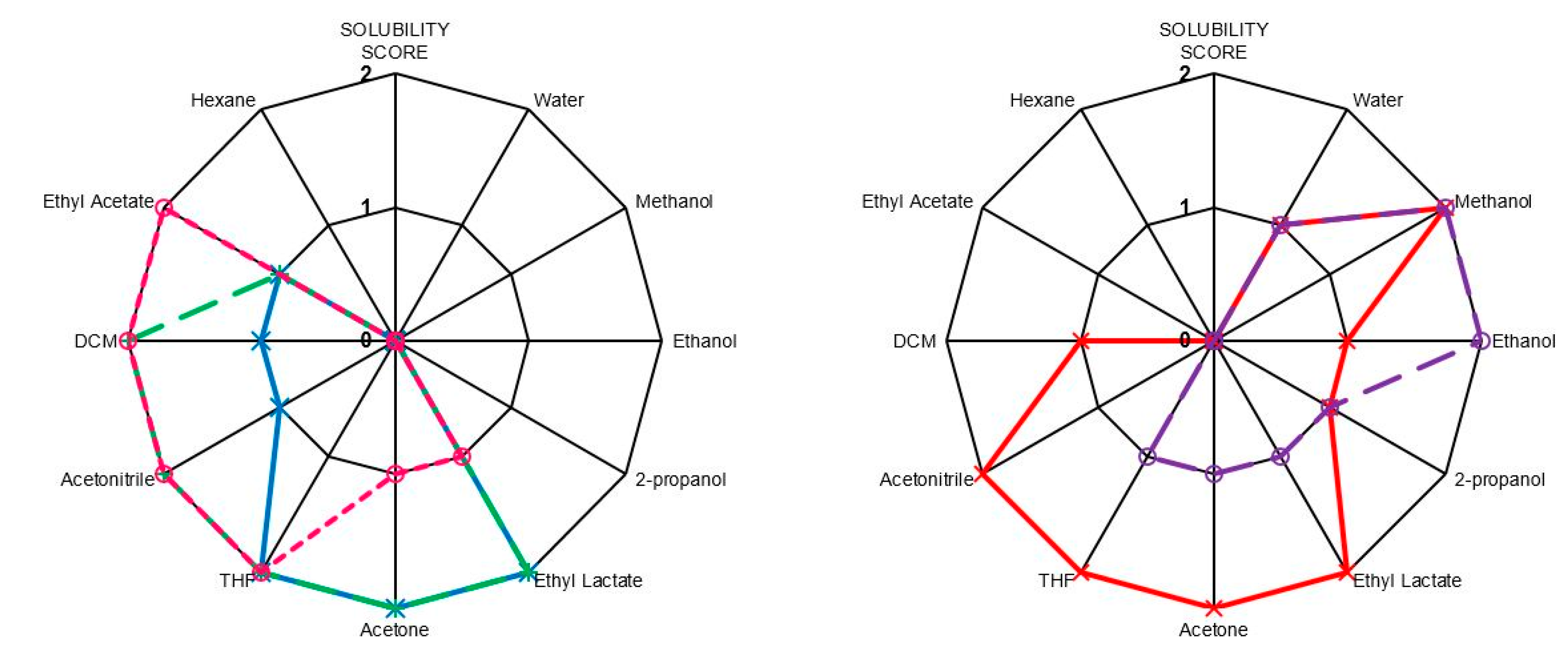
2.5. Solubilities
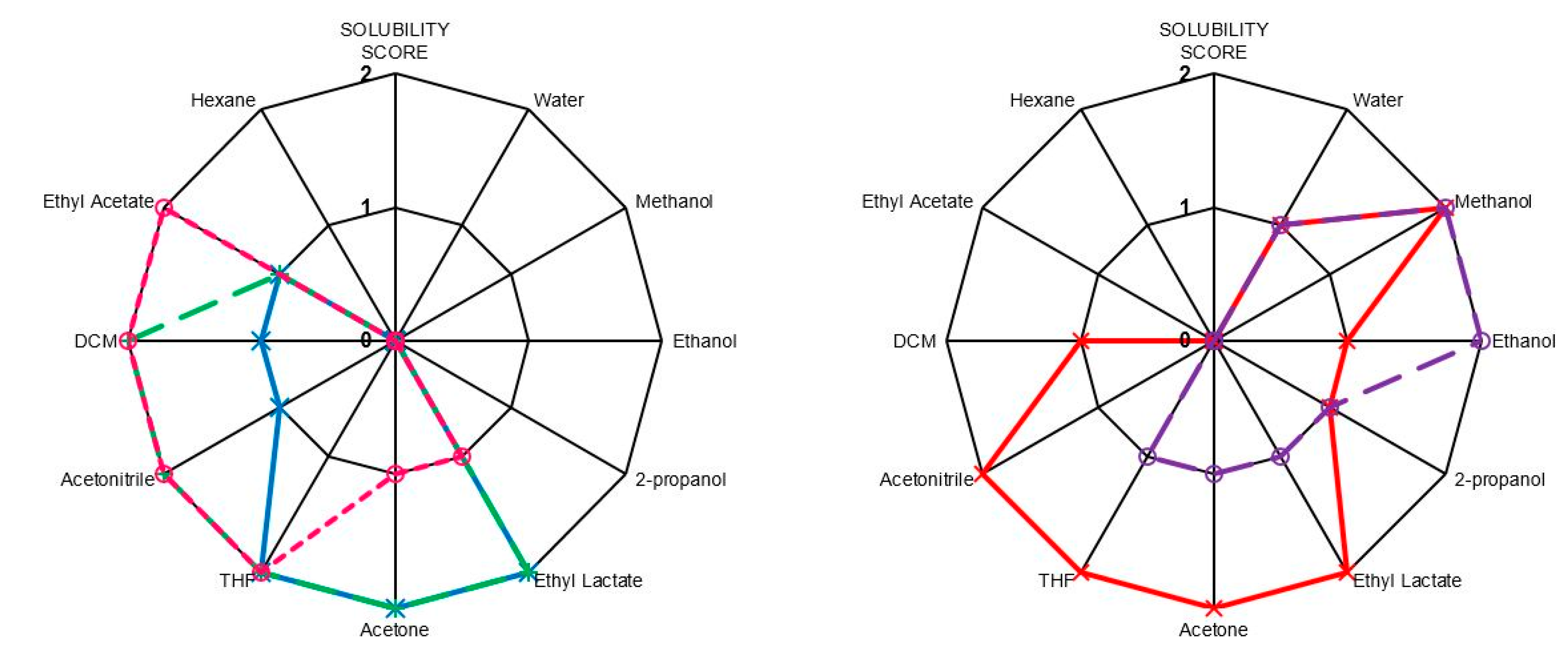
3. Experimental Section
3.1. Materials and Instrumentation
3.2. Synthesis of UPEs
3.2.1. Simple UPEs
3.2.2. PBIBS
3.2.3. PGI
3.2.4. PPISI
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tang, T.; Moyori, T.; Takasu, A. Isomerization-free polycondensations of cyclic anhydrides with diols and preparation of polyester gels containing cis or trans carbon double bonds via photo-cross-linking and isomerization in the gels. Macromolecules 2013, 46, 5464–5472. [Google Scholar] [CrossRef]
- Gowsika, J.; Nanthini, R. Synthesis, characterization and in vitro anti-cancer evaluation of itaconic acid based randon copolyester. J. Chem. 2014, 2014. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M. From monomers to polymers from renewable resources: Recent advances. Prog. Polym. Sci. 2015. [Google Scholar] [CrossRef]
- Okkerse, C.; van Bekkum, H. From fossil to green. Green Chem. 1999, 1, 107–114. [Google Scholar] [CrossRef]
- Farmer, T.; Mascal, M.; Clark, J.; Deswarte, F. Introduction to Chemicals from Biomass, 2nd ed.; John Wiley and Sons: Chichester, UK, 2015; pp. 89–156. [Google Scholar]
- Willke, T.; Vorlop, K.D. Biotechnological production of itaconic acid. Appl. Microbiol. Biotechnol. 2001, 56, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.H.; Farmer, T.J.; Macquarrie, D.J. The derivatisation of bio-platform molecules using KF-Alumina catalysis. Chem. Sus. Chem. 2009, 2, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Tokiwa, Y.; Calabia, B.P. Biodegradability and biodegradation of polyesters. J. Polym. Environ. 2007, 15, 259–267. [Google Scholar] [CrossRef]
- Barrett, D.G.; Merkel, T.J.; Luft, J.C.; Yousaf, M.N. One-step syntheses of photocurable polyesters based on a renewable resource. Macromolecules 2010, 43, 9660–9667. [Google Scholar] [CrossRef]
- Dai, J.; Ma, S.; Liu, X.; Han, L.; Wu, Y.; Dai, X.; Zhu, J. Synthesis of bio-based unsaturated polyester resins and their application in waterborne UV-curable coatings. Prog. Org. Coatings 2015, 78, 49–54. [Google Scholar] [CrossRef]
- Dai, J.; Ma, S.; Wu, Y.; Han, L.; Zhang, L.; Zhu, J.; Liu, X. Polyesters derived from itaconic acid for the properties and bio-based content enhancement of soybean oil-based thermosets. Green Chem. 2015, 17, 2383–2392. [Google Scholar] [CrossRef]
- Wang, R.; Ma, J.; Zhou, X.; Wang, Z.; Kang, H.; Zhang, L.; Hua, K.C.; Kulig, J. Design and preparation of a novel cross-linkable, high molecular weight, and bio-based elastomer by emulsion polymerization. Macromolecules 2012, 45, 6830–6839. [Google Scholar] [CrossRef]
- Guo, B.; Chen, Y.; Lei, Y.; Zhang, L.; Zhou, W.Y.; Rabie, A.B.M.; Zhao, J. Biobased poly(propylene sebacate) as shape memory polymer with tunable switching temperature for potential biomedical applications. Biomacromolecules 2011, 12, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Chanda, S.; Ramakrishnan, S. Poly(alkylene itaconate)s—An interesting class of polyesters with periodically located exo-chain double bonds susceptible to Michael addition. Polym. Chem. 2015, 6, 2108–2114. [Google Scholar] [CrossRef]
- Winkler, M.; Lacerda, T.M.; Mack, F.; Meier, M.A.R. Renewable polymers from itaconic acid by polycondensation and ring-opening-metathesis polymerization. Macromolecules 2015, 48, 1398–1403. [Google Scholar] [CrossRef]
- Tang, T.; Takasu, A. Facile synthesis of unsaturated polyester-based double-network gels via chemoselective cross-linking using Michael addition and subsequent UV-initiated radical polymerization. RSC Adv. 2015, 5, 819–829. [Google Scholar] [CrossRef]
- Lv, A.; Li, Z.-L.; Du, F.-S.; Li, Z.-C. Synthesis, functionalization, and controlled degradation of high molecular weight polyester from itaconic acid via ADMET polymerization. Macromolecules 2014, 47, 7707–7716. [Google Scholar] [CrossRef]
- Goerz, O.; Ritter, H. Polymers with shape memory effect from renewable resources: Crosslinking of polyesters based on isosorbide, itaconic acid and succinic acid. Polym. Int. 2013, 62, 709–712. [Google Scholar] [CrossRef]
- Paul, S.; Zhu, Y.; Romain, C.; Brooks, R.; Saini, P.K.; Williams, C.K. Ring-opening copolymerization (ROCOP): Synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51, 6459–6479. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Liu, X.; Jiang, Y.; Tang, Z.; Zhang, C.; Zhu, J. Bio-based epoxy resin from itaconic acid and its thermosets cured with anhydride and comonomers. Green Chem. 2013, 15, 245–254. [Google Scholar] [CrossRef]
- Goerz, O.; Ritter, H. N-Alkylated dinitrones from isosorbide as cross-linkers for unsaturated bio-based polyesters. Beilstein J. Org. Chem. 2014, 10, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, N.; Ozeki, M.; Fujiwara, I.; Shibata, M. Crosslinking and biodegradation of poly(butylene succinate) prepolymers containing itaconic or maleic acid units in the main chain. J. Appl. Polym. Sci. 2005, 95, 1473–1480. [Google Scholar] [CrossRef]
- Pellis, A.; Corici, L.; Sinigoi, L.; D’Amelio, N.; Fattor, D.; Ferrario, V.; Ebert, C.; Gardossi, L. Towards feasible and scalable solvent-free enzymatic polycondensations: Integrating robust biocatalysts with thin film reactions. Green Chem. 2015, 17, 1756–1766. [Google Scholar] [CrossRef] [Green Version]
- Naves, A.F.; Fernandes, H.T.C.; Immich, A.P.S.; Catalani, L.H. Enzymatic syntheses of unsaturated polyesters based on isosorbide and isomannide. J. Polym. Sci. 2013, 51, 3881–3891. [Google Scholar] [CrossRef]
- Ordelt, Z.; Novak, V.; Kratky, B. Reversibility of diol addition to olefin double bond of ethylene-1,2-dicarbonic acids in polycondensation in melts. Collect. Czechoslov. Chem. Commun. 1968, 33, 405–415. [Google Scholar] [CrossRef]
- Paci, M.; Crescenzi, V.; Supino, N.; Campana, F. Structural characterisation of unsaturated polyesters. Macromol. Chem. Phys. 1982, 183, 377–387. [Google Scholar] [CrossRef]
- Fradet, A.; Marechal, E. Study on models of double bond saturation during the synthesis of unsaturated polyesters. Macromol. Chem. Phys. 1982, 183, 319–329. [Google Scholar] [CrossRef]
- Lehtonen, J.; Salmi, T.; Immonen, K.; Paatero, E.; Nyholm, P. Kinetic model for the homogeneously catalyzed polyesterification of dicarboxylic acids with diols. Ind. Eng. Chem. Res. 1996, 35, 3951–3963. [Google Scholar] [CrossRef]
- Yang, Y.S.; Pascault, J.P. Modeling of unsaturated polyester prepolymer structures. II. Hydroxyl and carboxyl functionalities. J. Appl. Polym. Sci. 1996, 147–156. [Google Scholar] [CrossRef]
- Zetterlund, P.B.; Gosden, R.G.; Weaver, W.; Johnson, A.F. New aspects of unsaturated polyester resin synthesis. Part 2. Reactant sequence distribution and its effect on cure kinetics. Polym. Int. 2003, 52, 749–756. [Google Scholar] [CrossRef]
- Retuert, J.; Yazdani-Pedram, M.; Martinez, F.; Jeria, M. Soluble itaconic acid-ethylene glycol polyesters. Bull. Chem. Soc. Jpn. 1993, 66, 1707–1708. [Google Scholar] [CrossRef]
- Kharas, G.B.; Kamenetsky, M.; Simantirakis, J.; Beinlich, K.C.; Rizzo, A.T.; Caywood, G.A.; Watson, K. Synthesis and characterization of fumarate-based polyesters for use in bioresorbable bone cement composites. J. Appl. Polym. Sci. 1997, 66, 1123–1137. [Google Scholar] [CrossRef]
- Sakuma, T.; Kumagai, A.; Teramoto, N.; Shibata, M. Thermal and dynamic mechanical properties of organic-inorganic hybrid composited of itaconate-containing poly(butylene succinate) and methacrylate-substituted polysilsesquioxane. J. Appl. Polym. Sci. 2008, 107, 2159–2164. [Google Scholar] [CrossRef]
- Cassel, S.; Debaig, C.; Benvegnu, T.; Chaimbault, P.; Lafosse, M.; Plusquellec, D.; Rollin, P. Original synthesis of linear, branched and cyclic oligoglycerol standards. Eur. J. Org. Chem. 2001, 5, 875–896. [Google Scholar] [CrossRef]
- Billmeyer, F. Textbook of Polymer Science, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1984; pp. 320–323. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farmer, T.J.; Castle, R.L.; Clark, J.H.; Macquarrie, D.J. Synthesis of Unsaturated Polyester Resins from Various Bio-Derived Platform Molecules. Int. J. Mol. Sci. 2015, 16, 14912-14932. https://doi.org/10.3390/ijms160714912
Farmer TJ, Castle RL, Clark JH, Macquarrie DJ. Synthesis of Unsaturated Polyester Resins from Various Bio-Derived Platform Molecules. International Journal of Molecular Sciences. 2015; 16(7):14912-14932. https://doi.org/10.3390/ijms160714912
Chicago/Turabian StyleFarmer, Thomas J., Rachael L. Castle, James H. Clark, and Duncan J. Macquarrie. 2015. "Synthesis of Unsaturated Polyester Resins from Various Bio-Derived Platform Molecules" International Journal of Molecular Sciences 16, no. 7: 14912-14932. https://doi.org/10.3390/ijms160714912