
Stepchild or Prodigy? Neuroprotection in Multiple Sclerosis (MS) Research

Abstract
:1. Introduction
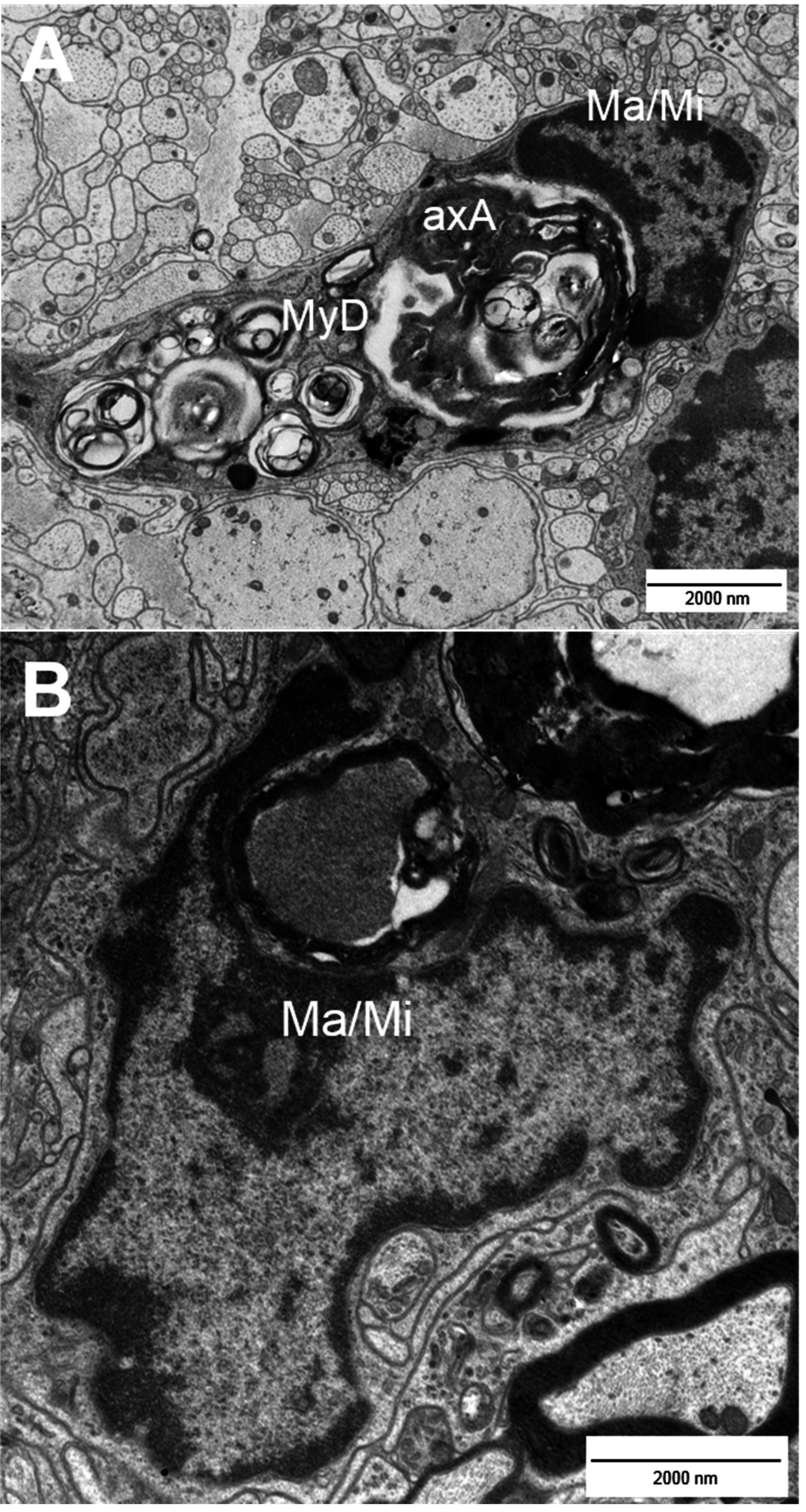


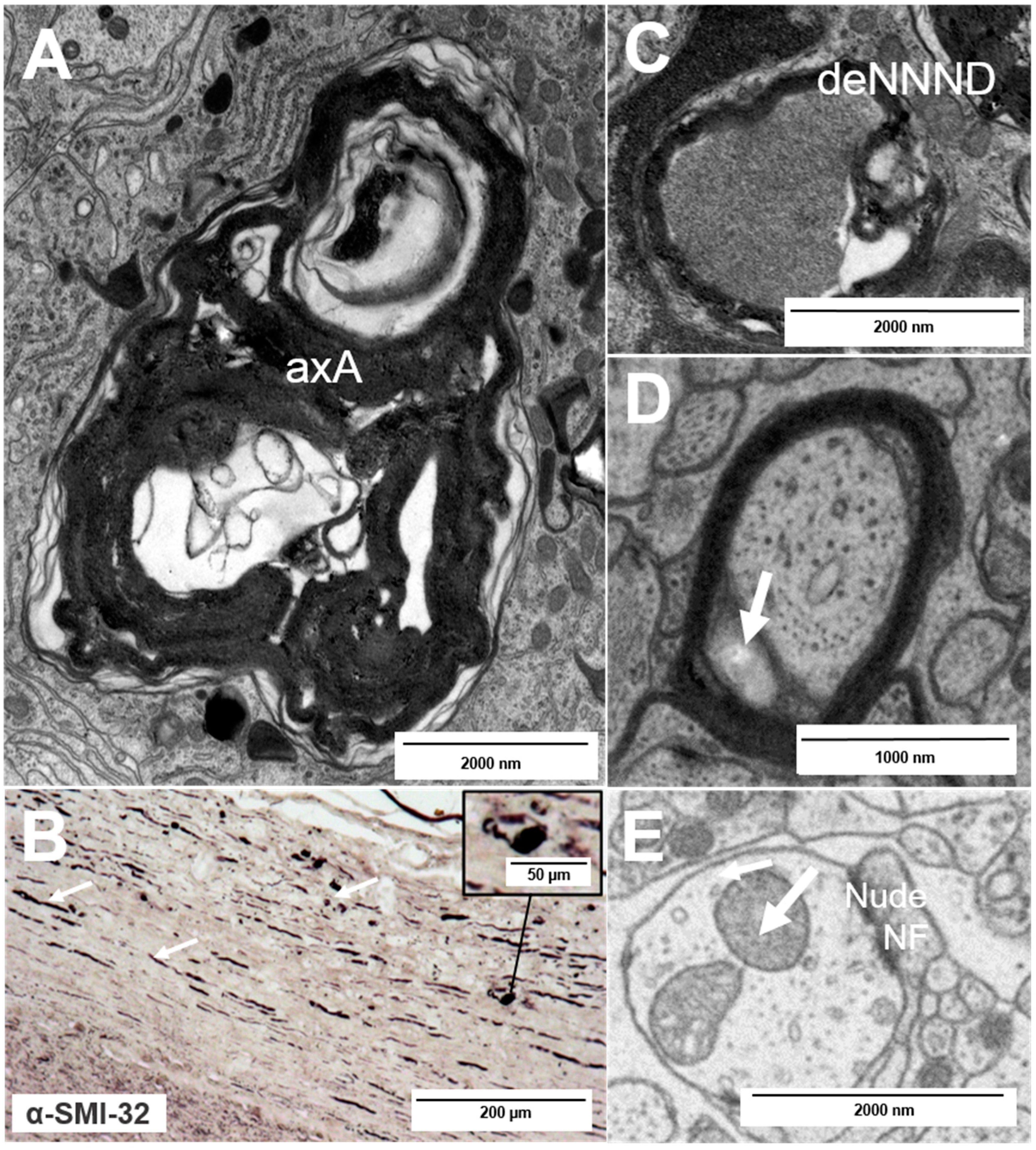
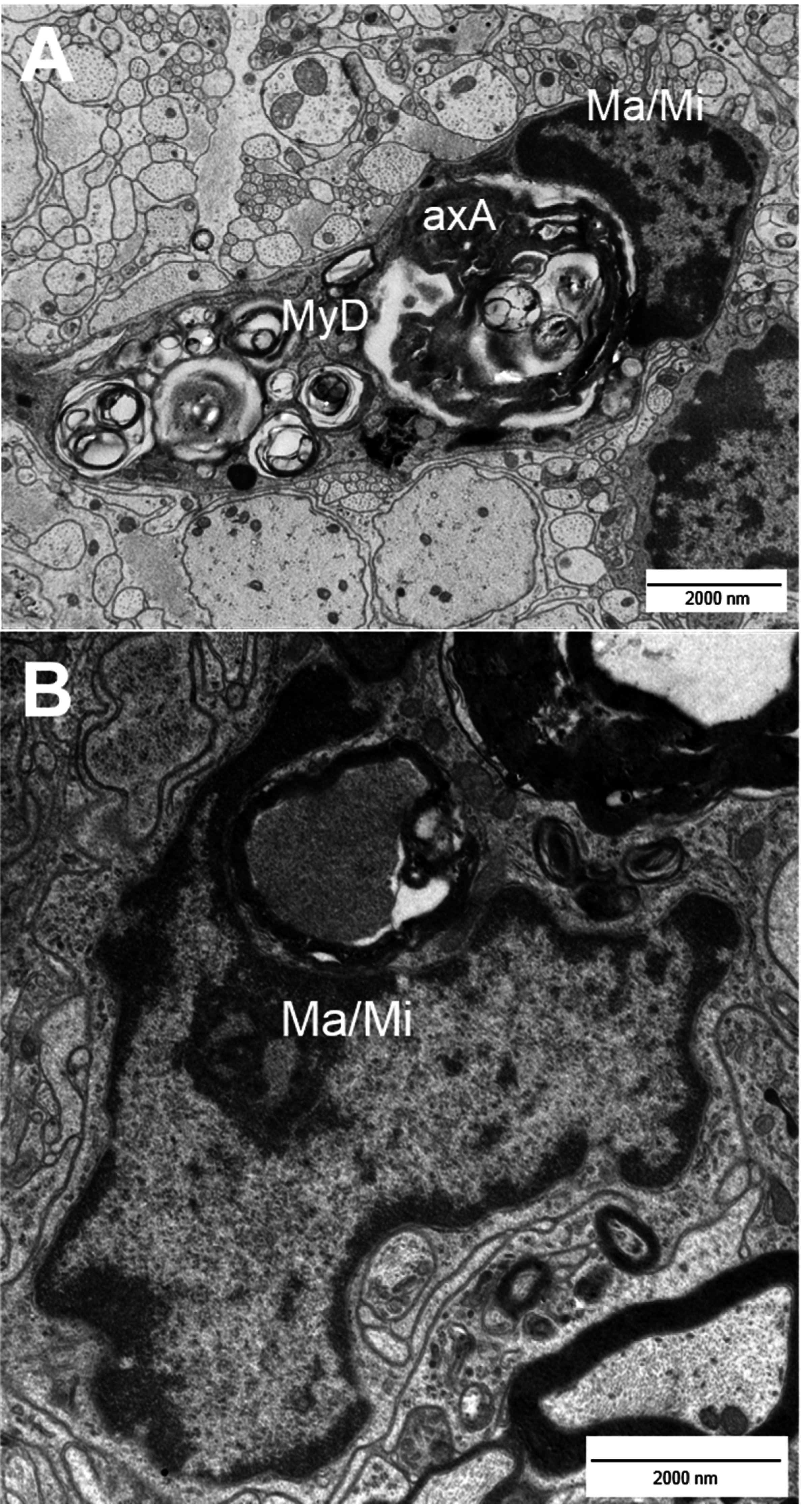
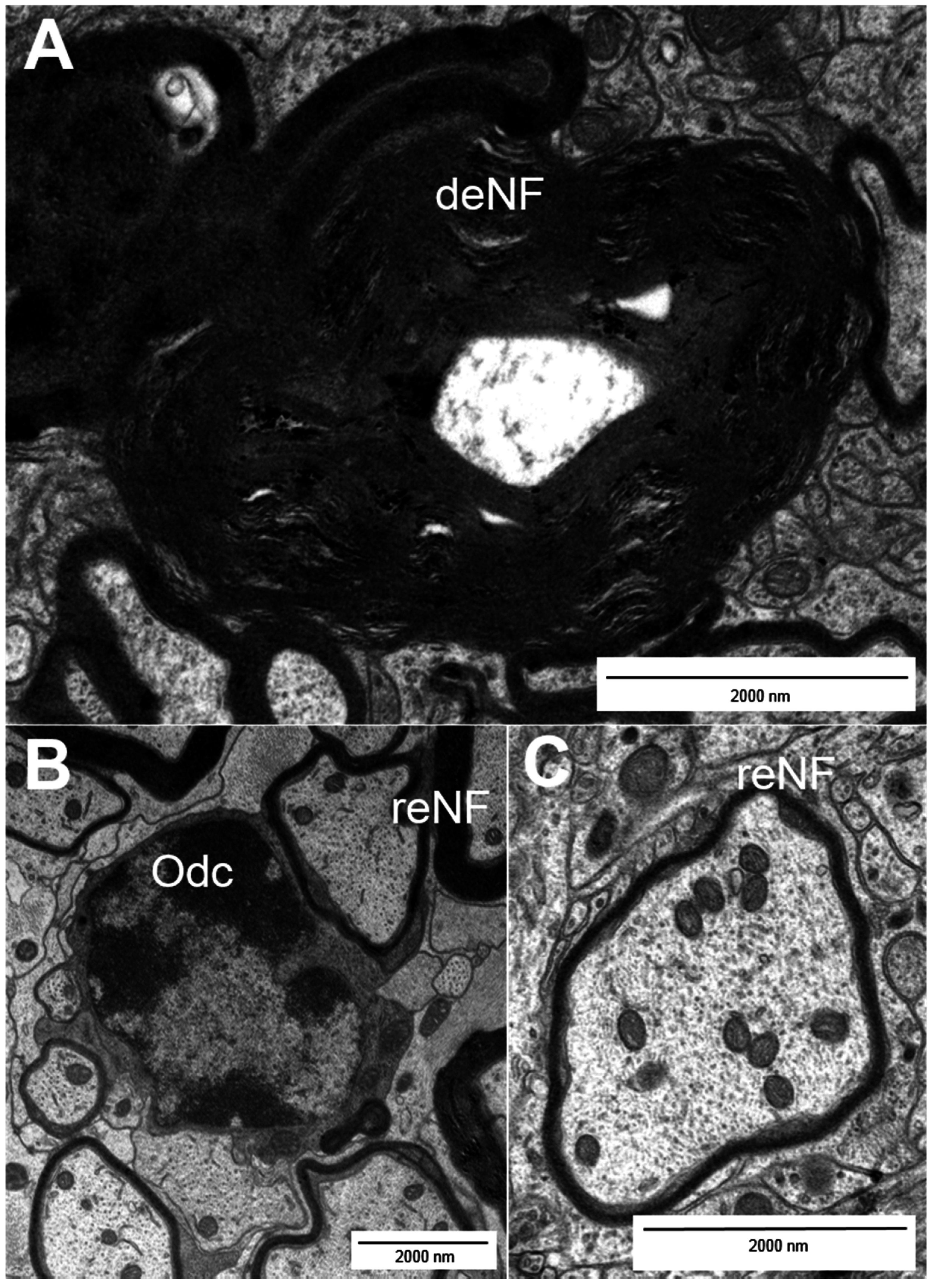
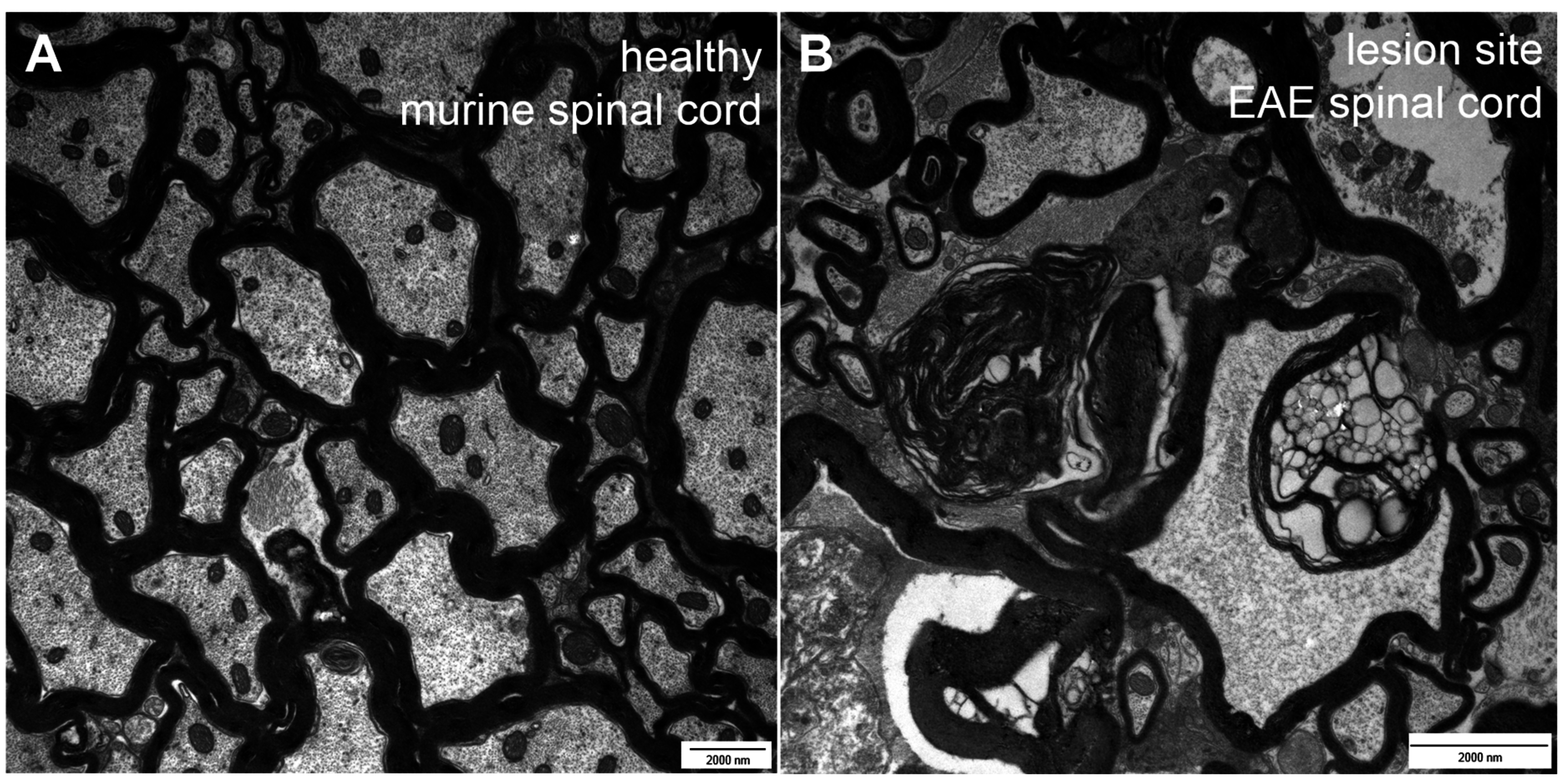
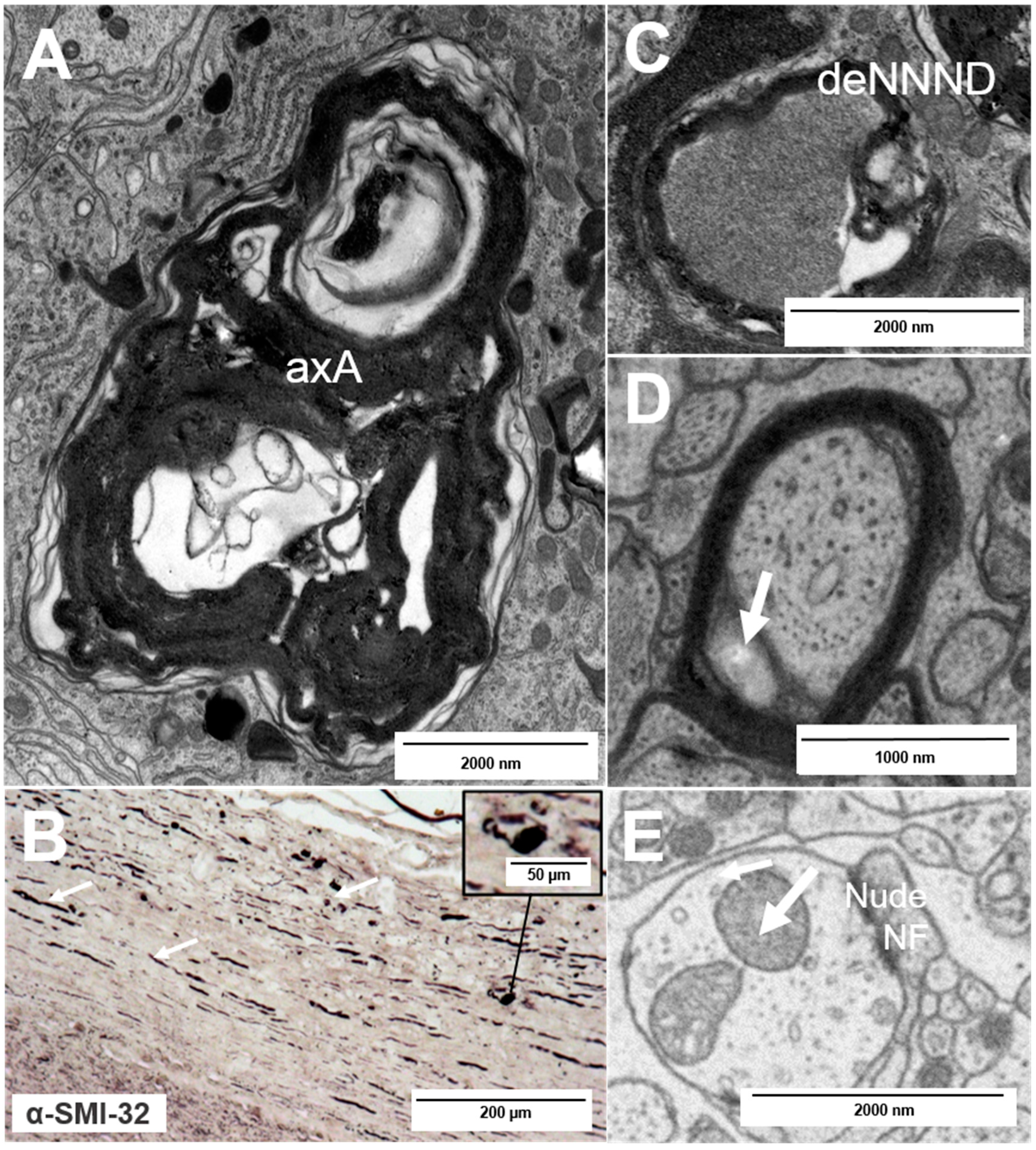
2. Types of Nerve Fiber Pathology
3. Mechanisms of Neuronal Damage in MS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Causes of Neurodegeneration | References |
|---|---|
| Injury by CD4+ and CD8+ T cells | [4,5,10] |
| Free radicals | [8] |
| Glutamate overload | [8] |
| Demyelination | [8] |
| Activation of microglia | [8] |
| NO | [6,7,9] |
| Ion accumulation/dysregulation | [6,7,9] |
| Oligodendrocyte/oligodendrocyte progenitor cell damage | [11,12] |
| Mitochondrial damage/collapse | [9,12,21] |
| Others: complement activation, cytokine expression etc. | [29–31] |
4. Physiological Neuroprotection and Remyelination
| Cell Types | References |
|---|---|
| TH2 cells: | [ 12 ] |
| activation of CNS-resident cells to produce trophic factors (e.g., GDNF by astrocytes), inhibition of TH1-mediated toxicity, production of neurotrophins and growth factors like NGFs and IGFs, vascular endothelial growth factors and platelet-derived growth factors | |
| Microglia: | [12,16,20,34] |
| expression of factors such as NTF, BDNF and GDNF as a “stepping stone” for sprouting axons, release of substances that induce an OPC switch towards a regenerative phenotype (activation) as well as recruitment of OPCs to the lesion site, clearance of myelin debris and lipid recycling (via apolipoprotein E) | |
| Astrocytes: | [12,20] |
| expression of GDNF, BDNF and factors that induce an OPC switch towards a regenerative phenotype | |
| Oligodendrocytes/OPCs: | [7,12,16,20,26] |
| expression of trophic factors, support of axonal stability and transport integrity, communication with the axon (e.g., via direct transfer of exosomes to neurons), remyelination | |
| Neuron: | [16] |
| increased expression of growth-associated proteins (GAPs, like GAP-43) in sprouting nerve fibers and of cell organelle-associated proteins (microtubule/neurofilament-associated proteins) | |
| Other factors: | [6,16,27,28] |
| strictly regulated ion levels |
5. Limitations of Regeneration
6. Therapeutic Options for MS
7. How to Evaluate Therapeutic Benefit?
8. Concluding Remarks
Key Points
- √
- Multiple mechanisms contribute to axonal pathology: After initial inflammatory damage mechanisms like dysfunction of mitochondria, trophic and ion imbalance, generation of free radicals, oligodendrocyte death, impaired OPC differentiation and remyelination failure lead to ongoing progression of neurodegeneration and disease over time.
- √
- Oligodendrocytes do not only remyelinate, but also support nerve fibers metabolically and regulate axonal motor proteins, cytoskeletal structure and integrity.
- √
- Increased intracellular ion levels lead to the disruption of mitochondrial function and degradation of proteins followed by cell death.
- √
- For the evaluation of therapeutic benefit, further clinical tools and biomarkers are needed and should be combined with common techniques to rate degenerative/regenerative processes and to provide a better overview of the disease processes in MS.
- √
- The availability of suitable biomarkers will also fuel future studies that are aimed at addressing the need for neuroprotective/-regenerative therapies in MS and other neurodegenerative diseases.
Conflicts of Interest
References
- Rottlaender, A.; Villwock, H.; Addicks, K.; Kuerten, S. Neuroprotective role of fibroblast growth factor-2 in experimental autoimmune encephalomyelitis. Immunology 2011, 133, 370–378. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Kuerten, S.; Lichtenegger, F.S.; Faas, F.; Angelov, D.N.; Tary-Lehmann, M.; Lehmann, P.V. MBP-PLP fusion protein-induced EAE in C57BL/6 mice. J. Neuroimmunol. 2006, 177, 99–111. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef]
- Lidster, K.; Jackson, S.J.; Ahmed, Z.; Munro, P.; Coffey, P.; Giovannoni, G.; Baker, M.D.; Baker, D. Neuroprotection in a novel mouse model of multiple sclerosis. PLoS ONE 2013, 8, e79188. [Google Scholar] [CrossRef]
- Hauser, S.L.; Chan, J.R.; Oksenberg, J.R. Multiple sclerosis: Prospects and promise. Ann. Neurol. 2013, 74, 317–327. [Google Scholar] [CrossRef]
- Lassmann, H. Axonal injury in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2003, 74, 695–697. [Google Scholar] [CrossRef]
- Waxman, S.G. Axonal conduction and injury in multiple sclerosis: The role of sodium channels. Nat. Rev. Neurosci. 2006, 7, 932–941. [Google Scholar] [CrossRef]
- Buddeberg, B.S.; Kerschensteiner, M.; Schwab, M.E. Die Bedeutung der axonalen Schädigung bei multipler Sklerose (in German). Schweiz. Med. Forum 2003, 38, 904–908. [Google Scholar]
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesions. Ann. Neurol. 2004, 55, 458–468. [Google Scholar] [CrossRef]
- Dhib-Jalbut, S.; Arnold, D.L.; Cleveland, D.W; Fischer, M.; Friedlander, R.M.; Mouradian, M.M.; Przedborski, S.; Trapp, B.D.; Wyss-Coray, T.; Yong, V.W. Neurodegeneration and neuroprotection in multiple sclerosis and other neurodegenerative diseases. J. Neuroimmunol. 2006, 176, 198–215. [Google Scholar] [CrossRef]
- Bo, L.; Vedele, C.A.; Nyland, H.I.; Trapp, B.D.; Mörk, S.J. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult. Scler. J. 2003, 9, 223–331. [Google Scholar] [CrossRef]
- De Stefano, N.; Matthews, P.M.; Filippi, M. Evidence of early cortical atrophy in MS: Relevance to white matter changes and disability. Neurology 2003, 60, 1157–1162. [Google Scholar] [CrossRef]
- Filippi, M.; Rocca, M.A.; de Stefano, N.; Enzinger, C.; Fisher, E.; Horsfield, M.A.; Inglese, M.; Pelletier, D.; Comi, G. Magnetic Resonance Techniques in Multiple Sclerosis. Arch. Neurol. 2001, 68, 1514–1520. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; ffrench-Constant, C.; Edgar, J.M.; Smith, K.J. Neuroprotection and repair in multiple sclerosis. Nat. Rev. Neurol. 2012, 8, 624–634. [Google Scholar] [CrossRef]
- Charcot, J.M. Histologie de la sclérose en plaques (in French). Gazette des Hôpitaux 1868, 41, 554–555. [Google Scholar]
- Recks, M.S.; Stormanns, E.R.; Bader, J.; Arnold, S.; Addicks, K.; Kuerten, S. Early axonal damage and progressive myelin pathologydefine the kinetics of CNS histopathology in a mouse model of multiple sclerosis. Clin. Immunol. 2013, 149, 32–45. [Google Scholar] [CrossRef]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmuneencephalomyelitis: A comparative quantitative study of axonal injury in active, inactive and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Batchelor, P.E.; Howells, D.W. CNS regeneration: Clinical possibility or basic science fantasy? J. Clin. Neurosci. 2003, 10, 523–534. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Suppression of mitochondrialoxidative stress provides long-term neuroprotection in experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 2007, 48, 681–691. [Google Scholar] [CrossRef]
- Soellner, I.A.; Rabe, J.; Mauri, V.; Kaufmann, J.; Addicks, K.; Kuerten, S. Differential aspects of immune cell infiltration and neurodegeneration in acute and relapse experimental autoimmune encephalomyelitis. Clin. Immunol. 2013, 149, 519–529. [Google Scholar] [CrossRef]
- Guy, J.; Ellis, E.A; Hope, G.M.; Emerson, S. Maintenance of myelinated fibre g ratio in acute experimental allergic encephalomyelitis. Brain 1991, 114, 281–294. [Google Scholar]
- Scherer, S. Axonal pathology in demyelinating diseases. Ann. Neurol. 1999, 45, 6–7. [Google Scholar] [CrossRef]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechnisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar]
- Stys, P.K.; Waxman, S.G.; Ransom, B.R. Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na+ channels and Na+-Ca2+ exchanger. J. Neurosci. 1992, 12, 430–439. [Google Scholar]
- Craner, M.J.; Damarjian, T.G.; Liu, S.; Hains, B.C.; Lo, A.C.; Black, J.A.; Newcombe, J.; Cuzner, M.L.; Waxman, S.G. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 2005, 9, 220–229. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Ingram, G.; Loveless, S.; Howell, O.W.; Hakobyan, S.; Dancey, B.; Harris, C.L.; Robertson, N.P.; Neal, J.W.; Morgan, B.P. Complement activation in multiple sclerosis plaques: An immunohistochemical analysis. Acta Neuropathol. Commun. 2014, 2. [Google Scholar] [CrossRef]
- Kemper, C.; Atkinson, J.P. T cell regulation: With complements to innate immunity. Nat. Rev. Immunol. 2007, 7, 9–18. [Google Scholar] [CrossRef]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef]
- Rouhani, S.J.; Eccles, J.D.; Tewalt, E.F.; Engelhard, V.H. Regulation of T cell tolerance by lymphatic endothelial cells. J. Clin. Cell Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Olah, M.; Amor, S.; Brouwer, N.; Vinet, J.; Eggen, B.; Biber, K.; Boddeke, H.W.G.M. Identification of a microglia phenotype supportive of remyelination. Glia 2012, 60, 306–321. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural und functional features of central nervous system lymphatic vessels. Nature 2015. [Google Scholar] [CrossRef]
- Yong, V.W.; Giuliani, F.; Xue, M.; Bar-Or, A.; Metz, L.M. Experimental models of neuroprotection relevant to multiple sclerosis. Neurology 2007, 68 (Suppl. 3), 32–37. [Google Scholar] [CrossRef]
- Aharoni, R.; Eilam, R.; Domey, H.; Labunskay, G.; Sela, M.; Arnon, R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains for experimental autoimmune encephalomyelitis mice. Proc. Natl. Acad. Sci. USA 2005, 102, 19045–19050. [Google Scholar] [CrossRef]
- Gago, N.; Akwa, Y.; Sananes, N.; Guennoun, R.; Baulieu, E.E.; Etr, M.; Schumacher, M. Progesterone and the oligodenroglial lineage: Stage-dependent biosynthesis and metabolism. Glia 2001, 36, 295–308. [Google Scholar] [CrossRef]
- Marin-Hussstege, M.; Muggironi, M.; Raban, D.; Skoff, R.P.; Casaccia-Bonnefil, P. Oligodendrocyte progenitor proliferation and maturation in differentially regulated by male and female sex steroid hormones. Dev. Neurosci. 2004, 26, 245–254. [Google Scholar] [CrossRef]
- Kipp, M.; Beyer, C. Impact of sex steroids on neuroinflammatory processes and experimental multiple sclerosis. Front. Neuroendocrinol. 2009, 30, 188–200. [Google Scholar] [CrossRef]
- Tekkok, S.B.; Goldberg, M.P. AMPA/kainate receptor activation mediates hypotoxic oligodendrocyte death and axonal injury in cerebral white matter. J. Neurosci. 2001, 21, 4237–4248. [Google Scholar]
- Dulamea, A. Mesenchymal stem cells in multiple sclerosis-translation to clinical trials. J. Med. Life 2015, 8, 24–27. [Google Scholar]
- Ruckh, J.M.; Zhao, J.W.; Shadrach, J.L.; van Wijngaardeb, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J. Rejuvination of regeneration in the aging central nervous system. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef]
- Huang, J.K.; Fancy, S.P.; Zhao, C.; Rowitch, D.H.; ffrench-Constant, C.; Franklin, R.J. Myelin regeneration in multiple sclerosis: Targeting endogenous stem cells. Neurotherapeutics 2011, 8, 650–658. [Google Scholar] [CrossRef]
- Schnell, L.; Schwab, M.E. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 1990, 343, 269–272. [Google Scholar] [CrossRef]
- Frank, J.A.; Richert, N.; Lewis, B.; Bash, C.; Howard, T.; Civil, R.; Stone, R.; Eaton, J.; McFarland, H.; Leist, T. A pilot study of recombinant insulin-like growth factor-1 in seven multiple sclerosis patients. Mult. Scler. J. 2002, 8, 24–29. [Google Scholar]
- Polman, C.H; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rottlaender, A.; Kuerten, S. Stepchild or Prodigy? Neuroprotection in Multiple Sclerosis (MS) Research. Int. J. Mol. Sci. 2015, 16, 14850-14865. https://doi.org/10.3390/ijms160714850
Rottlaender A, Kuerten S. Stepchild or Prodigy? Neuroprotection in Multiple Sclerosis (MS) Research. International Journal of Molecular Sciences. 2015; 16(7):14850-14865. https://doi.org/10.3390/ijms160714850
Chicago/Turabian StyleRottlaender, Andrea, and Stefanie Kuerten. 2015. "Stepchild or Prodigy? Neuroprotection in Multiple Sclerosis (MS) Research" International Journal of Molecular Sciences 16, no. 7: 14850-14865. https://doi.org/10.3390/ijms160714850