
A Plasmacytoid Dendritic Cells-Type I Interferon Axis Is Critically Implicated in the Pathogenesis of Systemic Lupus Erythematosus

{kind=link}
Abstract
:1. Introduction
2. DCs and Immune Tolerance
2.1. DC Subsets & Characteristics
2.2. DCs as Guards against Autoimmunity
3. The Role of DCs in SLE
3.1. pDCs in SLE
3.2. Role of Type I IFN in SLE
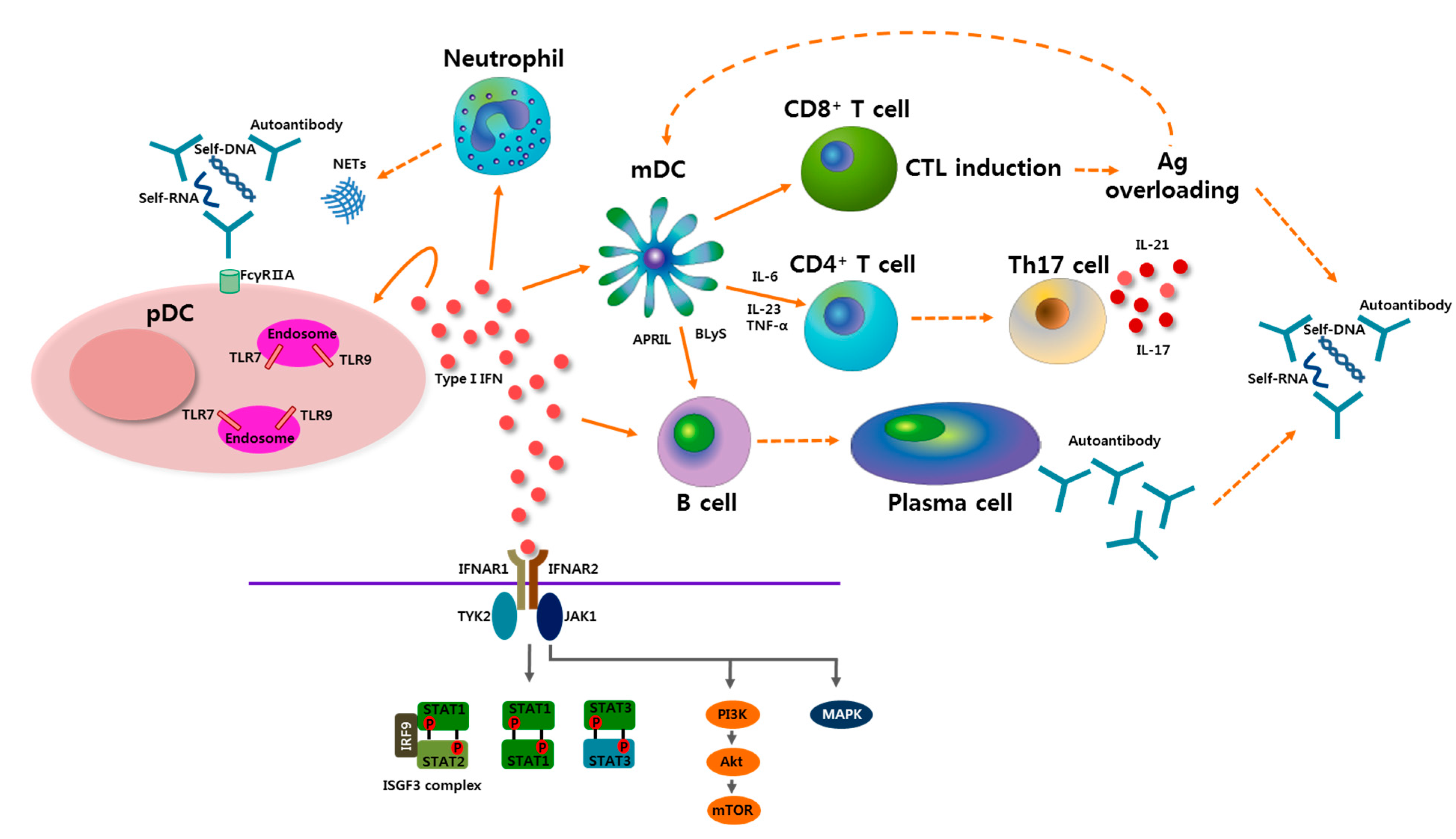
4. pDCs-Type I IFN Axis as a Therapeutic Target in SLE
5. Conclusions
Conflicts of Interest
References
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Munoz, L.E.; Lauber, K.; Schiller, M.; Manfredi, A.A.; Herrmann, M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat. Rev. Rheumatol. 2010, 6, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, A.T.; Wu, X.; Albring, J.C.; Murphy, K.M. Re(de)fining the dendritic cell lineage. Nat. Immunol. 2012, 13, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.P.; Munster, D.J.; Clark, G.J.; Dzionek, A.; Schmitz, J.; Hart, D.N. Characterization of human blood dendritic cell subsets. Blood 2002, 100, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Palucka, A.K.; Blanck, J.P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type I interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T. Pathogen sensors and chemokine receptors in dendritic cell subsets. Vaccine 2012, 30, 7652–7657. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral t cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory t cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Scimone, M.L.; Schaerli, P.; Grabie, N.; Lichtman, A.H.; von Andrian, U.H. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat. Immunol. 2006, 7, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Proietto, A.I.; van Dommelen, S.; Zhou, P.; Rizzitelli, A.; D’Amico, A.; Steptoe, R.J.; Naik, S.H.; Lahoud, M.H.; Liu, Y.; Zheng, P.; et al. Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc. Natl. Acad. Sci. USA 2008, 105, 19869–19874. [Google Scholar] [CrossRef] [PubMed]
- Probst, H.C.; McCoy, K.; Okazaki, T.; Honjo, T.; van den Broek, M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 2005, 6, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Sela, U.; Olds, P.; Park, A.; Schlesinger, S.J.; Steinman, R.M. Dendritic cells induce antigen-specific regulatory T cells that prevent graft vs. host disease and persist in mice. J. Exp. Med. 2011, 208, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Suffner, J.; Hochweller, K.; Kuhnle, M.C.; Li, X.; Kroczek, R.A.; Garbi, N.; Hammerling, G.J. Dendritic cells support homeostatic expansion of Foxp3+ regulatory t cells in Foxp3.Lucidtr mice. J. Immunol. 2010, 184, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal cd103+ dcs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, L.; Birnberg, T.; Kim, K.W.; Jung, S. Dendritic cell-restricted CD80/86 deficiency results in peripheral regulatory T-cell reduction but is not associated with lymphocyte hyperactivation. Eur. J. Immunol. 2011, 41, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, L.L.; Ols, M.L.; Kashgarian, M.; Reizis, B.; Kaplan, D.H.; Shlomchik, M.J. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity 2010, 33, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Stranges, P.B.; Watson, J.; Cooper, C.J.; Choisy-Rossi, C.M.; Stonebraker, A.C.; Beighton, R.A.; Hartig, H.; Sundberg, J.P.; Servick, S.; Kaufmann, G.; et al. Elimination of antigen-presenting cells and autoreactive T cells by fas contributes to prevention of autoimmunity. Immunity 2007, 26, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Carreno, L.J.; Pacheco, R.; Gutierrez, M.A.; Jacobelli, S.; Kalergis, A.M. Disease activity in systemic lupus erythematosus is associated with an altered expression of low-affinity Fc γ receptors and costimulatory molecules on dendritic cells. Immunology 2009, 128, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Miyashita, T.; Maeda, Y.; Kimura, H.; Nakamura, M.; Yatsuhashi, H.; Ishibashi, H.; Eguchi, K. Reduced blood BDCA-2+ (lymphoid) and CD11c+ (myeloid) dendritic cells in systemic lupus erythematosus. Clin. Exp. Immunol. 2005, 142, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Fiore, N.; Castellano, G.; Blasi, A.; Capobianco, C.; Loverre, A.; Montinaro, V.; Netti, S.; Torres, D.; Manno, C.; Grandaliano, G.; et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol. Immunol. 2008, 45, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Quatraro, C.; Lombardi, L.; Pellegrino, C.; Dammacco, F.; Silvestris, F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: Role of interleukin-18. Arthritis Rheum. 2008, 58, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Beiske, K.; Lund-Johansen, F.; Brandtzaeg, P.; Jahnsen, F.L. Plasmacytoid dendritic cells (natural interferon-α/β-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am. J. Pathol. 2001, 159, 237–243. [Google Scholar] [CrossRef]
- Blomberg, S.; Eloranta, M.L.; Cederblad, B.; Nordlin, K.; Alm, G.V.; Ronnblom, L. Presence of cutaneous interferon-α producing cells in patients with systemic lupus erythematosus. Lupus 2001, 10, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.K.; Lee, J.Y.; Park, S.H.; Cho, M.L.; Min, S.Y.; Park, S.H.; Kim, H.Y.; Cho, Y.G. Dysfunctional interferon-α production by peripheral plasmacytoid dendritic cells upon toll-like receptor-9 stimulation in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2008, 10. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.J.; Mok, M.Y.; Chan, G.C.; Chan, A.W.; Jin, O.U.; Kavikondala, S.; Lie, A.K.; Lau, C.S. Phenotypic and functional abnormalities of bone marrow-derived dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerl, V.; Lischka, A.; Panne, D.; Grossmann, P.; Berthold, R.; Hoyer, B.F.; Biesen, R.; Bruns, A.; Alexander, T.; Jacobi, A.; et al. Blood dendritic cells in systemic lupus erythematosus exhibit altered activation state and chemokine receptor function. Ann. Rheum. Dis. 2010, 69, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; le Poul, E.; Migeotte, I.; Brezillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Riboldi, E.; Wittamer, V.; Gentili, F.; Luini, W.; Marrelli, S.; Vecchi, A.; Franssen, J.D.; Communi, D.; Massardi, L.; et al. Role of chemr23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 2005, 201, 509–515. [Google Scholar] [CrossRef]
- Jin, O.; Kavikondala, S.; Mok, M.Y.; Sun, L.; Gu, J.; Fu, R.; Chan, A.; Yeung, J.; Nie, Y.; Lau, C.S. Abnormalities in circulating plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, S.L.; Riggs, J.M.; Gilfillan, S.; Bugatti, M.; Vermi, W.; Kolbeck, R.; Unanue, E.R.; Sanjuan, M.A.; Colonna, M. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med. 2014, 211, 1977–1991. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Lovgren, T.; Eloranta, M.L.; Bave, U.; Alm, G.V.; Ronnblom, L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004, 50, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Deane, J.A.; Pisitkun, P.; Barrett, R.S.; Feigenbaum, L.; Town, T.; Ward, J.M.; Flavell, R.A.; Bolland, S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007, 27, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Karrich, J.J.; Jachimowski, L.C.; Uittenbogaart, C.H.; Blom, B. The plasmacytoid dendritic cell as the swiss army knife of the immune system: Molecular regulation of its multifaceted functions. J. Immunol. 2014, 193, 5772–5778. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and rage. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Colonna, M.; Trinchieri, G.; Barrat, F.; Gilliet, M. Plasmacytoid dendritic cells: One-trick ponies or workhorses of the immune system? Nat. Rev. Immunol. 2011, 11, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Schilling, P.J.; Kurzrock, R.; Kantarjian, H.; Gutterman, J.U.; Talpaz, M. Development of systemic lupus erythematosus after interferon therapy for chronic myelogenous leukemia. Cancer 1991, 68, 1536–1537. [Google Scholar] [CrossRef]
- Ho, V.; McLean, A.; Terry, S. Severe systemic lupus erythematosus induced by antiviral treatment for hepatitis C. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2008, 14, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Dall’era, M.C.; Cardarelli, P.M.; Preston, B.T.; Witte, A.; Davis, J.C., Jr. Type I interferon correlates with serological and clinical manifestations of SLE. Ann. Rheum. Dis. 2005, 64, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Postal, M.; Sinicato, N.A.; Pelicari, K.O.; Marini, R.; Lavras Costallat, L.T.; Appenzeller, S. Clinical and serological manifestations associated with interferon-α levels in childhood-onset systemic lupus erythematosus. Clinics 2012, 67, 157–162. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Dong, C.; Cooper, M.D. Impairment of t and B cell development by treatment with a type I interferon. J. Exp. Med. 1998, 187, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Tyden, H.; Gullstrand, B.; Klint, C.; Wenglen, C.; Nielsen, C.T.; Heegaard, N.H.; Jonsen, A.; Kahn, R.; Bengtsson, A.A. Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PLoS ONE 2015, 10, e0125109. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.; Palucka, A.K.; Gill, M.; Pascual, V.; Banchereau, J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science 2001, 294, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Valenzuela, J.O.; Agarwal, P.; Lins, D.; Mescher, M.F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005, 174, 4465–4469. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Ann. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef]
- Yasuda, K.; Richez, C.; Maciaszek, J.W.; Agrawal, N.; Akira, S.; Marshak-Rothstein, A.; Rifkin, I.R. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF) 5 and IRF7 dependent and is required for ILl–6 production. J. Immunol. 2007, 178, 6876–6885. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Espinosa, A.; Wahren-Herlenius, M. IL-17: A new actor in IFN-driven systemic autoimmune diseases. Eur. J. Immunol. 2012, 42, 2274–2284. [Google Scholar] [CrossRef] [PubMed]
- Doreau, A.; Belot, A.; Bastid, J.; Riche, B.; Trescol-Biemont, M.C.; Ranchin, B.; Fabien, N.; Cochat, P.; Pouteil-Noble, C.; Trolliet, P.; et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat. Immunol. 2009, 10, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. An overview of IL-17 function and signaling. Cytokine 2008, 43, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Spolski, R.; Ettinger, R.; Kim, H.P.; Wang, G.; Qi, C.F.; Hwu, P.; Shaffer, D.J.; Akilesh, S.; Roopenian, D.C.; et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J. Immunol. 2004, 173, 5361–5371. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, A.; McGuire, H.M.; Yu, D.; Sprent, J.; Mackay, C.R.; King, C. A fundamental role for interleukin-21 in the generation of t follicular helper cells. Immunity 2008, 29, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Nzeusseu Toukap, A.; Galant, C.; Theate, I.; Maudoux, A.L.; Lories, R.J.; Houssiau, F.A.; Lauwerys, B.R. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.S.; Huang, J.F.; Zhu, J.; D’Agati, V.; Liu, X.; Miller, N.; Erlander, M.G.; Jackson, M.R.; Winchester, R.J. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J. Clin. Investig. 2004, 113, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bethunaickan, R.; Huang, W.; Lodhi, U.; Solano, I.; Madaio, M.P.; Davidson, A. Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011, 63, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, A.; Angelotti, M.L.; Mulay, S.R.; Kulkarni, O.O.; Demleitner, J.; Dietrich, A.; Sagrinati, C.; Ballerini, L.; Peired, A.; Shankland, S.J.; et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: Implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am. J. Pathol. 2013, 183, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Thacker, S.; Mehta, H.; Somers, E.C.; Dodick, T.; Barrat, F.J.; McCune, W.J.; Kaplan, M.J. Interferon-α promotes abnormal vasculogenesis in lupus: A potential pathway for premature atherosclerosis. Blood 2007, 110, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Thacker, S.G.; Zhao, W.; Smith, C.K.; Luo, W.; Wang, H.; Vivekanandan-Giri, A.; Rabquer, B.J.; Koch, A.E.; Pennathur, S.; Davidson, A.; et al. Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum. 2012, 64, 2975–2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santer, D.M.; Yoshio, T.; Minota, S.; Moller, T.; Elkon, K.B. Potent induction of IFN-α and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J. Immunol. 2009, 182, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Wallace, D.J.; Petri, M.; Kirou, K.A.; Yao, Y.; White, W.I.; Robbie, G.; Levin, R.; Berney, S.M.; Chindalore, V.; et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: A phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis. 2011, 70, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Wallace, D.J.; Spindler, A.; Chindalore, V.; Kalunian, K.; Mysler, E.; Neuwelt, C.M.; Robbie, G.; White, W.I.; Higgs, B.W.; et al. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus: A phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013, 65, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Lauwerys, B.R.; Hachulla, E.; Spertini, F.; Lazaro, E.; Jorgensen, C.; Mariette, X.; Haelterman, E.; Grouard-Vogel, G.; Fanget, B.; Dhellin, O.; et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthritis Rheum. 2013, 65, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zhang, F.; Yin, H. Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol. Ther. 2013, 138, 441–451. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-M.; Park, S.-H.; Kim, H.-Y.; Kwok, S.-K. A Plasmacytoid Dendritic Cells-Type I Interferon Axis Is Critically Implicated in the Pathogenesis of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2015, 16, 14158-14170. https://doi.org/10.3390/ijms160614158
Kim J-M, Park S-H, Kim H-Y, Kwok S-K. A Plasmacytoid Dendritic Cells-Type I Interferon Axis Is Critically Implicated in the Pathogenesis of Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2015; 16(6):14158-14170. https://doi.org/10.3390/ijms160614158
Chicago/Turabian StyleKim, Ji-Min, Sung-Hwan Park, Ho-Youn Kim, and Seung-Ki Kwok. 2015. "A Plasmacytoid Dendritic Cells-Type I Interferon Axis Is Critically Implicated in the Pathogenesis of Systemic Lupus Erythematosus" International Journal of Molecular Sciences 16, no. 6: 14158-14170. https://doi.org/10.3390/ijms160614158