
Transcriptional Profiling Reveals Differential Gene Expression of Amur Ide (Leuciscus waleckii) during Spawning Migration

Abstract
:1. Introduction
2. Results
2.1. Sequencing of Short Expressed Reads

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Reads | Clean Reads | Mapped Reads | Mapping Ratio (%) |
|---|---|---|---|---|
| FW | 351,560,104 | 232,819,696 | 194,404,446 | 83.5 |
| SM | 285,674,776 | 234,097,720 | 199,451,257 | 85.2 |
| Total | 637,234,880 | 466,917,416 | 393,855,703 | 84.4 |
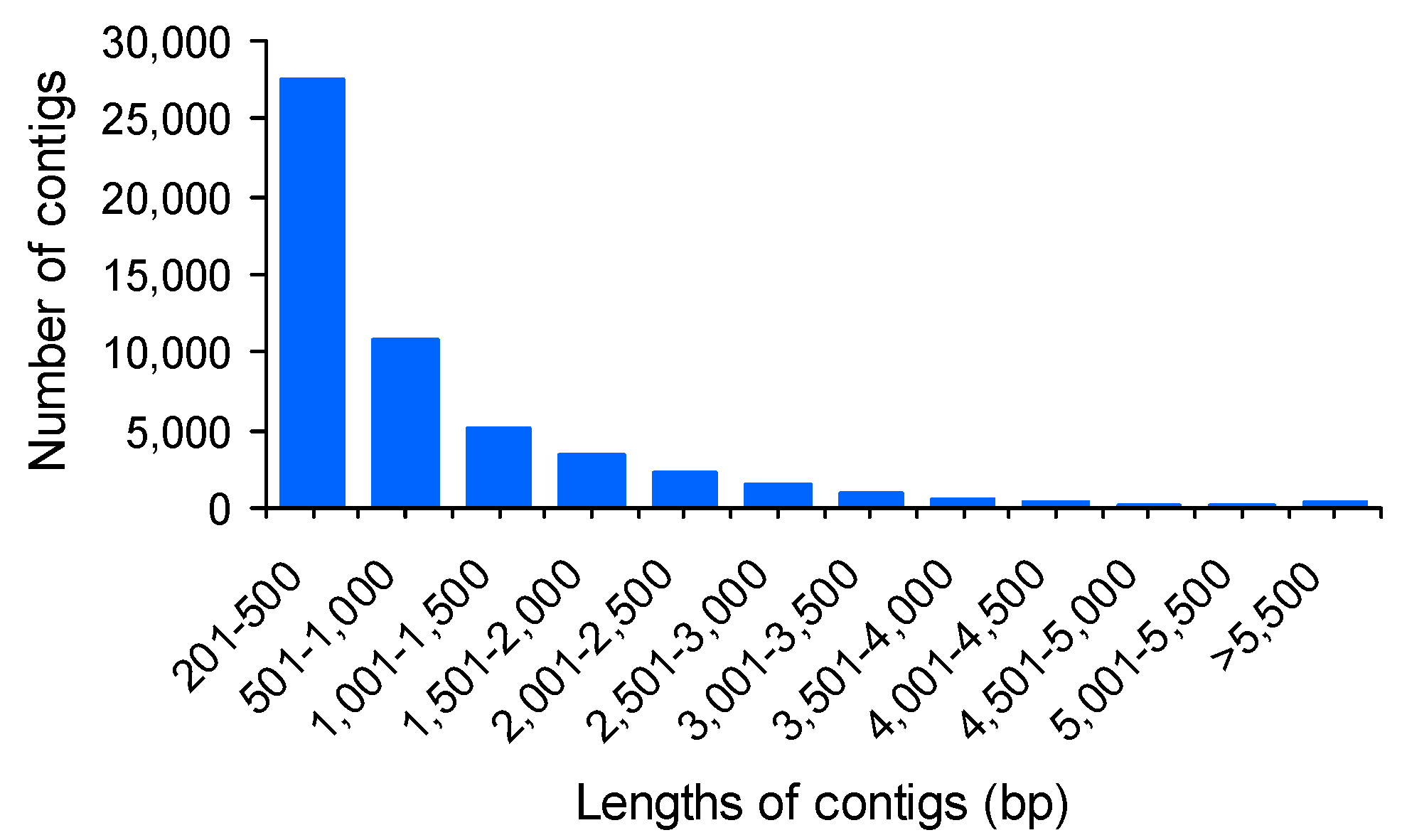
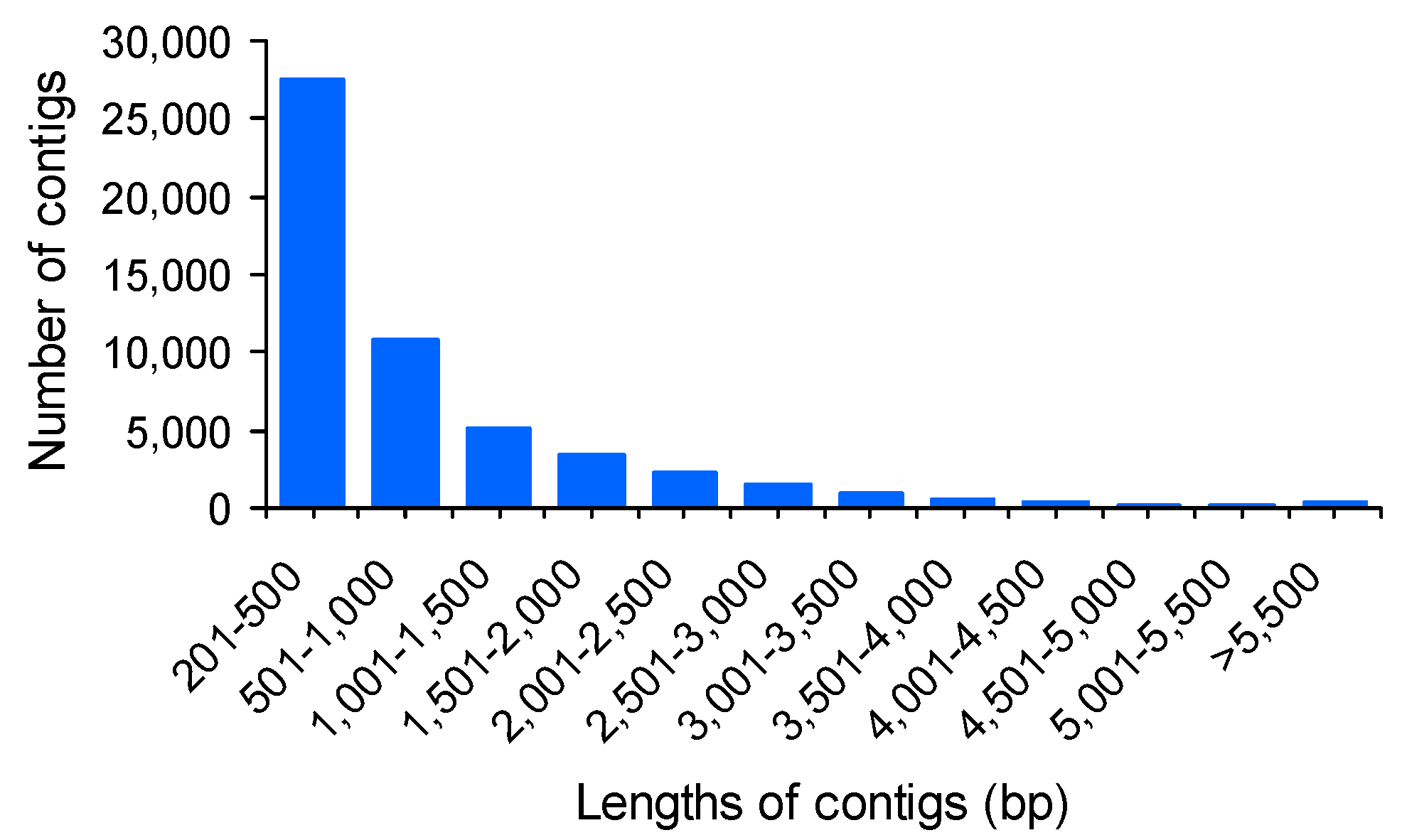
2.2. Reference Assembly and Annotation
| Steps | Categories | Number/Length |
|---|---|---|
| Assembly | Number of contigs | 53,440 |
| Maximum contig length | 13,690 bp | |
| Minimum contig length | 201 bp | |
| Average contig length | 917 bp | |
| Median contig length | 476 bp | |
| N50 length | 1686 bp | |
| Annotation | Contigs with blast hits to NR * | 38,382 |
| Contigs with blast hits to UniProt | 37,809 | |
| Contigs with blast hits to Zebrafish proteins | 38,559 | |
| Unigenes predicted | 19,972 | |
| Unigenes with GO terms | 10,988 |


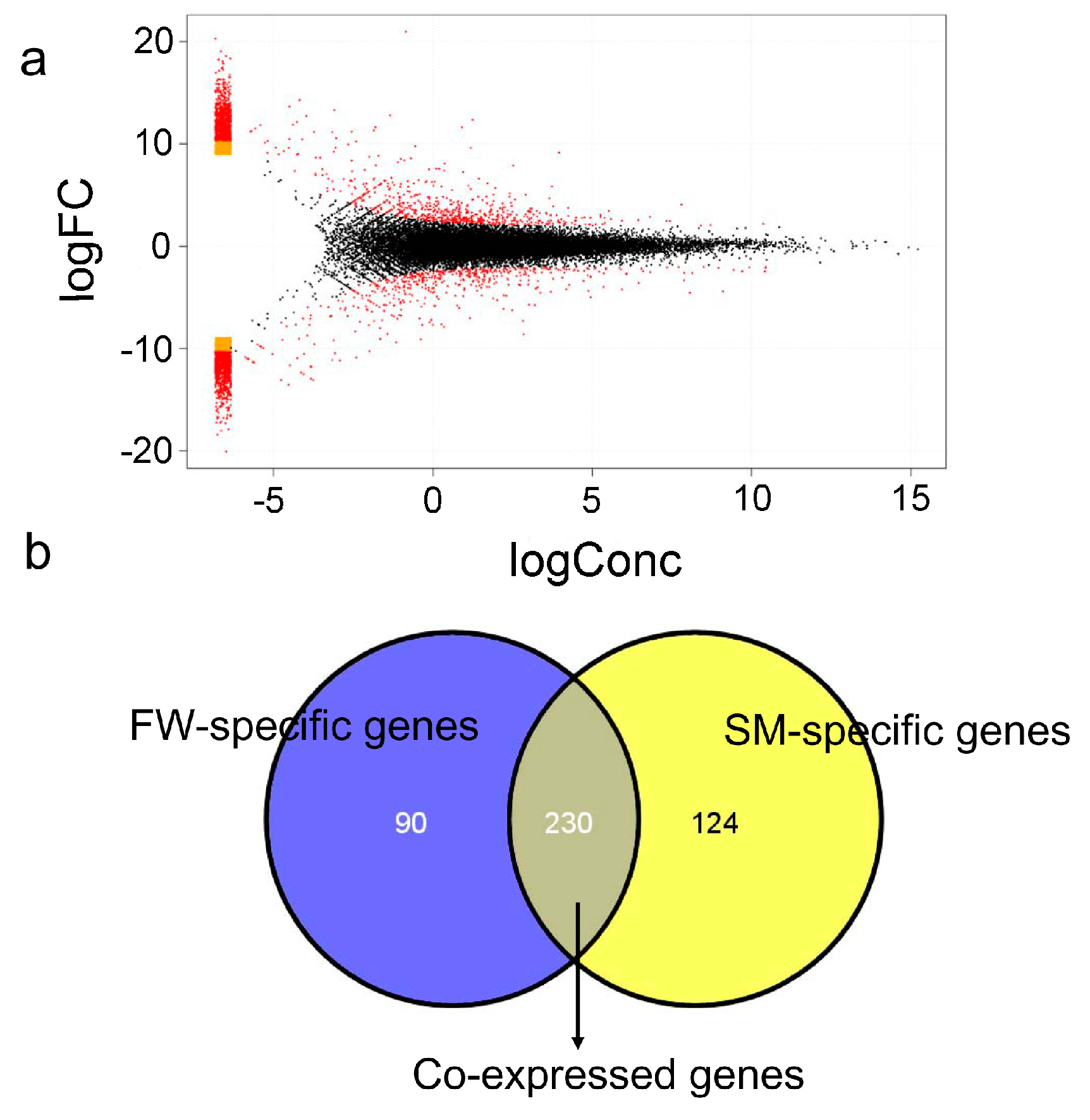
2.3. Differential Gene Expression (DEG) Identification
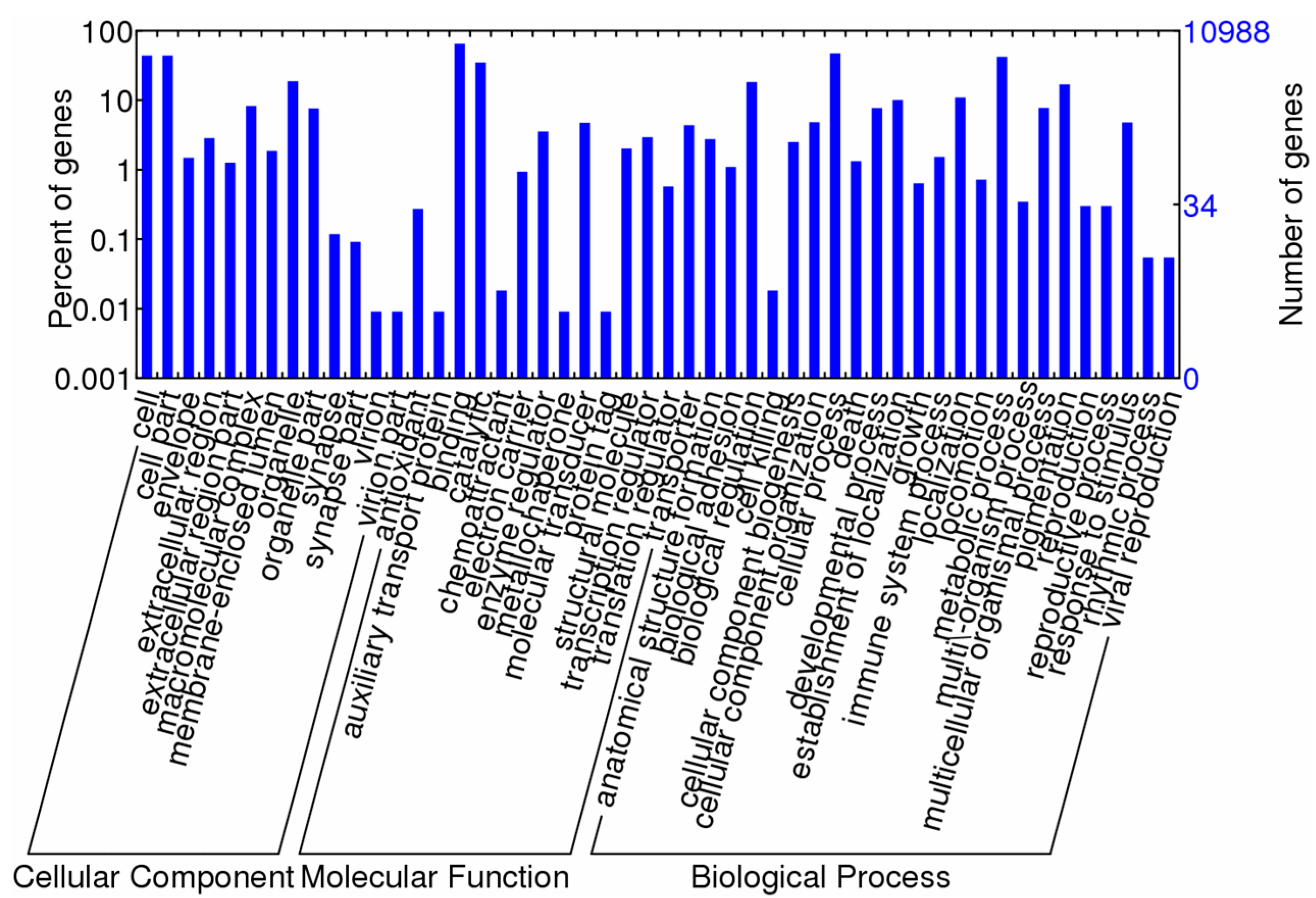
2.4. Enrichment and Pathway Analysis
| GO ID | GO Term | p-Value | Counts Reference/Differential Genes |
|---|---|---|---|
| GO: 0005576 | Extracellular region | 0.001 | 312/20 |
| GO: 0005623 | Cell | <0.001 | 4817/115 |
| GO: 0032991 | Macromolecular complex | 0.007 | 907/14 |
| GO: 0043226 | Organelle | 0.019 | 2062/46 |
| GO: 0044422 | Organelle part | 0.031 | 836/15 |
| GO: 0044464 | Cell part | <0.001 | 4817/115 |
| GO: 0044085 | Cellular component biogenesis | 0.026 | 274/2 |
| GO: 0005488 | Binding | 0.001 | 7161/249 |
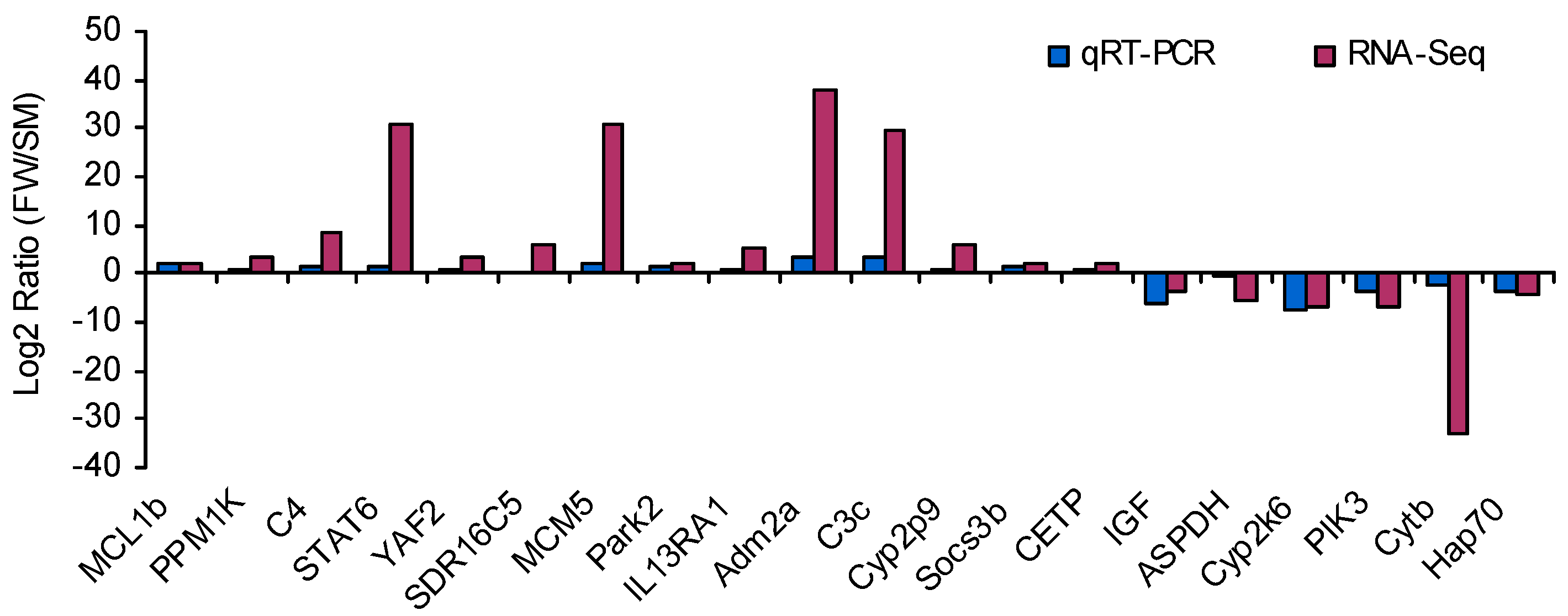
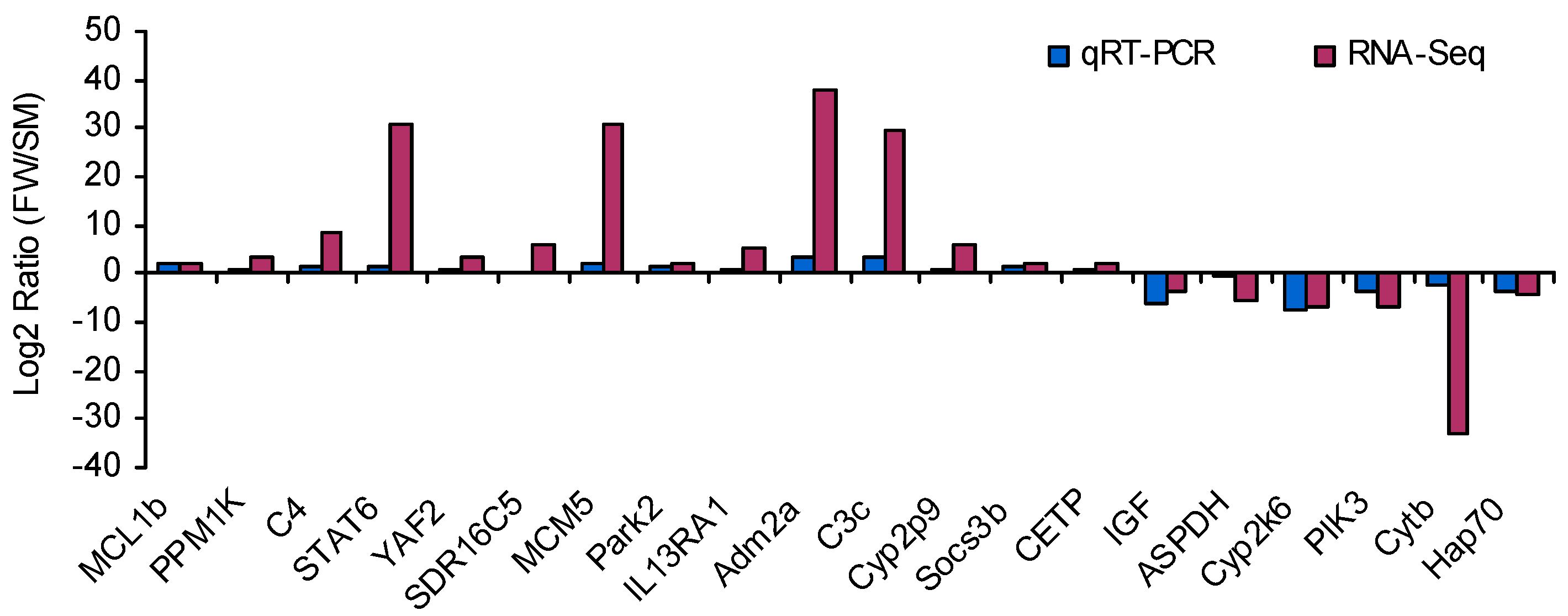
| Pathway Involved | Gene Name | Unigene ID | Fold Change |
|---|---|---|---|
| mTOR signaling pathway | PIK3 | comp102544_c0 | −6.51 |
| RPS6Kba1 | comp102058_c22 | −30.97 | |
| AKT2 | comp102966_c5 | −29.68 | |
| IGF | comp60482_c0 | −3.79 | |
| DDIT4 | comp101409_c1 | −31.58 | |
| JAK-STAT signaling pathway | IL13RA1 | comp102860_c0 | 5.05 |
| PIK3 | comp102544_c0 | −6.51 | |
| IL7R | comp101289_c0 | −2.52 | |
| socs3b | comp102087_c6 | 2.25 | |
| AKT2 | comp102966_c5 | −29.68 | |
| STAT6 | comp102564_c2 | 30.61 | |
| Oxidative phosphorylation pathway | COX1 | comp101302_c0 | −32.02 |
| Cytb | comp98419_c0 | −32.82 | |
| COX3 | comp99121_c0 | −33.75 | |
| COX2 | comp125637_c0 | −33.06 | |
| ATP6V1F | comp89974_c0 | −33.00 | |
| ATP6 | comp126569_c0 | −32.62 |
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Sample Collection
4.3. RNA Extraction, Library Construction, and Sequencing
4.4. Sequence Data Processing and De Novo Assembly
4.5. Transcriptome Annotation and Ontology
4.6. DEG Analysis
4.7. Experimental Validation by qRT-PCR
4.8. GO Enrichment Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xu, J.; Li, Q.; Xu, L.; Wang, S.; Jiang, Y.; Zhao, Z.; Zhang, Y.; Li, J.; Dong, C.; Xu, P.; et al. Gene expression changes leading extreme alkaline tolerance in Amur ide (Leuciscus waleckii) inhabiting soda lake. BMC Genomics 2013, 14, 682. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, P.; Wang, B.; Zhao, L.; Wang, J.; Zhao, Z.; Zhang, Y.; Li, J.; Xu, P.; Sun, X. Transcriptome sequencing and analysis of wild Amur Ide (Leuciscus waleckii) inhabiting an extreme alkaline-saline lake reveals insights into stress adaptation. PLoS ONE 2013, 8, e59703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, L.; Jiang, P.; Li, D.; Lu, C.; Sun, X. Genome evolution trend of common carp (Cyprinus carpio L.) as revealed by the analysis of microsatellite loci in a gynogentic family. J. Genet. Genomics 2008, 35, 97–103. [Google Scholar] [CrossRef]
- Chi, B.; Chang, Y.; Yan, X.; Cao, D.; Li, Y.; Gao, Y.; Liu, Y.; Liang, L. Genetic variability and genetic structure of Leuciscus waleckii Dybowski in Wusuli River and Dali Lake. J. Fish. Sci. China 2010, 17, 228–235. [Google Scholar]
- Liu, J.; Chang, Y.; Xu, L.; Liu, C.; Liang, L.; Han, G.; Gao, Y.; Liu, Y. Isolation and characterization of microsatellite from genome of Leuciscus waleckii Dybowski. Acta Agric. Boreali-Sin. 2011, 26, 87–93. [Google Scholar]
- Chang, Y.; Tang, R.; Sun, X.; Liang, L.; Chen, J.; Huang, J.; Dou, X.; Tao, R. Genetic analysis of population differentiation and adaptation in Leuciscus waleckii. Genetica 2013, 141, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ji, P.; Xu, J.; Sun, J.; Yang, J.; Xu, P.; Sun, X. Complete mitochondrial genome of Leuciscus waleckii (Cypriniformes: Cyprinidae: Leuciscus). Mitochondrial DNA 2013, 24, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.M.; Tang, R.; Dou, X.J.; Tao, R.; Sun, X.W.; Liang, L.Q. Transcriptome and expression profiling analysis of Leuciscus waleckii: An exploration of the alkali-adapted mechanisms of a freshwater teleost. Mol. Biosyst. 2014, 10, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, Z.; Lu, J.; Sun, F.; Wang, S.; Liu, H.; Jiang, Y.; Kucuktas, H.; Kaltenboeck, L.; Peatman, E.; et al. Generation of genome-scale gene-associated SNPs in catfish for the construction of a high-density SNP array. BMC Genomics 2011, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Zhou, Z.; Waldbieser, G.; Sun, F.; Lu, J.; Zhang, J.; Jiang, Y.; Zhang, H.; Wang, X.; et al. Efficient assembly and annotation of the transcriptome of catfish by RNA-Seq analysis of a doubled haploid homozygote. BMC Genomics 2012, 13, 595. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, X.; Sun, F.; Zhang, J.; Feng, J.; Liu, H.; Rajendran, K.V.; Sun, L.; Zhang, Y.; Jiang, Y.; et al. RNA-Seq reveals expression signatures of genes involved in oxygen transport, protein synthesis, folding, and degradation in response to heat stress in catfish. Physiol. Genomics 2013, 45, 462–476. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, P.; Zhao, Z.; Zhang, Y.; Feng, J.; Wang, J.; Li, J.; Zhang, X.; Zhao, L.; Liu, G.; et al. Genome-wide SNP discovery from transcriptome of four common carp strains. PLoS ONE 2012, 7, e48140. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; McIntyre, L.M.; Scardina, J.; Wheeler, P.A.; Thorgaard, G.H.; Nichols, K.M. Transcriptome profiling of embryonic development rate in rainbow trout advanced backcross introgression lines. Mar. Biotechnol. 2011, 13, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Peatman, E.; Li, C.; Liu, S.; Jiang, Y.; Zhou, Z.; Liu, Z. Transcriptomic signatures of attachment, NF-κB suppression and IFN stimulation in the catfish gill following columnaris bacterial infection. Dev. Comp. Immunol. 2012, 38, 69–180. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Wang, R.; Lu, J.; Nandi, S.; Mohanty, S.; Terhune, J.; Liu, Z.; Peatman, E. RNA-Seq analysis of mucosal immune responses reveals signatures of intestinal barrier disruption and pathogen entry following Edwardsiella ictaluri infection in channel catfish, Ictalurus punctatus. Fish Shellfish Immunol. 2012, 32, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, H.; Liu, S.; Zhu, L.; Qiu, X.; Jiang, Z.; Wang, X.; Liu, Z. SNP discovery from transcriptome of the swimbladder of Takifugu rubripes. PLoS ONE 2014, 9, e92502. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, S.; Zhang, B.; Wang, H.; Sun, H.; Song, S.; Qiu, X.; Liu, Y.; Wang, X.; Jiang, Z.; et al. Transciptome analysis of the gill and swimbladder of Takifugu rubripes by RNA-Seq. PLoS ONE 2014, 9, e85505. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, H.; Liu, S.; Qiu, X.; Jiang, Z.; Wang, X. Transcriptome analysis of the gill of Takifugu rubripes using Illumina sequencing for discovery of SNPs. Comp. Biochem. Physiol. Part D Genomics Proteomics 2014, 10, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Liu, S.; Gao, X.; Jiang, Y.; Perera, D.; Wang, X.; Li, C.; Sun, L.; Zhang, J.; Kaltenboeck, L.; et al. Male-biased genes in catfish as revealed by RNA-Seq analysis of the testis transcriptome. PLoS ONE 2013, 8, e68452. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Li, J.T.; Kuang, Y.Y.; Xu, R.; Zhao, Z.X.; Hou, G.Y.; Liang, H.W.; Sun, X.W. The transcriptomes of the Crucian carp complex (Carassius auratus) provide insights into the distinction between unisexual triploids and sexual diploids. Int. J. Mol. Sci. 2014, 15, 9386–9406. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, S.; Xu, J.; Feng, J.; Mahboob, S.; Al-Ghanim, K.A.; Sun, X.; Xu, P. Comparative transcriptome analysis reveals the genetic basis of skin color variation in common carp. PLoS ONE 2014, 9, e108200. [Google Scholar] [CrossRef] [PubMed]
- Lavado, R.; Aparicio-Fabre, R.; Schlenk, D. Effects of salinity acclimation on the pesticide-metabolizing enzyme flavin-containing monooxygenase (FMO) in rainbow trout (Oncorhynchus mykiss). Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2013, 157, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Arstikaitis, J.; Gagné, F.; Cyr, D.G. Exposure of fathead minnows to municipal wastewater effluent affects intracellular signaling pathways in the liver. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2014, 164C, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Seli, E.; Babayev, E.; Collins, S.C.; Nemeth, G.; Horvath, T.L. Minireview: Metabolism of female reproduction: Regulatory mechanisms and clinical implications. Mol. Endocrinol. 2014, 28, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, A.; Larsson, L.; Kiessling, K.H.; Lutes, P.B.; Storebakken, T.; Hung, S.S. Spawning induces a shift in energy metabolism from glucose to lipid in rainbow trout white muscle. Fish Physiol. Biochem. 1995, 14, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, A.M.; Nieminen, P.; Hyvärinen, H. Leptin, ghrelin, and energy metabolism of the spawning burbot (Lota lota, L.). J. Exp. Zool. 2002, 293, 119–126. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.D. Endocrine control of osmoregulation in teleost fish. Am. Zool. 2001, 41, 781–794. [Google Scholar] [CrossRef]
- Sakamoto, T.; Hirano, T.; Madsen, S.S.; Nishioka, R.S.; Bern, H.A. Insulin-like growth factor I gene expression during parr-smolt transformation of coho salmon. Zool. Sci. 1995, 12, 249–252. [Google Scholar] [CrossRef]
- Seidelin, M.; Madsen, S.S.; Byrialsen, A.; Kristiansen, K. Effects of insulin-like growth factor-I and cortisol on Na+, K+-ATPase expression in osmoregulatory tissues of brown trout (Salmo trutta). Gen. Comp. Endocrinol. 1999, 113, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.H. Cell signaling and ion transport across the fish gill epithelium. J. Exp. Zool. 2002, 293, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D. Research progress of insulin-like growth factors of fish. J. Shanghai Fish. Univ. 2005, 14, 66–71. [Google Scholar]
- Li, J.; Chu, L.; Sun, X.; Liu, Y.; Cheng, C.H. IGFs mediate the action of LH on oocyte maturation in zebrafish. Mol. Endocrinol. 2015, 29, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Buchmaier, B.S.; Bibi, A.; Müller, G.A.; Dihazi, G.H.; Eltoweissy, M.; Kruegel, J.; Dihazi, H. Renal cells express different forms of vimentin: The independent expression alteration of these forms is important in cell resistance to osmotic stress and apoptosis. PLoS ONE 2013, 8, e68301. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, M.; Kaneko, T. Recruitment and degeneration of mitochondrion-rich cells in the gills of Mozambique tilapia Oreochromis mossambicus during adaptation to a hyperosmotic environment. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2012, 162, 245–251. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Li, L.; Xu, Q.; Ren, J.; Zhang, F. Postconditioning reduces infarct size and cardiac myocyte apoptosis via the opioid receptor and JAK-STAT signaling pathway. Mol. Biol. Rep. 2011, 38, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Kinowaki, K. PI3K–AKT network roles in infectious diseases. Kansenshogaku Zasshi 2008, 82, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, G. Computer-aided targeting of the PI3K/Akt/mTOR pathway: Toxicity reduction and therapeutic opportunities. Int. J. Mol. Sci. 2014, 15, 18856–18891. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Xu, J.; Zhang, S.; Wang, K.; Jiang, Y.; Mahboob, S.; Al-Ghanim, K.A.; Xu, P. Transcriptional Profiling Reveals Differential Gene Expression of Amur Ide (Leuciscus waleckii) during Spawning Migration. Int. J. Mol. Sci. 2015, 16, 13959-13972. https://doi.org/10.3390/ijms160613959
Cui J, Xu J, Zhang S, Wang K, Jiang Y, Mahboob S, Al-Ghanim KA, Xu P. Transcriptional Profiling Reveals Differential Gene Expression of Amur Ide (Leuciscus waleckii) during Spawning Migration. International Journal of Molecular Sciences. 2015; 16(6):13959-13972. https://doi.org/10.3390/ijms160613959
Chicago/Turabian StyleCui, Jun, Jian Xu, Songhao Zhang, Kai Wang, Yanliang Jiang, Shahid Mahboob, Khalid A. Al-Ghanim, and Peng Xu. 2015. "Transcriptional Profiling Reveals Differential Gene Expression of Amur Ide (Leuciscus waleckii) during Spawning Migration" International Journal of Molecular Sciences 16, no. 6: 13959-13972. https://doi.org/10.3390/ijms160613959