
Genome-Wide Analysis of Sorbitol Dehydrogenase (SDH) Genes and Their Differential Expression in Two Sand Pear (Pyrus pyrifolia) Fruits

Abstract
:1. Introduction
2. Results
2.1. RNA-Seq of Fruit Tissue of Sand Pear and Identification of PpySDHs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Count/Percentage |
|---|---|
| Raw reads | 48,085,640 |
| Clean reads | 46,379,236 |
| Clean bases (bp) | 4,505,546,201 |
| Frequency of any nucleotide (N) (%) | 0.30 |
| ≥Q20 (%) | 98.64 |
| Mapped reads | 37,763,167 |
| Mapped ratio (%) | 81.4 |
| Number of mapped genes | 23,955 |
2.2. Sequence Collection and Identification of Sorbitol Dehydrogenase (SDH) Genes
| Family Name | Common Name | Species Name | Chromosome Number | Release Version | Genome Gene Number | Identified SDH Genes | Gene Name Prefix |
|---|---|---|---|---|---|---|---|
| Rosaceae | Pear | Pyrus bretschneideri | 34 | NJAU, v1.0 | 42,341 | 15 | Pbr |
| Pyrus communis | 34 | GDR, v1.0 | 43,419 | 22 | TCONS | ||
| Apple | Malus domestica | 34 | GDR, v1.0 | 54,921 | 16 | MDP | |
| Peach | Prunus persica | 16 | GDR, v1.0 | 27,864 | 4 | EMJ | |
| Plum | Prunus mume | 16 | NCBI, v1.0 | 31,390 | 3 | XM | |
| Strawberry | Fragaria vesca | 14 | GDR, v1.1 | 32,831 | 1 | mrna | |
| Brassicaceae | Arabidopsis | Arabidopsis thaliana | 10 | TAIR, V10.0 | 27,416 | 2 | AT |
| Gramineae | Maize | Zea mays | 20 | NCBI, B73_RefGen_v3 | 39,475 | 2 | GRMZM |
| Rutaceae | Sweet orange | Citrus sinensis | 18 | GDR, v1.0 | 29,445 | 1 | Csi |
| Solanaceae | Tomato | Solanum lycopersicum | 24 | ITAG, V2.3 | 34,727 | 1 | Solyc |
| Vitaceae | Grape | Vitis vinifera | 38 | CNS, v1.0 | 26,346 | 2 | VIT |
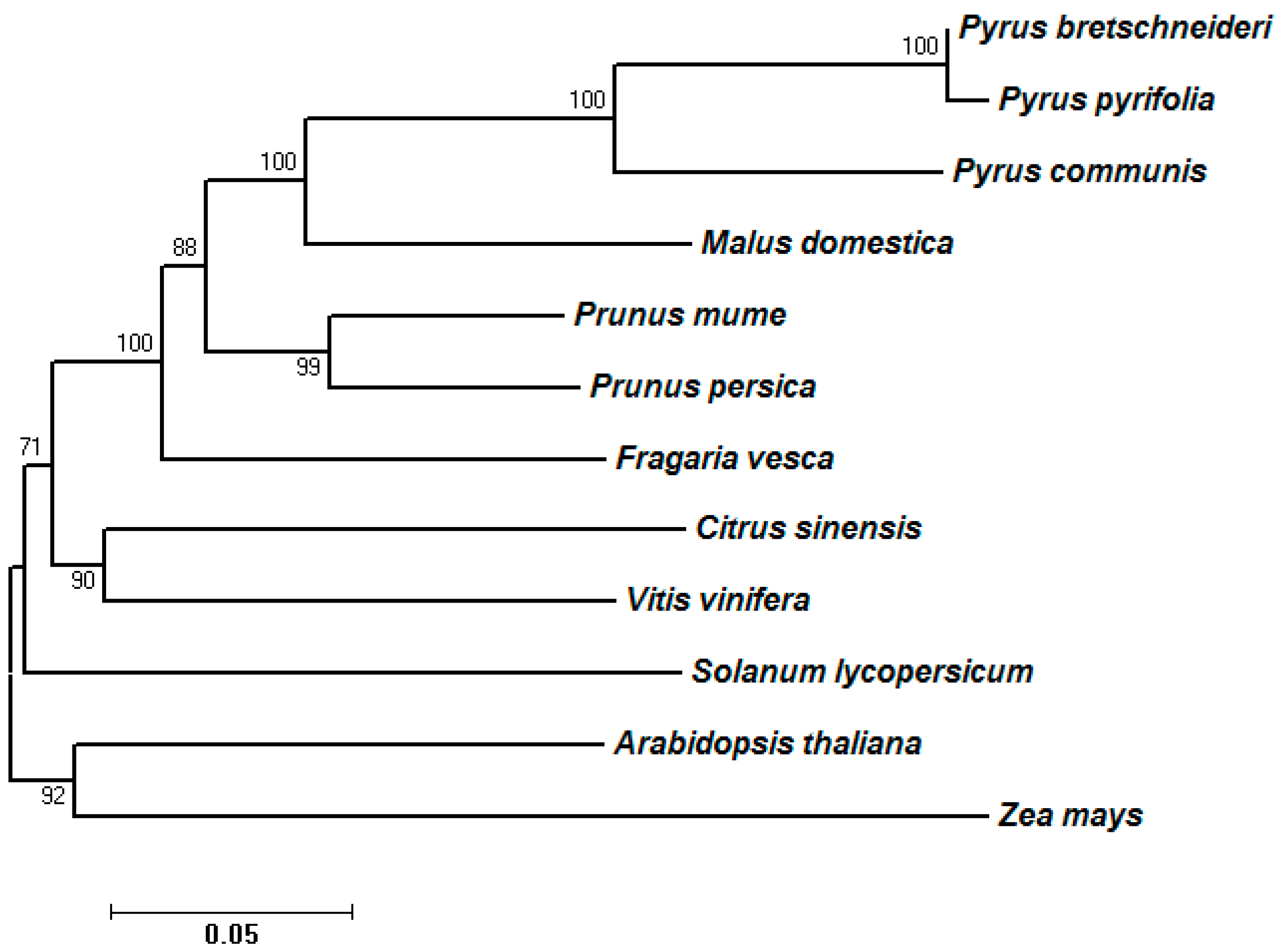
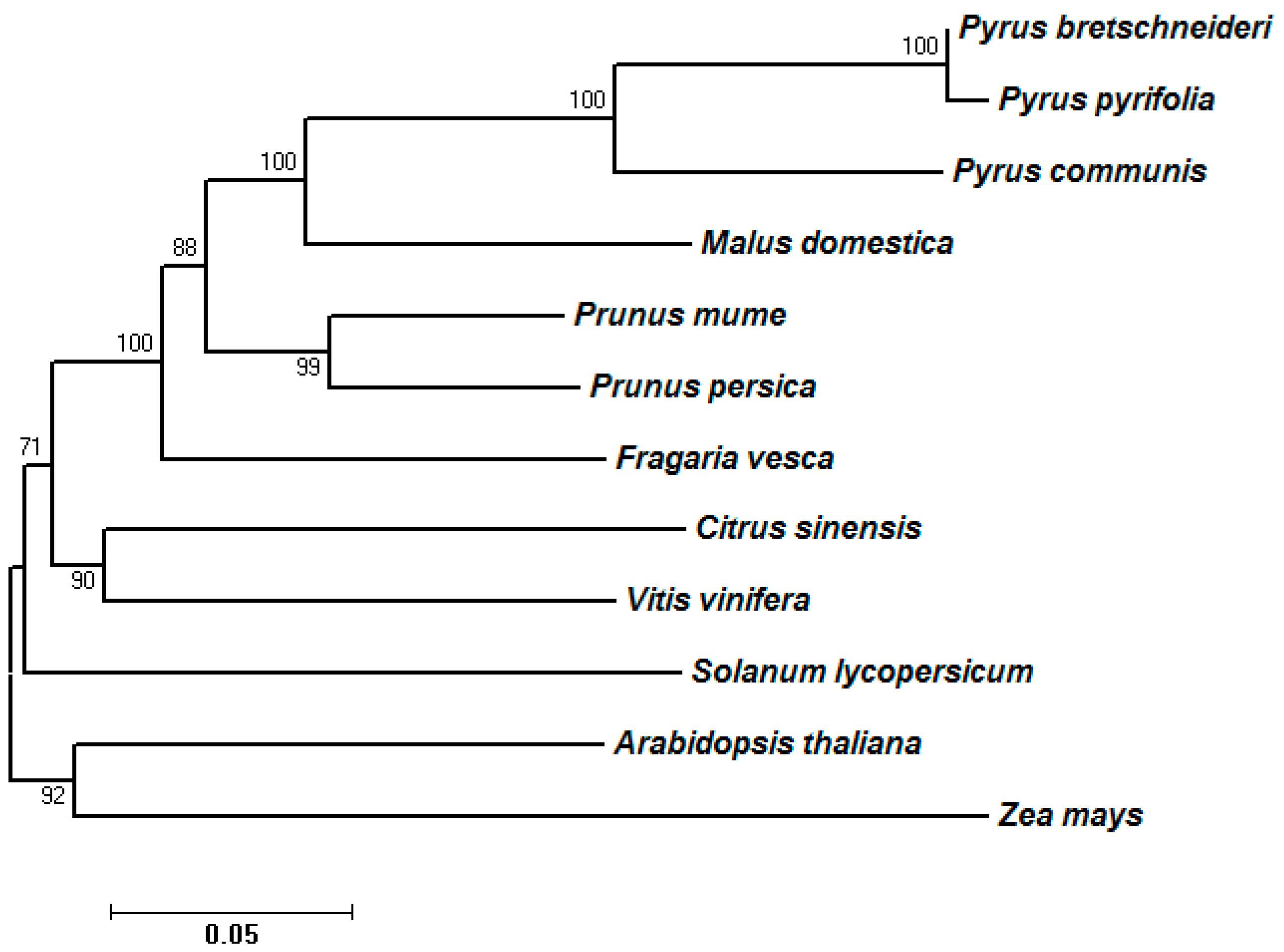
2.3. Phylogenetic Analysis of Predicted SDH Genes
2.4. Transcript Abundance of PpySDHs

| Gene Name | Corresponding Gene in Reference Genome | TPM | RPKM | Percentage in Total RPKM (%) |
|---|---|---|---|---|
| PpySDH1 | Pbr013912.1 | 8147.01 | 7244.58 | 0.93 |
| PpySDH2 | Pbr013913.1 | 270,655.77 | 240,675.52 | 30.99 |
| PpySDH3 | Pbr013914.1 | 7402.56 | 6582.59 | 0.85 |
| PpySDH4 | Pbr013915.1 | 125,806.21 | 111,870.79 | 14.40 |
| PpySDH5 | Pbr013916.1 | 3874.83 | 3445.62 | 0.44 |
| PpySDH6 | Pbr013917.1 | 360.01 | 320.13 | 0.04 |
| PpySDH7 | Pbr032770.1 | 0 | 0 | 0.00 |
| PpySDH8 | Pbr032771.1 | 41,652.83 | 37,039 | 4.77 |
| PpySDH9 | Pbr032772.1 | 642.55 | 571.37 | 0.07 |
| PpySDH10 | Pbr032773.1 | 4841.99 | 4305.65 | 0.55 |
| PpySDH11 | Pbr032774.1 | 4253.54 | 3782.38 | 0.49 |
| PpySDH12 | Pbr032775.1 | 174,199.23 | 154,903.37 | 19.95 |
| PpySDH13 | Pbr032776.1 | 21,448.95 | 19,073.08 | 2.46 |
| PpySDH14 | Pbr032777.1 | 207,687.56 | 184,682.23 | 23.78 |
| PpySDH15 | Pbr032778.1 | 2408.97 | 2142.13 | 0.28 |
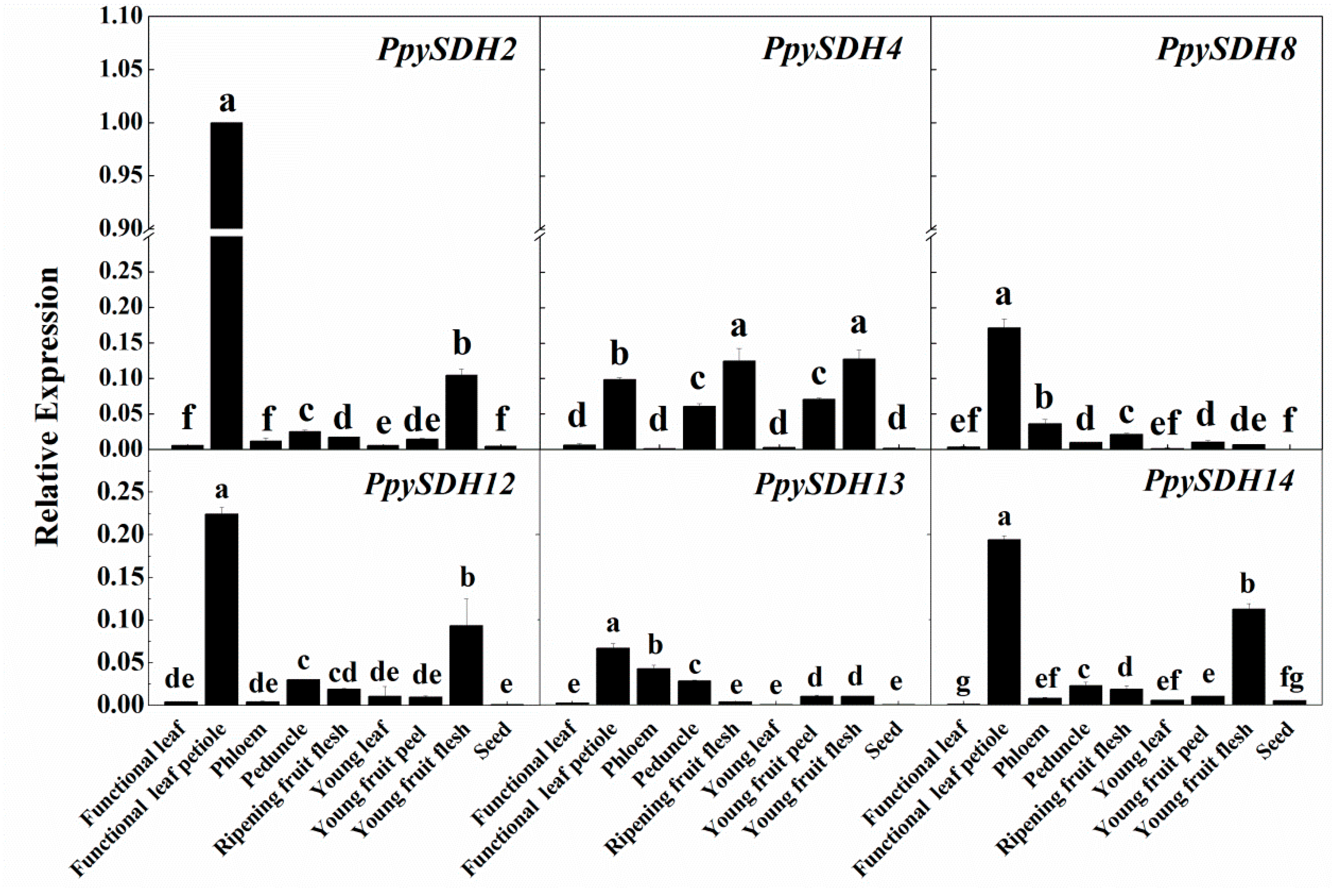
2.5. Tissue-Specific Expression of Six Selected PpySDHs

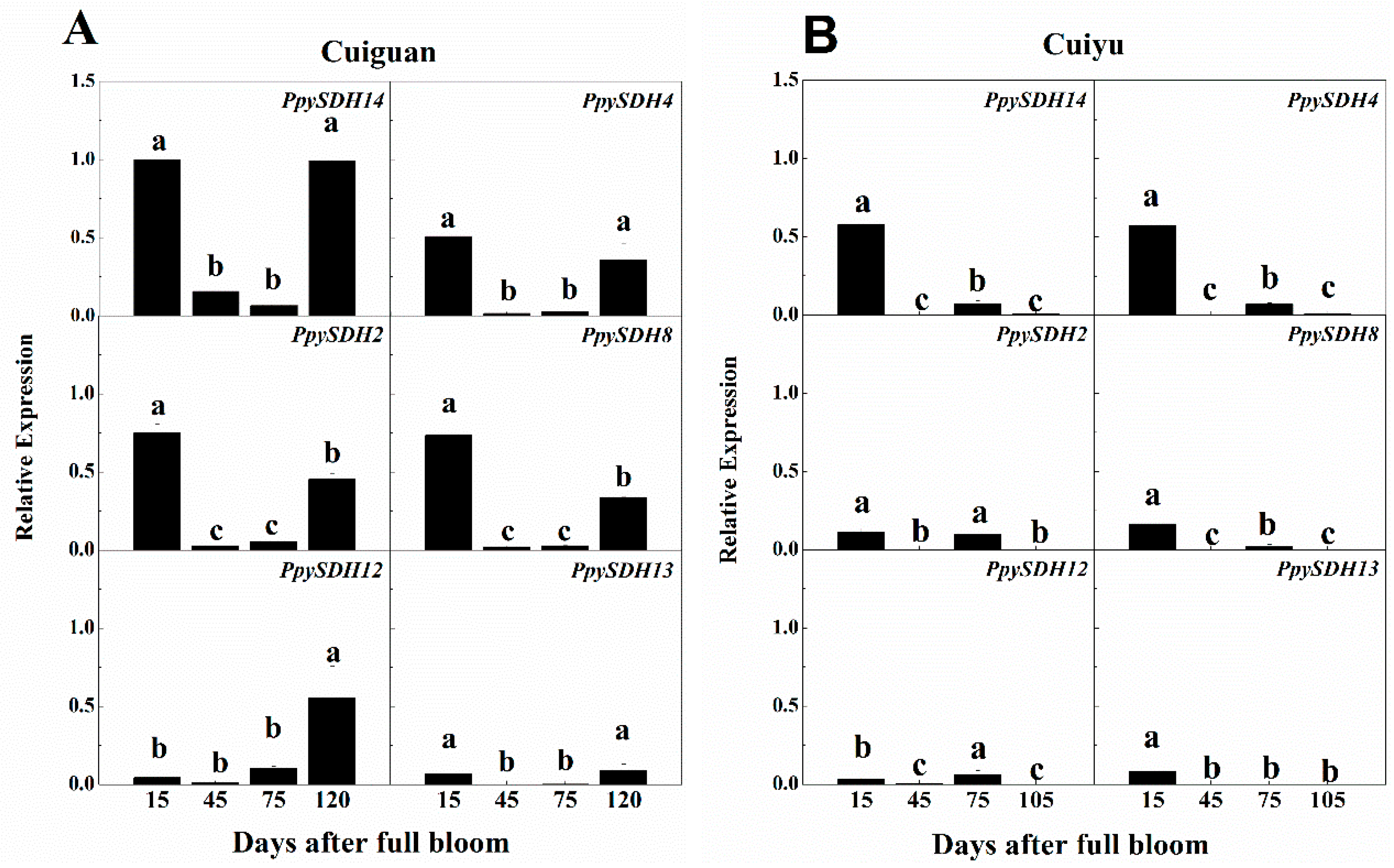
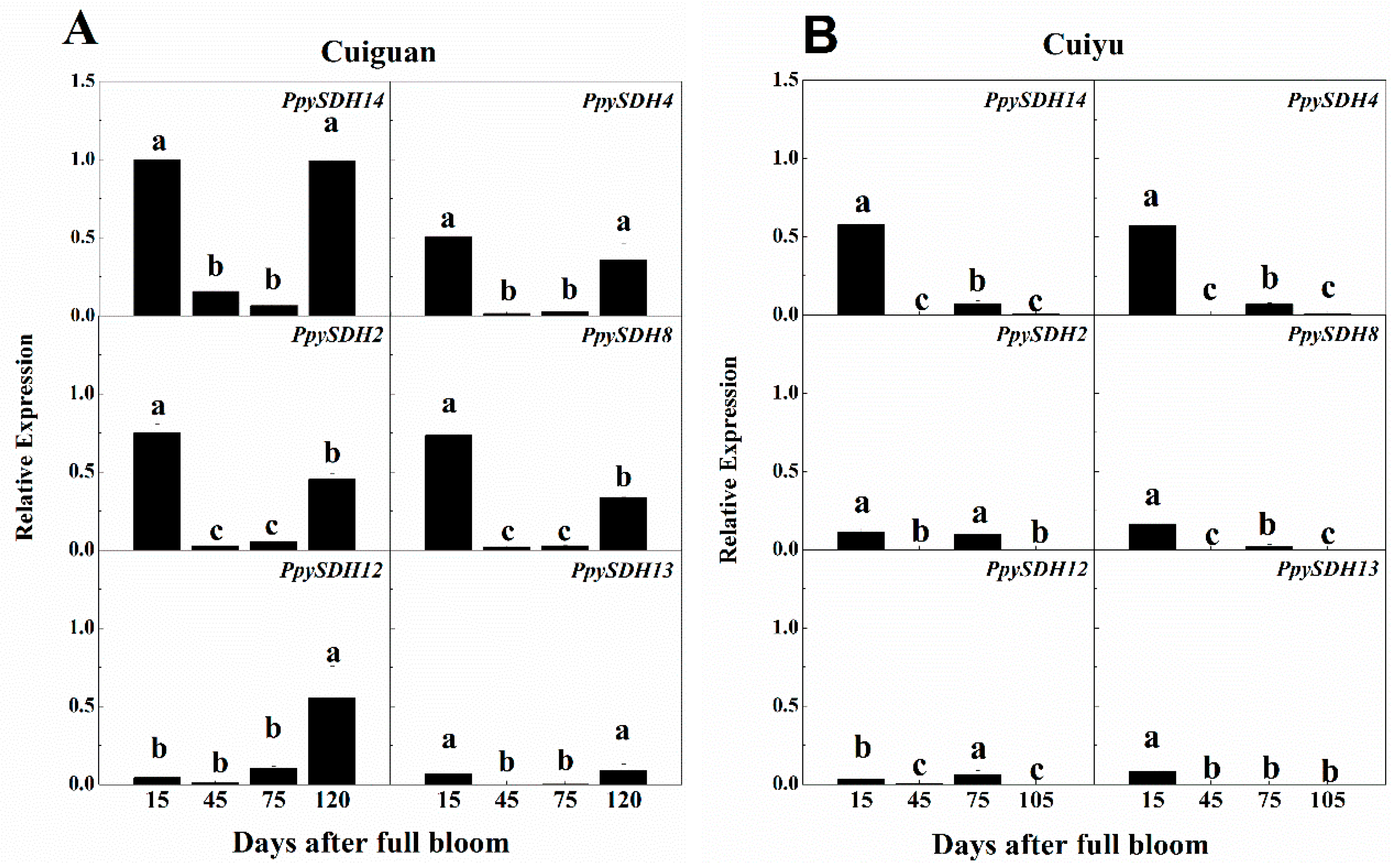
2.6. Expression of PpySDHs during Fruit Development
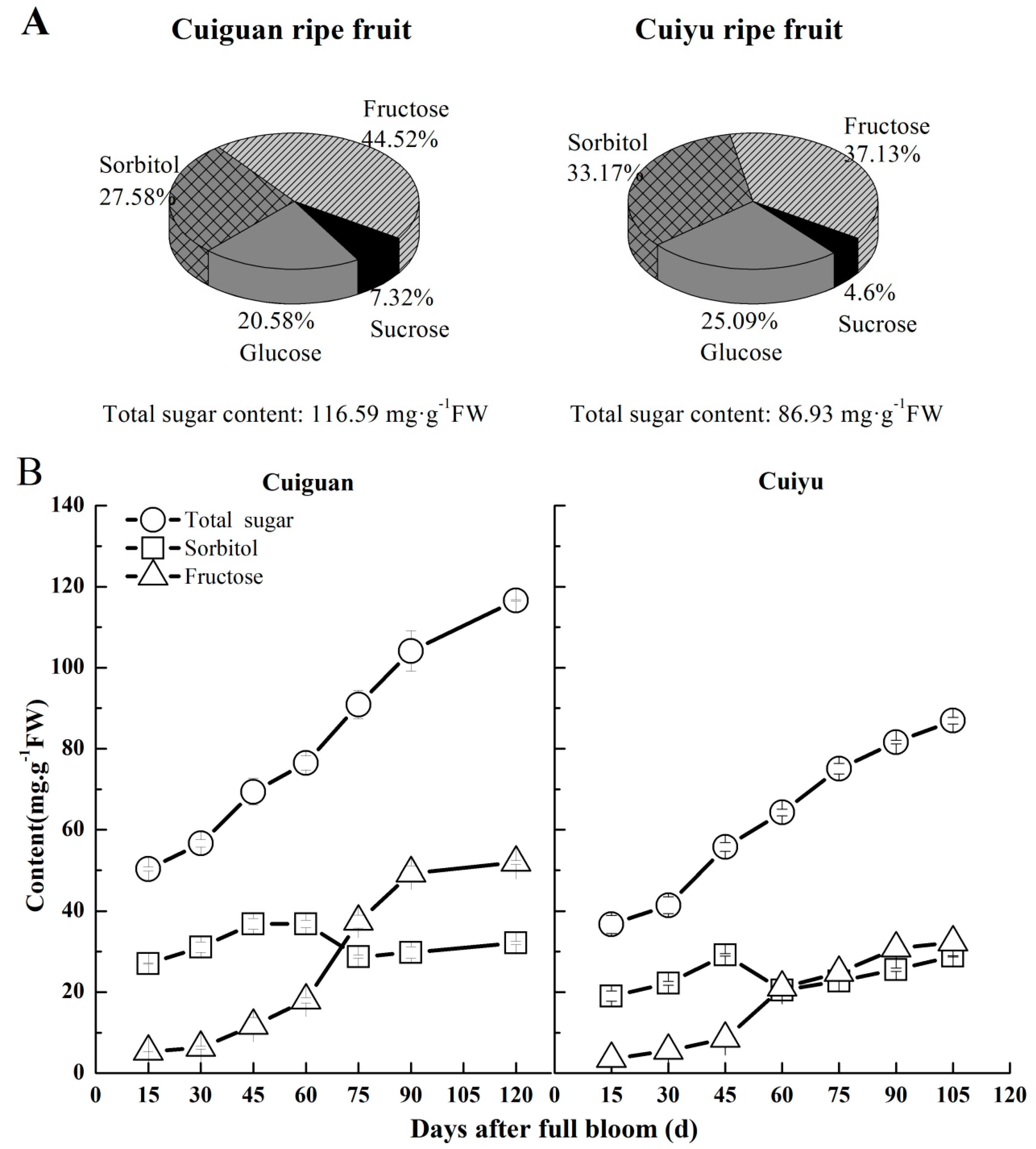
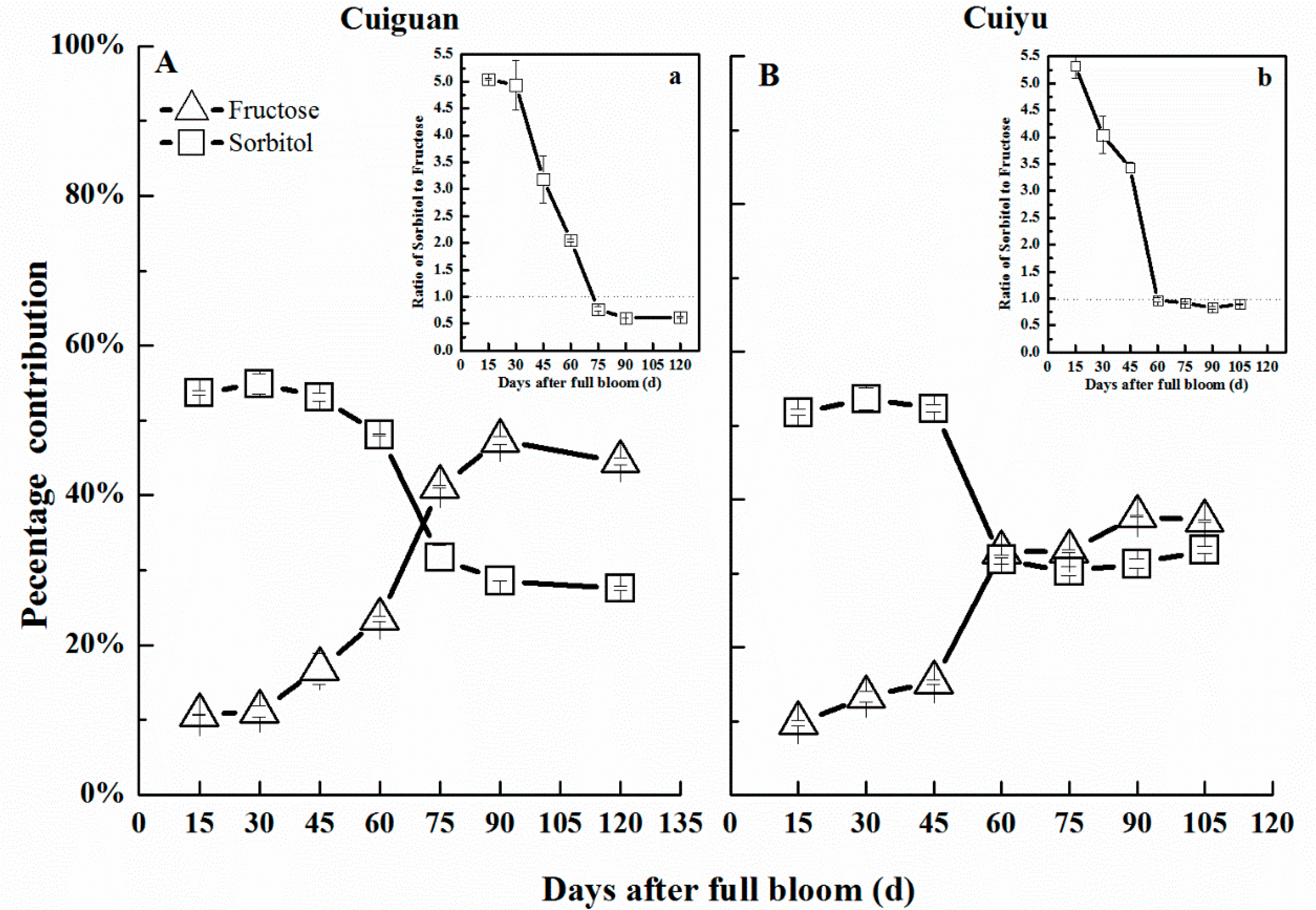
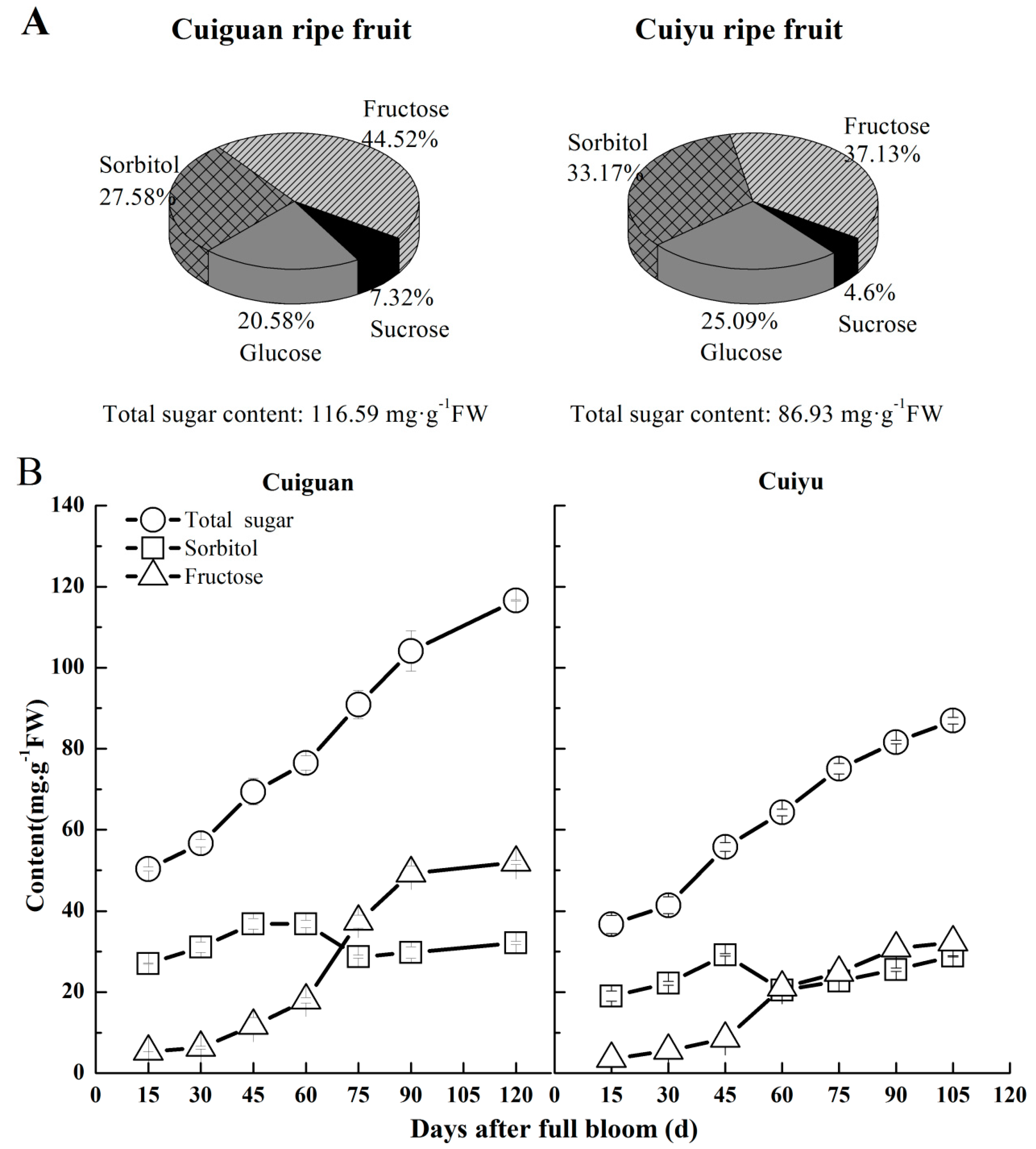
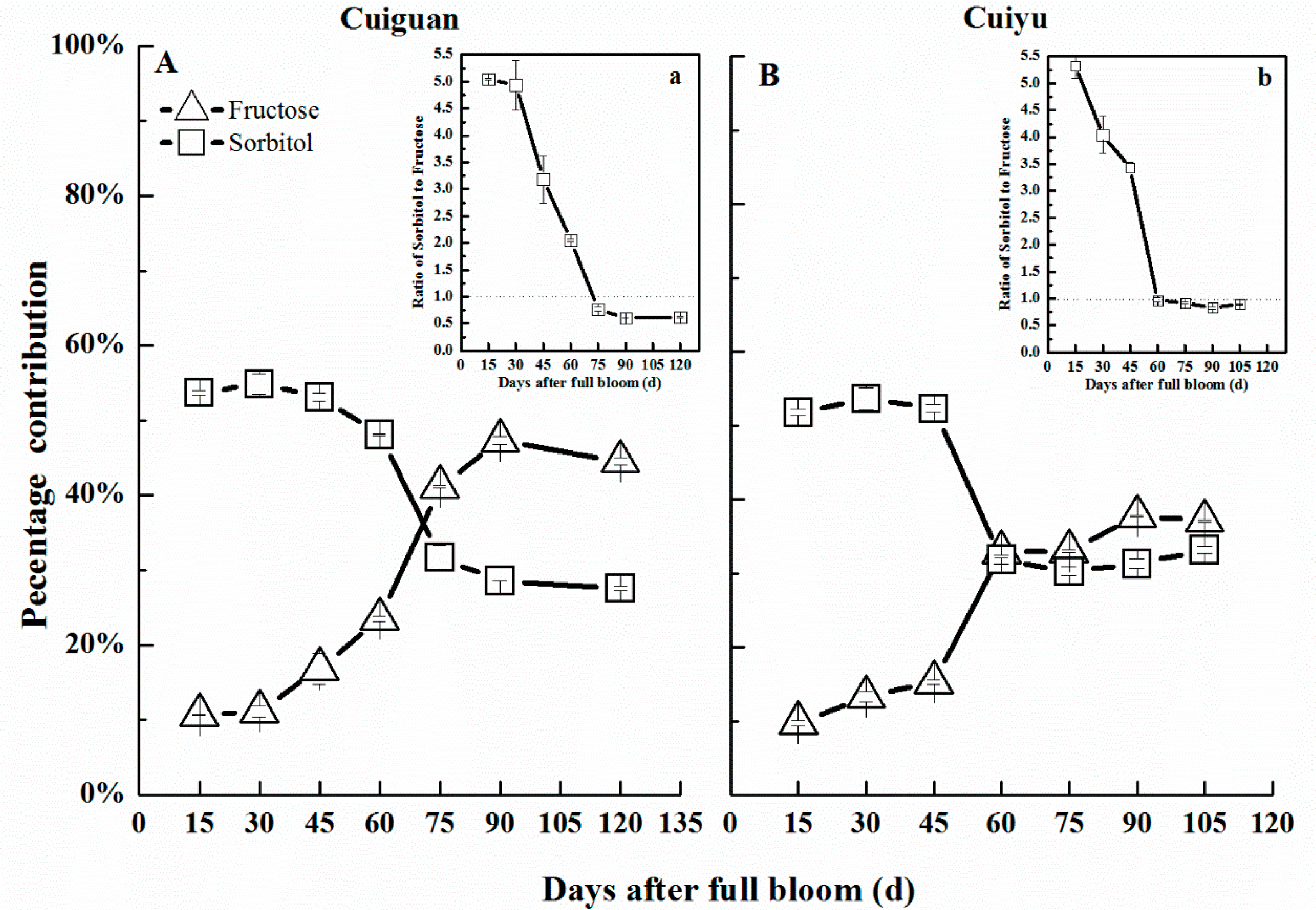
2.7. Changes in Sorbitol Content in Flesh during Fruit Development
| Cultivar | Commercial Harvest Date (D/M) | Average Fruit Weight (g) | Soluble Solids Content (%) | Firmness of Fruit Flesh (kg·cm−2) |
|---|---|---|---|---|
| Cuiguan | 27/7 | 302.5 ± 6.7 a | 13.08 ± 0.49 a | 2.65 ± 0.65 a |
| Cuiyu | 12/7 | 295.0 ± 11.1 a | 10.77 ± 0.80 b | 2.23 ± 0.32 a |
3. Discussion
4. Experimental Section
4.1. Plant Materials
4.2. Measurement of Soluble Solids Content (SSC) and Fruit Firmness
4.3. HPLC Analysis of Sugar Content
4.4. RNA Isolation and RNA-Seq Analysis
4.5. Construction of Phylogenetic Tree
4.6. Quantitative Reverse Transcriptase PCR (qRT-PCR) and Data Analysis
| Gene | Primers | Annealing Temperature (°C) | Amplicon Size (bp) |
|---|---|---|---|
| PpySDH2 | F 5′-GTCCGTTCCACTGTATGGTT-3′ | 58 | 115 |
| R 5′-GCAAAGGAGTGGAGGAGTC-3′ | 58 | ||
| PpySDH4 | F 5′-GTTGATGTACAGAGACCATTG-3′ | 58 | 115 |
| R 5′-GCATACGTACGCACACAATTAT-3′ | 58 | ||
| PpySDH8 | F 5′-GGTGGAAGAAGCCTTTGAAA-3′ | 58 | 241 |
| R 5′-TTACAAGAAGCGGAGGGTTT-3′ | 58 | ||
| PpySDH12 | F 5′-CCAGTGTAGTATAGCTTCCC-3′ | 58 | 135 |
| R 5′-AGGCCAACAGACTCGTGTC-3′ | 58 | ||
| PpySDH13 | F 5′-ATTAGAATGTAGAGAAAGGGA-3′ | 58 | 175 |
| R 5′-TTTGGTTCAATAGCCACCC-3′ | 58 | ||
| PpySDH14 | F 5′-AGGTCATCACAATAAGCACG-3′ | 58 | 91 |
| R 5′-AAACAAACAGAACGAGAAGCC-3′ | 58 | ||
| 18S rRNA | F 5′-CATGGCCGTTCTTAGTTGGTGGAG-3′ | 58 | 110 |
| R 5′-AAGAAGCTGGCCGCGAAGGGATAC-3′ | 58 |
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DAFB | days after full bloom |
| FW | fresh weight |
| MDR | medium-chain dehydrogenase/reductase |
| qRT-PCR | quantitative reverse transcription PCR |
| RPKM | reads per kilo-base per million |
| SDH | sorbitol dehydrogenase |
| SSC | soluble solids content |
| TPM | transcripts per million |
References
- Stoop, J.M.H.; Williamson, J.D.; Pharr, D.M. Mannitol metabolism in plants: A method for coping with stress. Trends Plant Sci. 1996, 1, 139–144. [Google Scholar] [CrossRef]
- Kanayama, Y. Physiological roles of polyols in horticultural crops. J. Jpn. Soc. Hortic. Sci. 2009, 78, 158–168. [Google Scholar] [CrossRef]
- Yamaki, S. Metabolism and accumulation of sugars translocated to fruit and their regulation. J. Jpn. Soc. Hortic. Sci. 2010, 79, 1–15. [Google Scholar] [CrossRef]
- Livesey, G. Health potential of polyols as sugar replacers, with emphasis on low glycaemic properties. Nutr. Res. Rev. 2003, 16, 163–191. [Google Scholar] [CrossRef] [PubMed]
- Eveland, A.L.; Jackson, D.P. Sugars, signalling, and plant development. J. Exp. Bot. 2011, 63, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Teo, G.; Suzuki, Y.; Uratsu, S.L.; Lampinen, B.; Ormonde, N.; Hu, W.K.; DeJong, T.M.; Dandekar, A.M. Silencing leaf sorbitol synthesis alters long-distance partitioning and apple fruit quality. Proc. Natl. Acad. Sci. USA 2006, 103, 18842–18847. [Google Scholar] [CrossRef] [PubMed]
- Loescher, W.H. Physiology and metabolism of sugar alcohols in higher plants. Physiol. Plant. 1987, 70, 553–557. [Google Scholar] [CrossRef]
- Estonius, M.; Danielsson, O.; Karlsson, C.; Persson, H.; Jornvall, H.; Hoog, J. Distribution of alcohol and sorbitol dehydrogenases. Eur. J. Biochem. 1993, 215, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Aguayo, M.F.; Ampuero, D.; Mandujano, P.; Parada, R.; Muñoz, R.; Gallart, M.; Altabella, T.; Cabrera, R.; Stange, C.; Handford, M. Sorbitol dehydrogenase is a cytosolic protein required for sorbitol metabolism in Arabidopsis thaliana. Plant Sci. 2013, 205–206, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Moriguchi, R.; Kanahama, K.; Yamaki, S.; Kanayama, Y. Molecular evidence of sorbitol dehydrogenase in tomato, a non-Rosaceae plant. Phytochemistry 2005, 66, 2822–2828. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, Z.W.; Shi, Z.B.; Zhang, S.; Ray, M.; Zhu, S.L.; Khan, M.A.; Tao, S.T.; Korban, S.S.; Wang, H.; et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013, 23, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.H.; Li, S.H.; Nosarzewski, M.; Archbold, D.D. Sorbitol dehydrogenase gene expression and enzyme activity in apple: Tissue specificity during bud development and response to rootstock vigor and growth manipulation. J. Am. Soc. Hortic. Sci. 2010, 135, 379–387. [Google Scholar]
- Nosarzewski, M.; Archbold, D.D. Tissue-specific expression of sorbitol dehydrogenase in apple fruit during early development. J. Exp. Bot. 2007, 58, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hayama, H.; Kashimura, Y. Partial cloning and expression analysis of genes encoding NAD+-dependent sorbitol dehydrogenase in pear bud during flower bud formation. Sci. Hortic. 2005, 103, 413–420. [Google Scholar] [CrossRef]
- Zhang, S.L. Pear, 1st ed.; China Agriculture Press: Beijing, China, 2013; p. 858. (in Chinese) [Google Scholar]
- Teng, Y.W.; Chai, M.L.; Li, X.G. A historic retrospect and the progress in the taxonomy of the genus Pyrus. J. Fruit Sci. 2004, 21, 252–257. (in Chinese). [Google Scholar]
- Xie, M.; Huang, Y.; Zhang, Y.P.; Wang, X.; Yang, H.; Yu, O.; Dai, W.H.; Fang, C.B. Transcriptome profiling of fruit development and maturation in Chinese white pear (Pyrus bretschneideri Rehd). BMC Genomics. 2013, 14, 823–843. [Google Scholar] [CrossRef] [PubMed]
- Conserved Domains and Protein Classification. Available online: http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml (accessed on 2 April 2015).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ahn, J.C.; Choi, J.; Hwang, B.; Choi, D. Expression and cloning of the full-length cDNA for sorbitol-6-phosphate dehydrogenase and NAD-dependent sorbitol dehydrogenase from pear (Pyrus pyrifolia N.). Sci. Hortic. 2007, 112, 406–412. [Google Scholar] [CrossRef]
- Shangguan, L.; Song, C.N.; Leng, X.P.; Kayesh, E.; Sun, X.; Fang, J.G. Mining and comparison of the genes encoding the key enzymes involved in sugar biosynthesis in apple, grape, and sweet orange. Sci. Hortic. 2014, 165, 311–318. [Google Scholar] [CrossRef]
- Persson, B.; Hedlund, J.; Jörnvall, H. The MDR superfamily. Cell. Mol. Life Sci. 2008, 65, 3879–3894. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Hua, Z.Y.; You, C.X.; Kong, X.Z.; Shi, X.P. Subcellular localization and vacuolar targeting of sorbitol dehydrogenase in apple seed. Plant Sci. 2013, 210, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Xu, Y.H.; Peng, C.C.; Fan, R.C.; Gao, X.Q. Ubiquitous distribution and different subcellular localization of sorbitol dehydrogenase in fruit and leaf of apple. J. Exp. Bot. 2009, 60, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Song, K.J.; Kim, M.Y.; Jeong-Hwan, H.; Shin, Y.U.; Kim, W.; Chung, W. Molecular cloning and characterization of four cDNAs encoding the isoforms of NAD-dependent sorbitol dehydrogenase from the Fuji apple. Plant Sci. 2002, 162, 513–519. [Google Scholar] [CrossRef]
- Oura, Y.; Yamada, K.; Shiratake, K.; Yamaki, S. Purification and characterization of a NAD+-dependent sorbitol dehydrogenase from Japanese pear fruit. Phytochemistry 2000, 54, 567–572. [Google Scholar] [CrossRef]
- Rothe, K.; Porzel, A.; Neumann, S.; Grimm, E. Characteristics of the phloem path: Analysis and distribution of carbohydrates in the petiole of Cyclamen. J. Exp. Bot. 1999, 50, 1807–1816. [Google Scholar] [CrossRef]
- De Sousa, S.M.; Paniago, M.D.G.; Arruda, P.; Yunes, J.A. Sugar levels modulate Sorbitol dehydrogenase expression in maize. Plant Mol. Biol. 2008, 68, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Sutsawat, D.; Kunio, Y.; Katsuhiro, S.; Yoshinori, K.; Yamaki, S. Properties of sorbitol dehydrogenase in strawberry fruit and enhancement of the activity by fructose and auxin. J. Jpn. Soc. Hortic. Sci. 2008, 77, 318–323. [Google Scholar] [CrossRef]
- Nookaraju, A.; Upadhyaya, C.P.; Pandey, S.K.; Young, K.E.; Hong, S.J.; Park, S.K.; Park, S.W. Molecular approaches for enhancing sweetness in fruits and vegetables. Sci. Hortic. 2010, 127, 1–15. [Google Scholar] [CrossRef]
- Dai, M.S.; Sun, T.L.; Wang, Y.Z.; Zhang, S.J.; Shi, Z.B. Breeding report of a new early maturing pear cultivar “Cuiyu”. J. Fruit Sci. 2013, 30, 175–176. (in Chinese). [Google Scholar]
- Zhang, C.X.; Tanabe, K.; Tamura, F.; Itai, A.; Wang, S.P. Spur characteristics, fruit growth and carbon partitioning in two late-maturing Japanese pear (Pyrus pyrifolia) cultivars with contrasting fruit size. J. Am. Soc. Hortic. Sci. 2005, 130, 252–260. [Google Scholar]
- Ho, L.C. Metabolism and compartmentation of imported sugars in sink organs in relation to sink strength. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1988, 39, 355–378. [Google Scholar] [CrossRef]
- Zhang, S.L.; Chen, K.S. Molecular Physiology of Fruit Quality Development and Regulation, 1st ed.; China Agriculture Press: Beijing, China, 2007; p. 252. (in Chinese) [Google Scholar]
- Hubbard, N.L.; Pharr, D.M.; Huber, S.C. Sucrose phosphate synthase and other sucrose metabolizing enzymes in fruits of various species. Physiol. Plant. 1991, 82, 191–196. [Google Scholar] [CrossRef]
- Lingle, S.E.; Dunlap, J.R. Sucrose metabolism in netted muskmelon fruit during development. Plant Physiol. 1987, 84, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.F.; Zhang, S.L.; Wu, J.; Cao, Y.F.; Liu, J.; Han, K.; Yang, Z.J. Analysis of components and contents of soluble sugars and organic acids in ten cultivars of pear by high performance liquid chromatography. J. Nanjing Agric. Univ. 2011, 34, 25–31. (in Chinese). [Google Scholar]
- Wang, J.Y.; Zhu, S.G.; Xu, C.F. Biochemistry (Part I), 3rd ed.; Higher Education Press: Beijing, China, 2002; p. 626. (in Chinese) [Google Scholar]
- Pommerrenig, B.; StalPapini-Terzi, F.; Sauer, N. Differential regulation of sorbitol and sucrose loading into the phloem of Plantago major in response to salt stress. Plant Physiol. 2007, 144, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Acuña, M.G.; Biasi, W.V.; Flores, S.; Jiang, C.Z.; Reid, M.S.; Willits, N.H.; Mitcham, E.J. Effect of maturity and cold storage on ethylene biosynthesis and ripening in “Bartlett” pears treated after harvest with 1-MCP. Postharvest Biol. Technol. 2011, 59, 1–9. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, M.; Shi, Z.; Xu, C. Genome-Wide Analysis of Sorbitol Dehydrogenase (SDH) Genes and Their Differential Expression in Two Sand Pear (Pyrus pyrifolia) Fruits. Int. J. Mol. Sci. 2015, 16, 13065-13083. https://doi.org/10.3390/ijms160613065
Dai M, Shi Z, Xu C. Genome-Wide Analysis of Sorbitol Dehydrogenase (SDH) Genes and Their Differential Expression in Two Sand Pear (Pyrus pyrifolia) Fruits. International Journal of Molecular Sciences. 2015; 16(6):13065-13083. https://doi.org/10.3390/ijms160613065
Chicago/Turabian StyleDai, Meisong, Zebin Shi, and Changjie Xu. 2015. "Genome-Wide Analysis of Sorbitol Dehydrogenase (SDH) Genes and Their Differential Expression in Two Sand Pear (Pyrus pyrifolia) Fruits" International Journal of Molecular Sciences 16, no. 6: 13065-13083. https://doi.org/10.3390/ijms160613065