
Urotensin II Protects Cardiomyocytes from Apoptosis Induced by Oxidative Stress through the CSE/H2S Pathway

Abstract
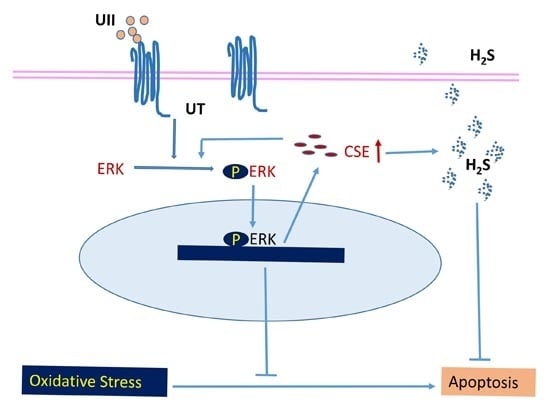
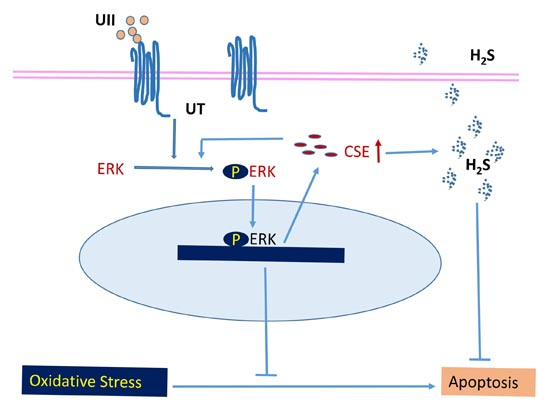
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
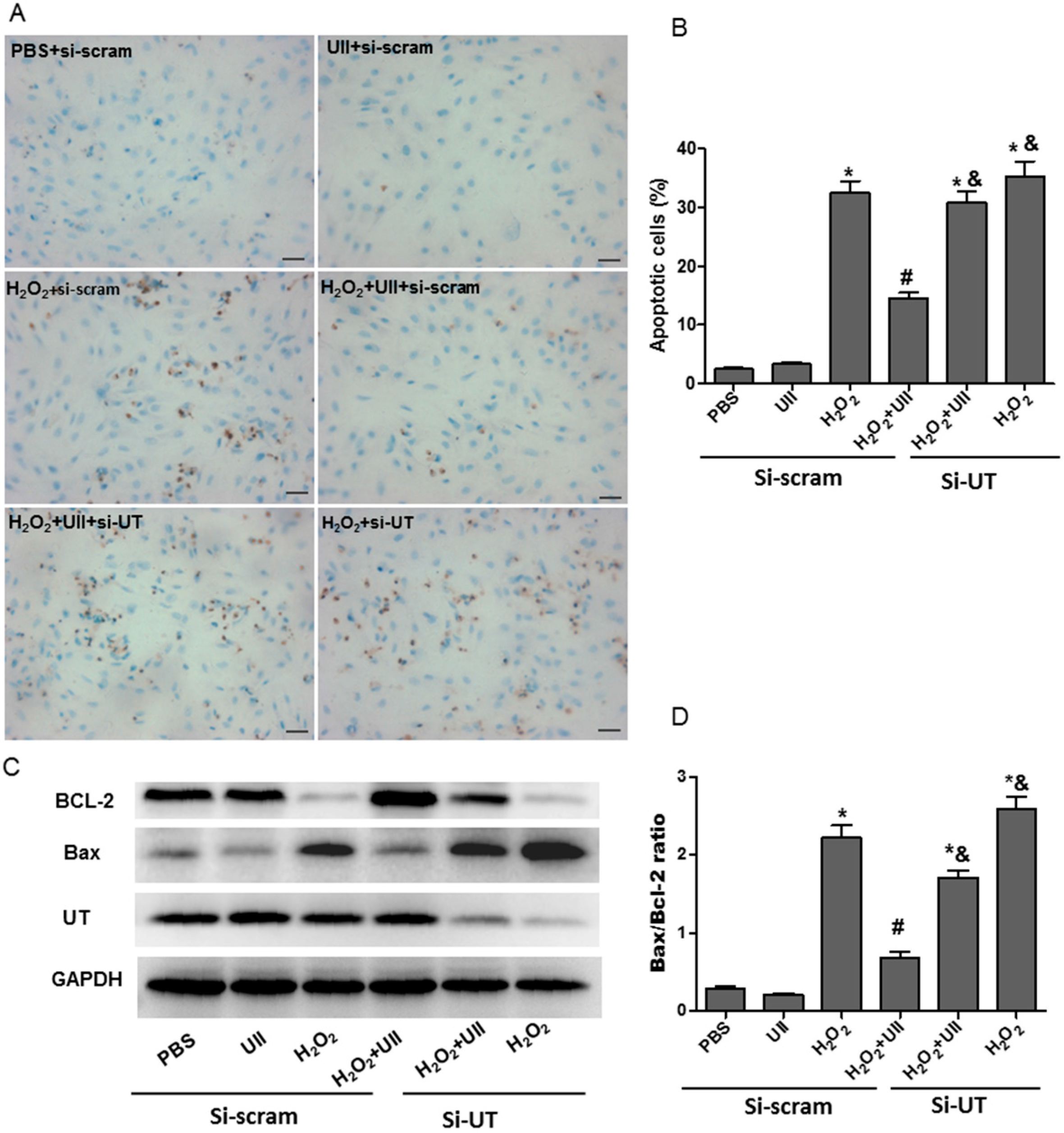
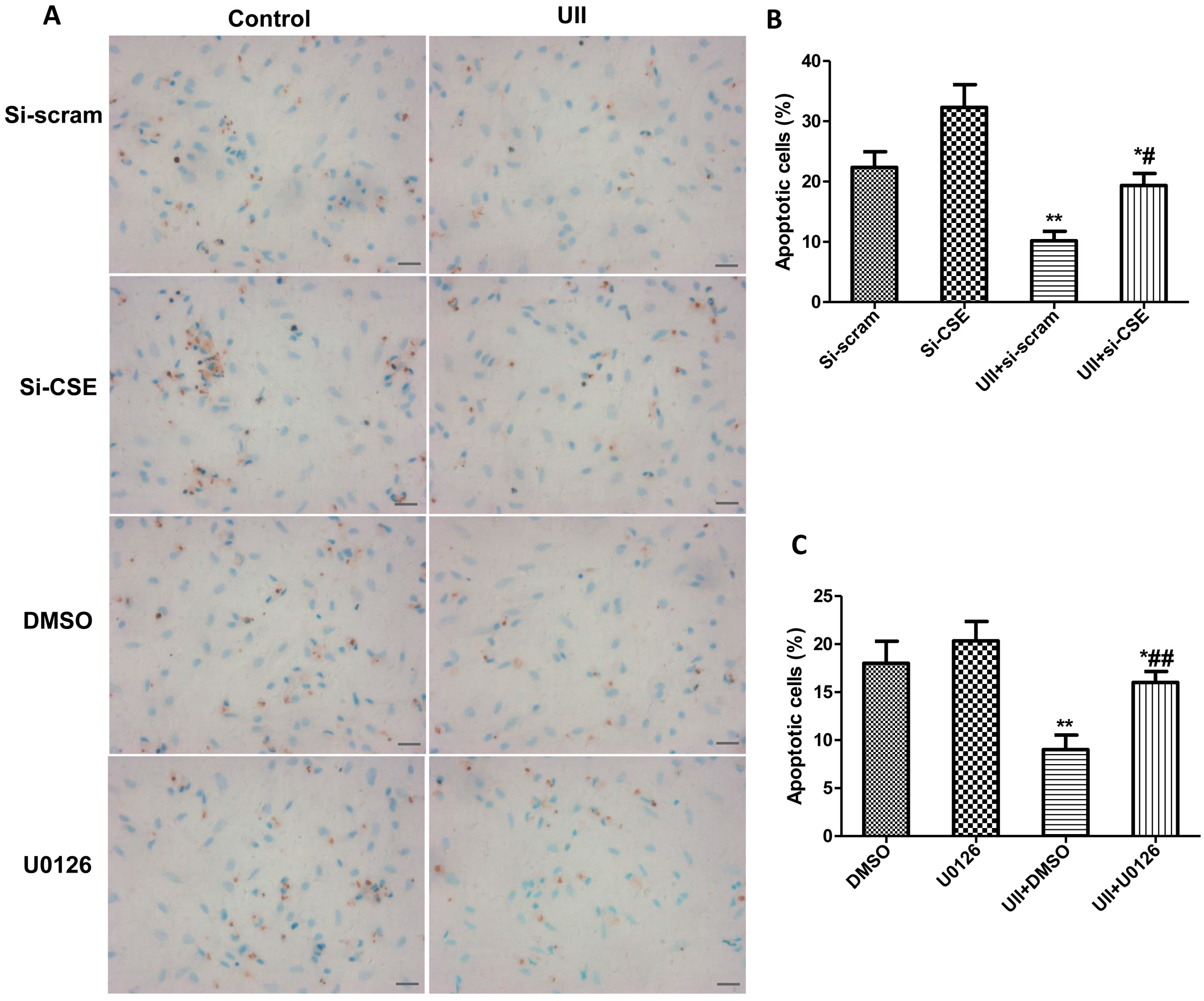
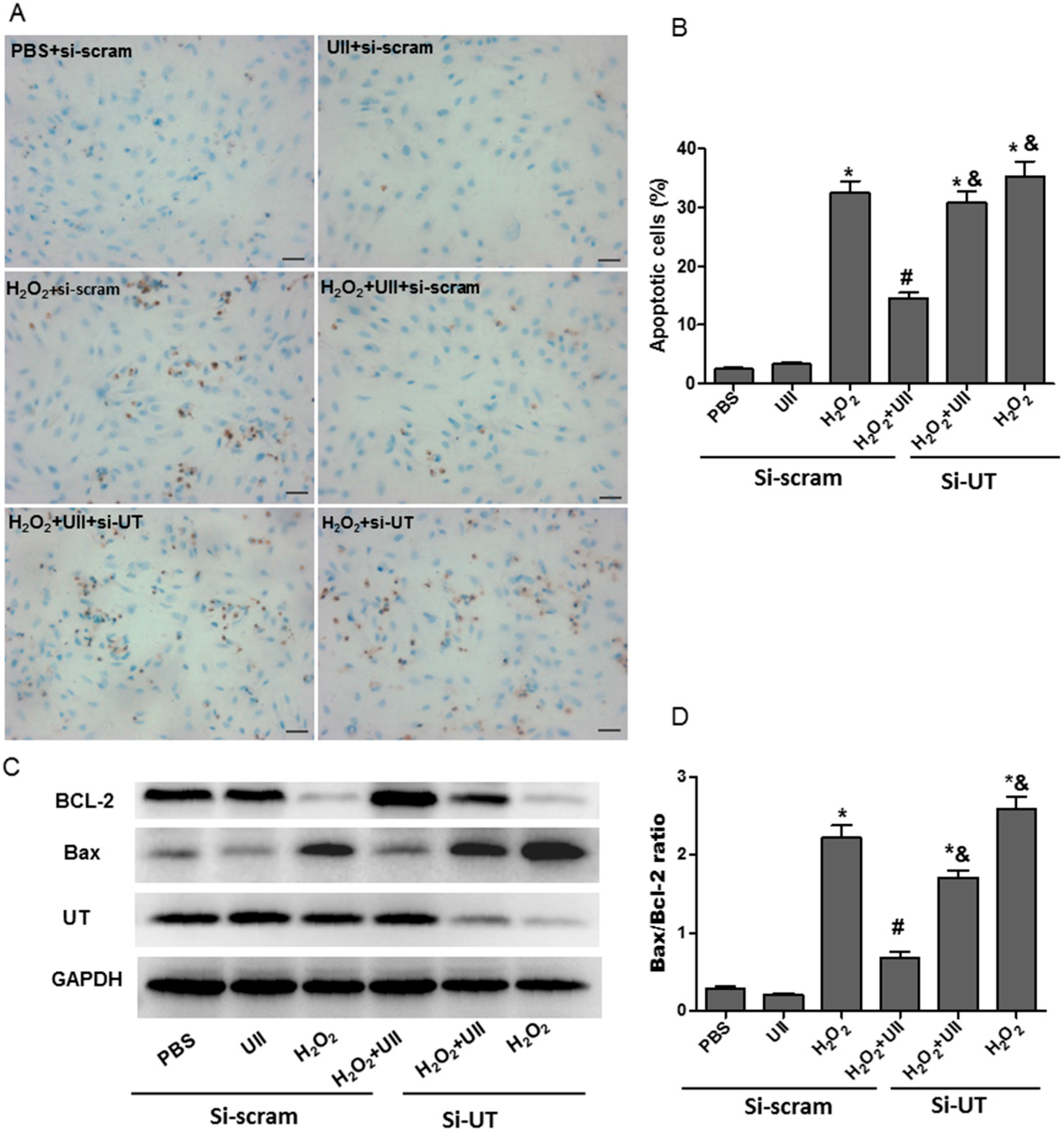
2.1. UII Prevents Cardiomyocytes from Apoptosis Induced by H2O2
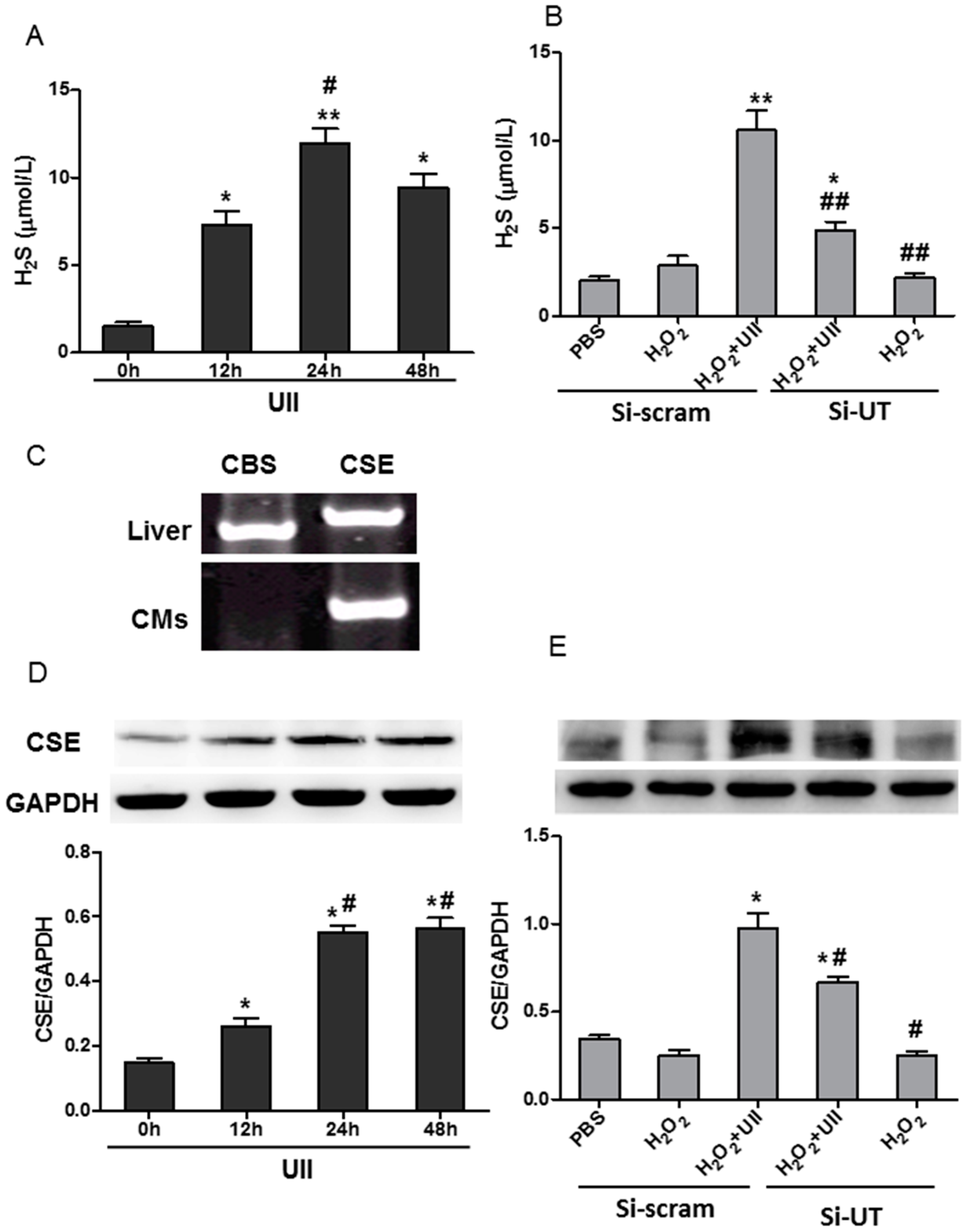
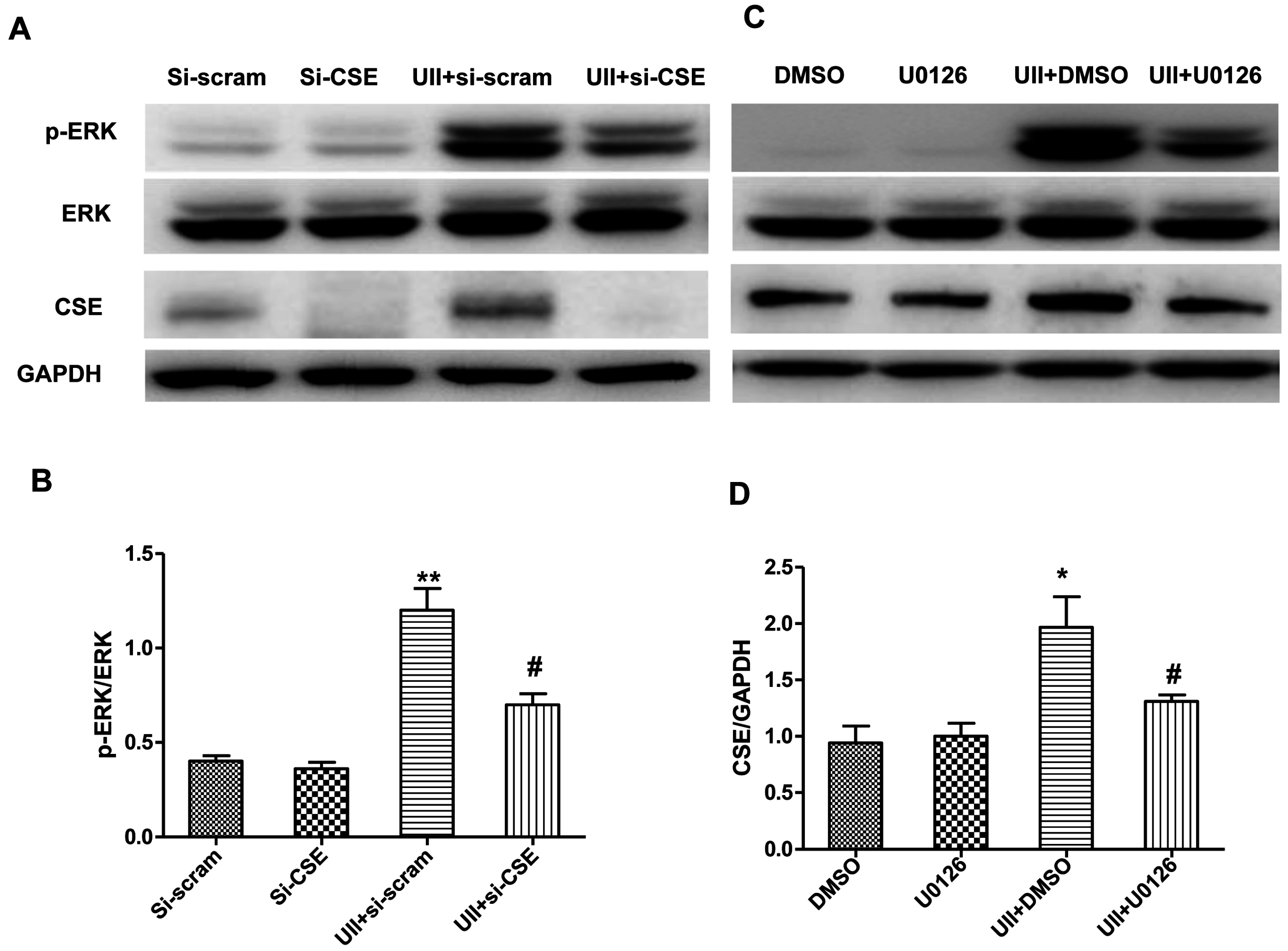
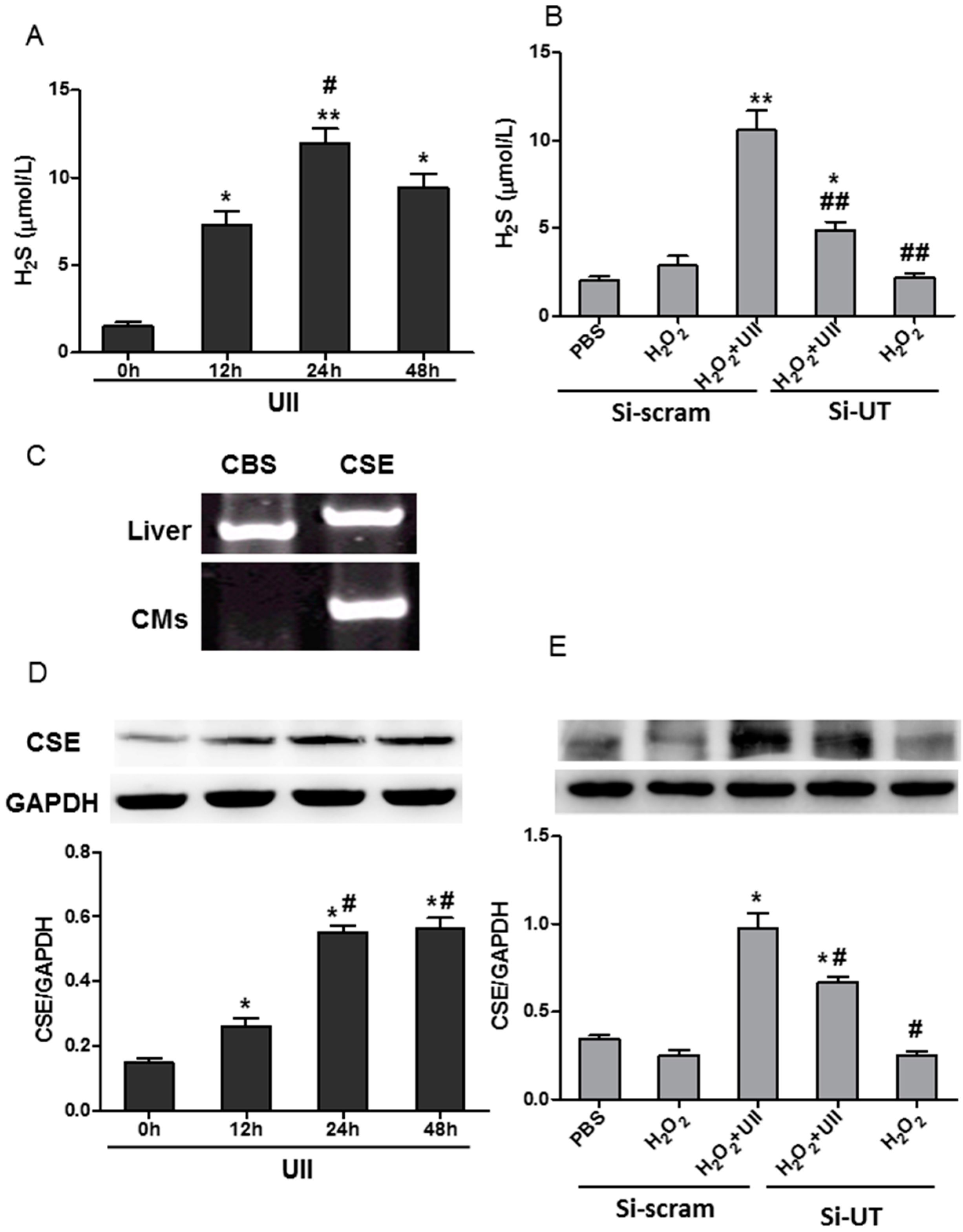
2.2. UII Increases the H2S Production by Enhancing the Level of CSE (Cystathionine-γ-Lyase) in Cardiomyocytes Exposed to H2O2
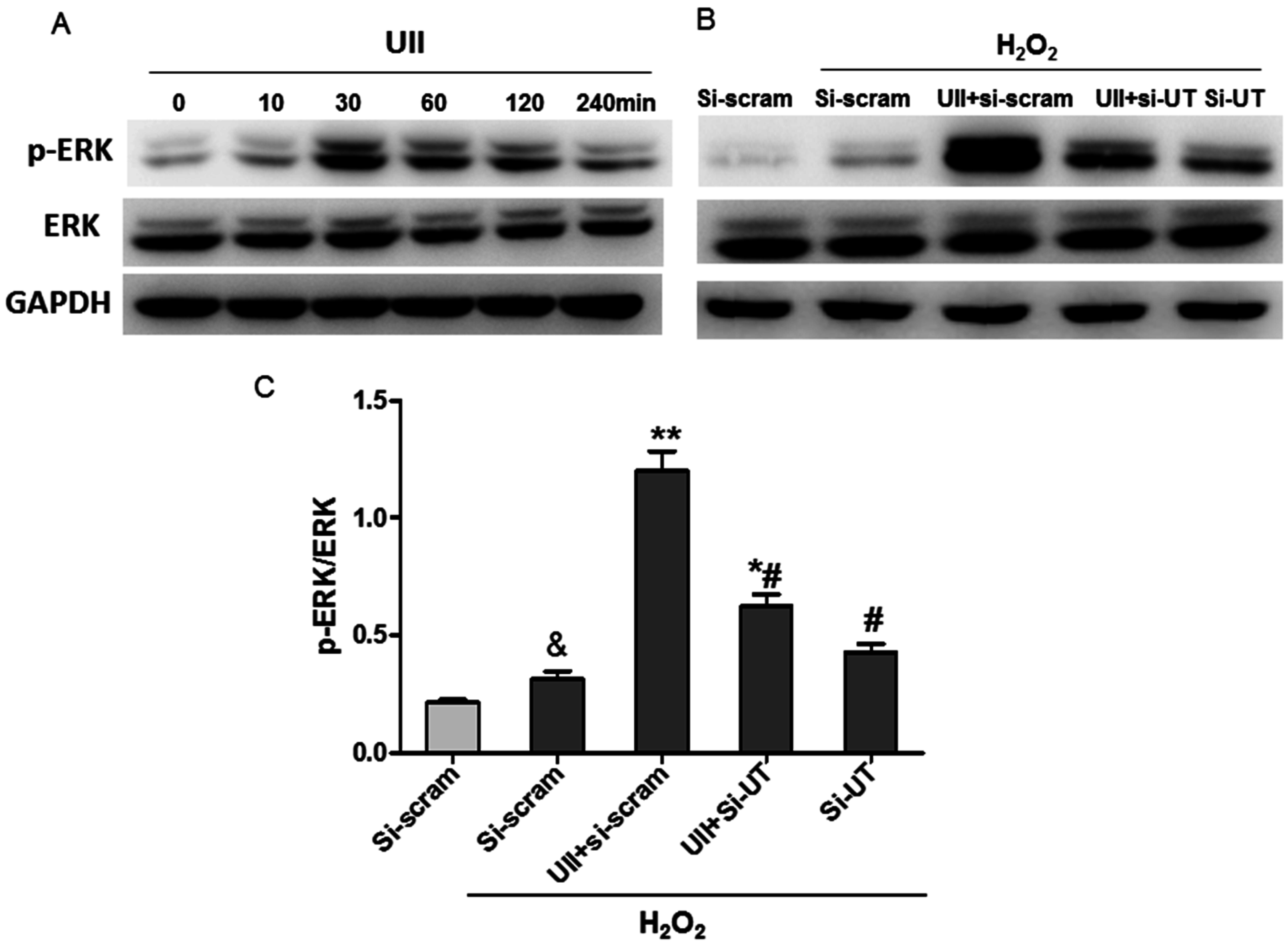
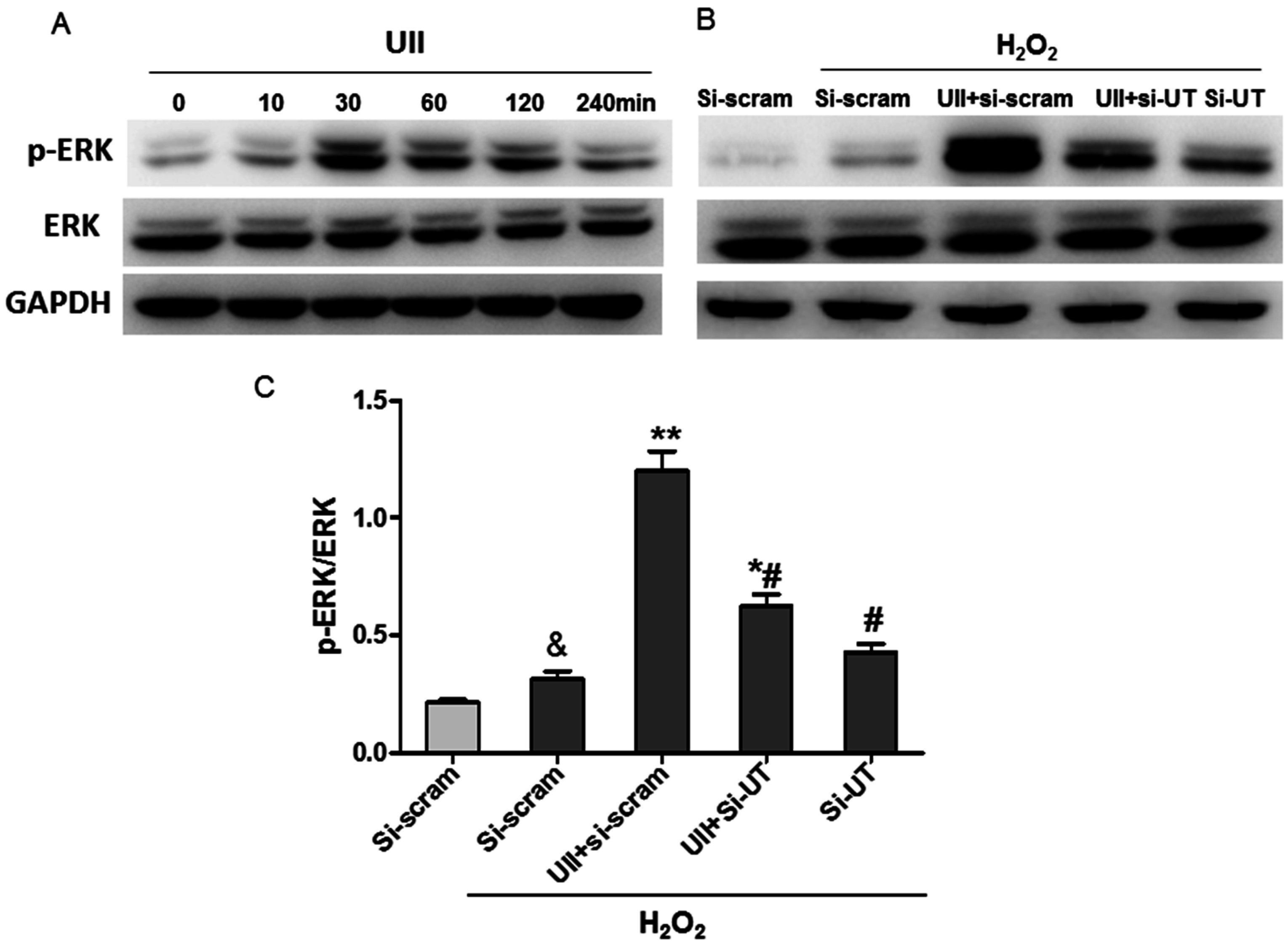
2.3. UII Promotes the Activation of ERK in Cultured Cardiomyocytes Exposed to H2O2
2.4. UII Induces the Reciprocal Regulation of CSE and ERK in Cardiomyocytes Exposed to H2O2
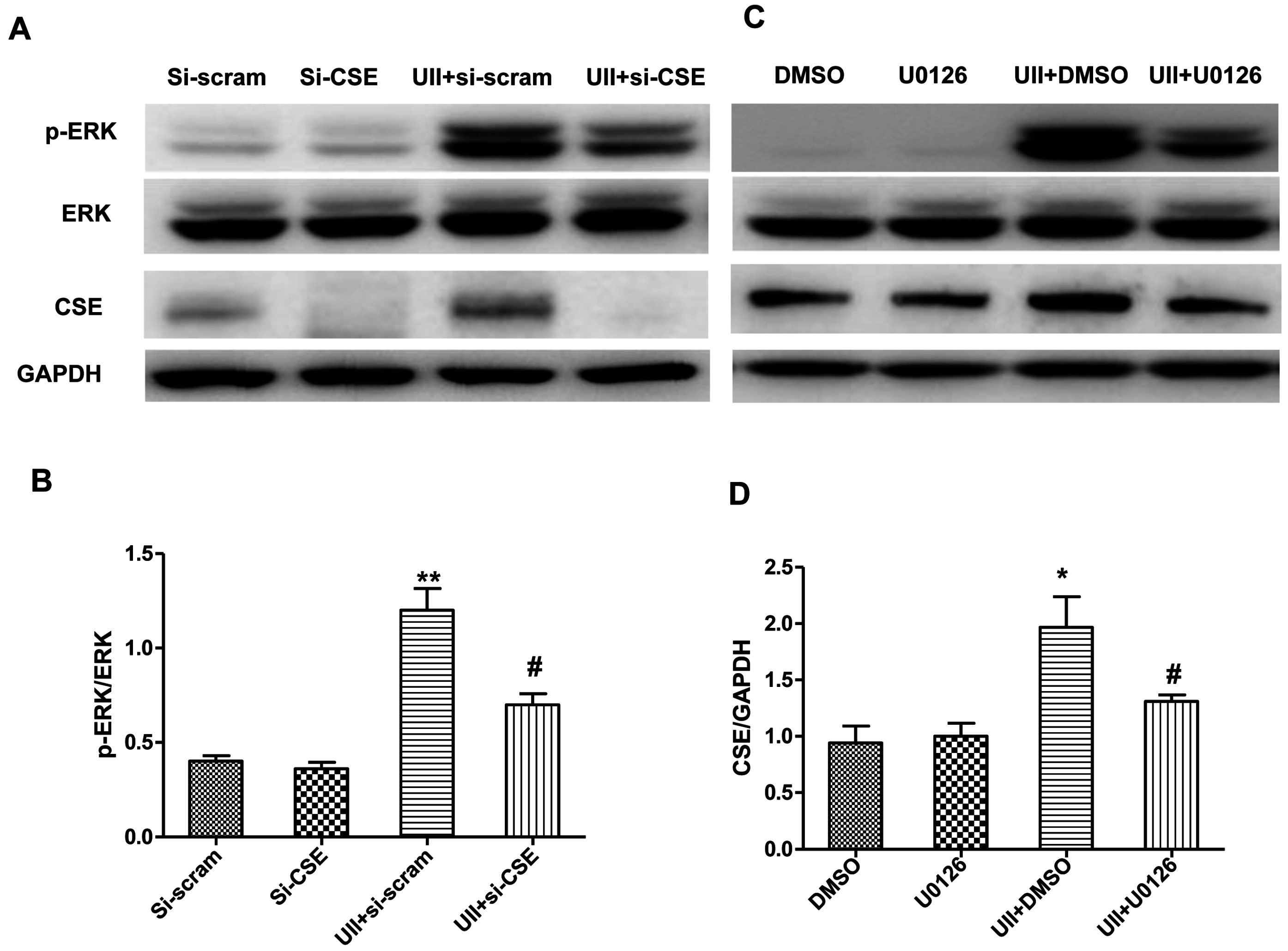
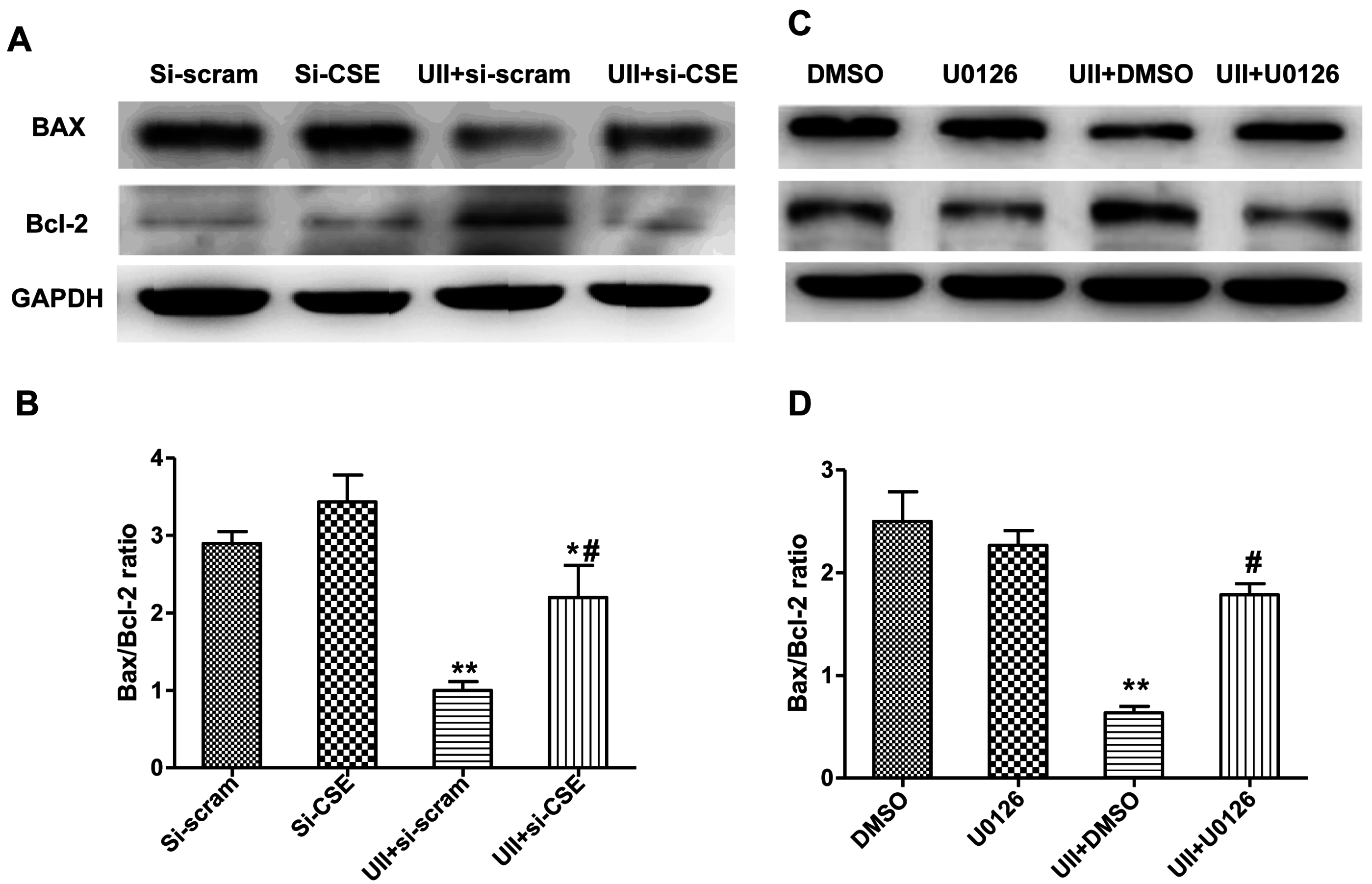
2.5. Si-CSE or ERK Inhibitor Partly Abolishes Anti-Apoptotic Effects of UII on H2O2-Treated Cardiomyocytes
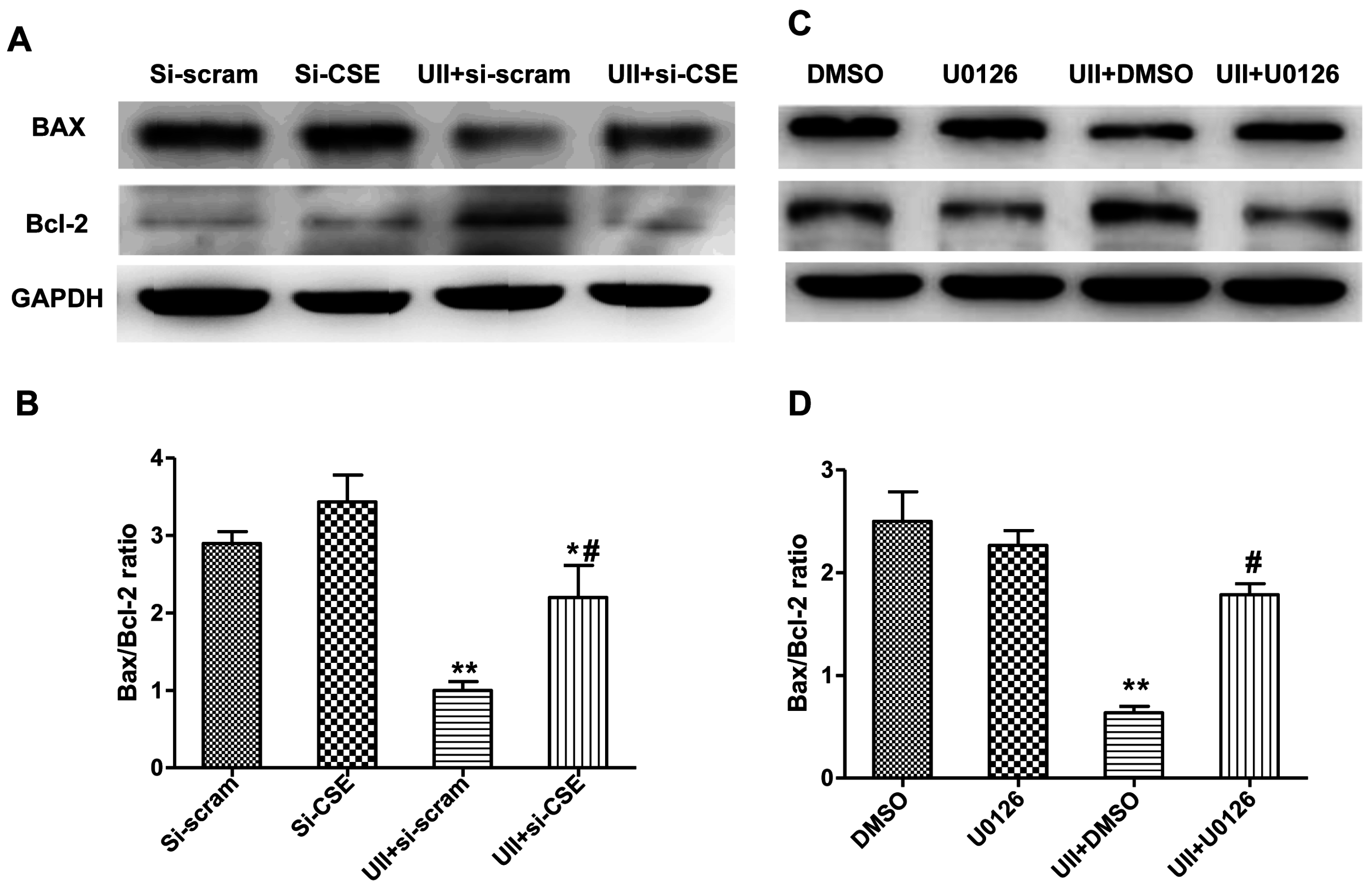
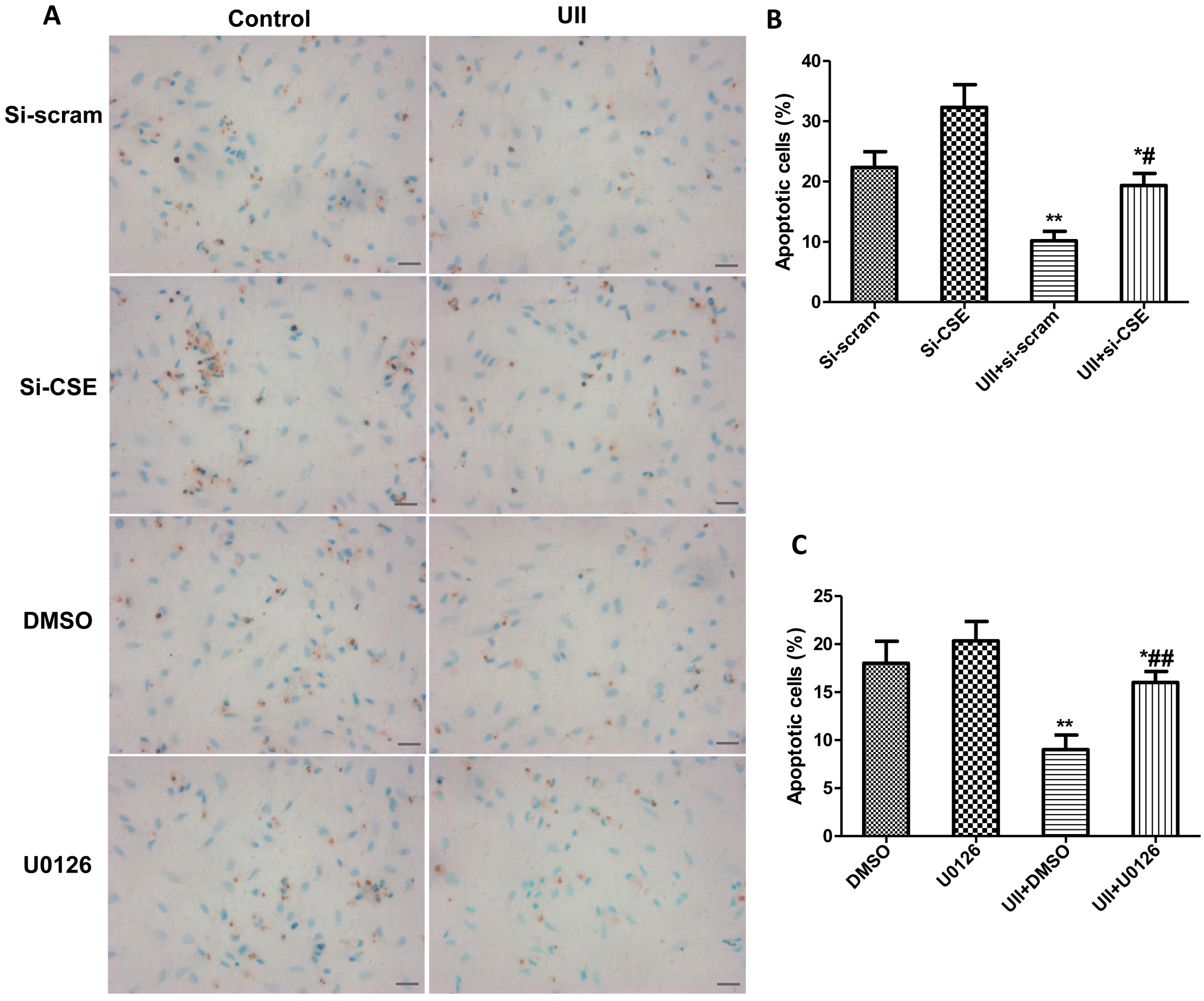
3. Experimental Section
3.1. Primary Culture of Neonatal Rat Cardiomyocytes
3.2. Apoptotic Cell Analysis by TUNEL Labeling
3.3. H2S Measurement
3.4. Reverse Transcription PCR (RT-PCR)
3.5. SiRNA and in Vitro Transfection
3.6. Western Blot Analysis
3.7. Myocardial Infarction Model of Mouse and Real-Time Polymerase Chain Reaction (RT-PCR)
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Russell, F.D.; Meyers, D.; Galbraith, A.J.; Bett, N.; Toth, I.; Kearns, P.; Molenaar, P. Elevated plasma levels of human urotensin-II immunoreactivity in congestive heart failure. Am. J. Physiol. 2003, 285, H1576–H1581. [Google Scholar] [CrossRef] [PubMed]
- Tzanidis, A.; Hannan, R.D.; Thomas, W.G.; Onan, D.; Autelitano, D.J.; See, F.; Kelly, D.J.; Gilbert, R.E.; Krum, H. Direct actions of urotensin II on the heart: Implications for cardiac fibrosis and hypertrophy. Circ. Res. 2003, 93, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Cao, J.; Chen, J.; Yang, J.; Zhang, Z.; Du, J.; Tang, C. Effect of chronic hypoxia on contents of urotensin II and its functional receptors in rat myocardium. Heart Vessel. 2002, 16, 64–68. [Google Scholar] [CrossRef]
- Djordjevic, T.; BelAiba, R.S.; Bonello, S.; Pfeilschifter, J.; Hess, J.; Gorlach, A. Human urotensin II is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.G.; Ao, Z.; Naselsky, D.; Herold, C.L.; Maniscalco, K.; Sarov-Blat, L.; Steplewski, K.; Aiyar, N.; Douglas, S.A. Urotensin-II-mediated cardiomyocyte hypertrophy: Effect of receptor antagonism and role of inflammatory mediators. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Russell, F.D. Emerging roles of urotensin-II in cardiovascular disease. Pharmacol. Ther. 2004, 103, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Q.; Bhandari, S.S.; Quinn, P.; Davies, J.E.; Ng, L.L. Urotensin II is raised in acute myocardial infarction and low levels predict risk of adverse clinical outcome in humans. Int. J. Cardiol. 2007, 117, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Prosser, H.C.; Forster, M.E.; Richards, A.M.; Pemberton, C.J. Urotensin II and urotensin II-related peptide (URP) in cardiac ischemia-reperfusion injury. Peptides 2008, 29, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Oh, Y.B.; Park, B.M.; Park, W.H.; Kim, S.H. Urotensin II protects ischemic reperfusion injury of hearts through ROS and antioxidant pathway. Peptides 2012, 36, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1215–H1224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Yang, W.; Cao, G. H2S inhibits the activation of hepatic stellate cells and downregulates the expression of urotensin II. Hepatol. Res. 2013, 43, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Gong, H.; Chen, Z.; Liang, Y.; Yuan, J.; Zhang, G.; Wu, J.; Ye, Y.; Yang, C.; Nakai, A.; et al. Association of Stat3 with HSF1 plays a critical role in G-CSF-induced cardio-protection against ischemia/reperfusion injury. J. Mol. Cell Cardiol. 2012, 52, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Shan, P.; Pu, J.; Yuan, A.; Shen, L.; Shen, L.; Chai, D.; He, B. RXR agonists inhibit oxidative stress-induced apoptosis in H9c2 rat ventricular cells. Biochem. Biophys. Res. Commun. 2008, 375, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Nagai, R.; Yamazaki, T. Urotensin II induces hypertrophic responses in cultured cardiomyocytes from neonatal rats. FEBS Lett. 2001, 508, 57–60. [Google Scholar] [CrossRef]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Coulouarn, Y.; Lihrmann, I.; Jegou, S.; Anouar, Y.; Tostivint, H.; Beauvillain, J.C.; Conlon, J.M.; Bern, H.A.; Vaudry, H. Cloning of the cDNA encoding the urotensin II precursor in frog and human reveals intense expression of the urotensin II gene in motoneurons of the spinal cord. Proc. Natl. Acad. Sci. USA 1998, 95, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Ames, R.S.; Sarau, H.M.; Chambers, J.K.; Willette, R.N.; Aiyar, N.V.; Romanic, A.M.; Louden, C.S.; Foley, J.J.; Sauermelch, C.F.; Coatney, R.W.; et al. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature 1999, 401, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.C.; Zhu, Y.Z.; Moore, P.K. The role of urotensin II in cardiovascular and renal physiology and diseases. Br. J. Pharmacol. 2006, 148, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Wang, Y.X.; Zhu, Y.Z.; Wang, W.W.; Wang, M.J.; Yao, T.; Zhu, Y.C. Cellular distribution of GPR14 and the positive inotropic role of urotensin II in the myocardium in adult rat. J. Appl. Physiol. 2004, 97, 2228–2235. [Google Scholar] [CrossRef] [PubMed]
- Russell, F.D.; Molenaar, P.; O’Brien, D.M. Cardiostimulant effects of urotensin-II in human heart in vitro. Br. J. Pharmacol. 2001, 132, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Le Mellionnec, E.; Bertoglio, J.; Scalbert, E.; Pacaud, P.; Loirand, G. Human urotensin II-induced contraction and arterial smooth muscle cell proliferation are mediated by RhoA and Rho-kinase. Circ. Res. 2001, 88, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Liu, J.C.; Loh, S.H.; Chen, C.H.; Hong, C.Y.; Chen, J.J.; Cheng, T.H. Involvement of reactive oxygen species in urotensin II-induced proliferation of cardiac fibroblasts. Eur. J. Pharmacol. 2008, 593, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.; McKendy, K.; Giaid, A. Role of urotensin II in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1156–R1172. [Google Scholar] [CrossRef] [PubMed]
- Russell, F.D. Urotensin II in cardiovascular regulation. Vasc. Health Risk Manag. 2008, 4, 775–785. [Google Scholar] [PubMed]
- Gao, Y.; Liu, C.Y.; Zhang, X.J.; Gao, J.; Yang, C.Y. Circulating endothelial cells as potential markers of atherosclerosis. Can. J. Neurol. Sci. 2008, 35, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Xing, J.; Zhang, X.; Dong, S.; Zhao, Y.; Wang, L.; Li, H.; Yang, F.; Xu, C.; Zhang, W. Exogenous hydrogen sulfide prevents cardiomyocyte apoptosis from cardiac hypertrophy induced by isoproterenol. Mol. Cell. Biochem. 2013, 381, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Tyagi, N.; Sen, U.; Givvimani, S.; Tyagi, S.C. H2S ameliorates oxidative and proteolytic stresses and protects the heart against adverse remodeling in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H451–H456. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, Q.; Liu, X.; Pan, L.; Xiong, Q.; Zhu, Y.Z. Hydrogen sulfide protects against apoptosis under oxidative stress through SIRT1 pathway in H9c2 cardiomyocytes. Nitric Oxide 2015, 46, 204–212. [Google Scholar] [CrossRef] [PubMed]
- King, A.L.; Lefer, D.J. Cytoprotective actions of hydrogen sulfide in ischaemia-reperfusion injury. Exp. Physiol. 2011, 96, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Yuan, A.; Shan, P.; Gao, E.; Wang, X.; Wang, Y.; Lau, W.B.; Koch, W.; Ma, X.L.; He, B. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur. Heart J. 2013, 34, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.T.; Feng, Z.N.; Lee, S.W.; Moore, P.K.; Bian, J.S. Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. J. Mol. Cell. Cardiol. 2006, 40, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.T.; Neo, K.L.; Hu, L.F.; Yong, Q.C.; Bian, J.S. H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am. J. Physiol. Cell Physiol. 2008, 294, C169–C177. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Z.; Wang, Z.J.; Ho, P.; Loke, Y.Y.; Zhu, Y.C.; Huang, S.H.; Tan, C.S.; Whiteman, M.; Lu, J.; Moore, P.K. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J. Appl. Physiol. 2007, 102, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, T.; Li, D.; Zhu, S.; Chen, Q.; Hu, W.; Pan, D.; Zhu, H.; Sun, H. ERK/PP1a/PLB/SERCA2a and JNK pathways are involved in luteolin-mediated protection of rat hearts and cardiomyocytes following ischemia/reperfusion. PLoS ONE 2013, 8, e82957. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Guan, Y.; Duan, J.; Wei, G.; Zhu, Y.; Quan, W.; Guo, C.; Zhou, D.; Wang, Y.; Xi, M.; Wen, A. Cardioprotective effect of Danshensu against myocardial ischemia/reperfusion injury and inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2 phosphorylation. Eur. J. Pharmacol. 2013, 699, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Yoshiyama, M.; Omura, T.; Hanatani, A.; Kim, S.; Takeuchi, K.; Iwao, H.; Yoshikawa, J. Activation of mitogen-activated protein kinases and activator protein-1 in myocardial infarction in rats. Cardiovasc. Res. 1998, 38, 116–124. [Google Scholar] [CrossRef]
- Laatikainen, L.E.; Incoronato, M.; Castellone, M.D.; Laurila, J.P.; Santoro, M.; Laukkanen, M.O. SOD3 decreases ischemic injury derived apoptosis through phosphorylation of Erk1/2, Akt, and FoxO3a. PLoS ONE 2011, 6, e24456. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M. Activation and targeting of mitogen-activated protein kinases by G-protein-coupled receptors. Can. J. Physiol. Pharmacol. 2002, 80, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Perrino, C.; Cannavo, A.; Schiattarella, G.G.; Borgia, F.; Sannino, A.; Pironti, G.; Gargiulo, G.; di Serafino, L.; Franzone, A.; et al. EGFR trans-activation by urotensin II receptor is mediated by beta-arrestin recruitment and confers cardioprotection in pressure overload-induced cardiac hypertrophy. Basic Res. Cardiol. 2011, 106, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Okazaki, M.; Tamura, M.; Isozumi, K.; Tasaki, H.; Nakashima, Y. Urotensin II-induced activation of extracellular signal-regulated kinase in cultured vascular smooth muscle cells: Involvement of cell adhesion-mediated integrin signaling. Life Sci. 2003, 72, 1049–1060. [Google Scholar] [CrossRef]
- Arcucci, A.; Ruocco, M.R.; Albano, F.; Granato, G.; Romano, V.; Corso, G.; Bancone, C.; de Vendittis, E.; della Corte, A.; Montagnani, S. Analysis of extracellular superoxide dismutase and Akt in ascending aortic aneurysm with tricuspid or bicuspid aortic valve. Eur. J. Histochem. 2014, 58, 2383. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Wang, R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006, 20, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Kimura, T.; Umeki, T.; Kimura, Y.; Kimura, H.; Ishii, I.; Itoh, N.; Naito, Y.; Yamamoto, H.; Niki, I. Protein phosphorylation involved in the gene expression of the hydrogen sulphide producing enzyme cystathionine gamma-lyase in the pancreatic β-cell. Mol. Cell. Endocrinol. 2012, 350, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; O’Connell, M.A.; Pawlinski, R.; Hollis, A.; McGovern, P.; Yan, S.F.; Stern, D.; Mackman, N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 2001, 98, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tang, Z.; Cong, B.; Du, J.; Wang, C.; Wang, L.; Ni, X.; Lu, J. Estrogens increase cystathionine-gamma-lyase expression and decrease inflammation and oxidative stress in the myocardium of ovariectomized rats. Menopause 2013, 20, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Chun, J.Y.; Kim, M.S. Insulin stimulates production of nitric oxide via ERK in osteoblast cells. Biochem. Biophys. Res. Commun. 2000, 278, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; He, X.; Wei, W.; Wang, J.; Zhang, T.; Shen, X. Tenascin-C induces resistance to apoptosis in pancreatic cancer cell through activation of ERK/NF-κB pathway. Apoptosis 2015, 20, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Mintz, G.S.; Biro, S.; Lee, J.B.; Sum, S.T.; Madden, S.P.; Burke, A.P.; Zhang, P.; He, B.; Goldstein, J.A.; et al. Insights into echo-attenuated plaques, echolucent plaques, and plaques with spotty calcification: novel findings from comparisons among intravascular ultrasound, near-infrared spectroscopy, and pathological histology in 2294 human coronary artery segments. J. Am. Coll. Cardiol. 2014, 63, 2220–2233. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, H.; Chen, Z.; Zhang, X.; Li, Y.; Zhang, J.; Chen, Y.; Ding, Y.; Zhang, G.; Yang, C.; Zhu, Y.; et al. Urotensin II Protects Cardiomyocytes from Apoptosis Induced by Oxidative Stress through the CSE/H2S Pathway. Int. J. Mol. Sci. 2015, 16, 12482-12498. https://doi.org/10.3390/ijms160612482
Gong H, Chen Z, Zhang X, Li Y, Zhang J, Chen Y, Ding Y, Zhang G, Yang C, Zhu Y, et al. Urotensin II Protects Cardiomyocytes from Apoptosis Induced by Oxidative Stress through the CSE/H2S Pathway. International Journal of Molecular Sciences. 2015; 16(6):12482-12498. https://doi.org/10.3390/ijms160612482
Chicago/Turabian StyleGong, Hui, Zhidan Chen, Xiaoyi Zhang, Yang Li, Jie Zhang, Ying Chen, Yingjiong Ding, Guoping Zhang, Chunjie Yang, Yichun Zhu, and et al. 2015. "Urotensin II Protects Cardiomyocytes from Apoptosis Induced by Oxidative Stress through the CSE/H2S Pathway" International Journal of Molecular Sciences 16, no. 6: 12482-12498. https://doi.org/10.3390/ijms160612482