
Anti-Fibrotic Effects of Class I HDAC Inhibitor, Mocetinostat Is Associated with IL-6/Stat3 Signaling in Ischemic Heart Failure

Abstract
:1. Introduction
2. Results
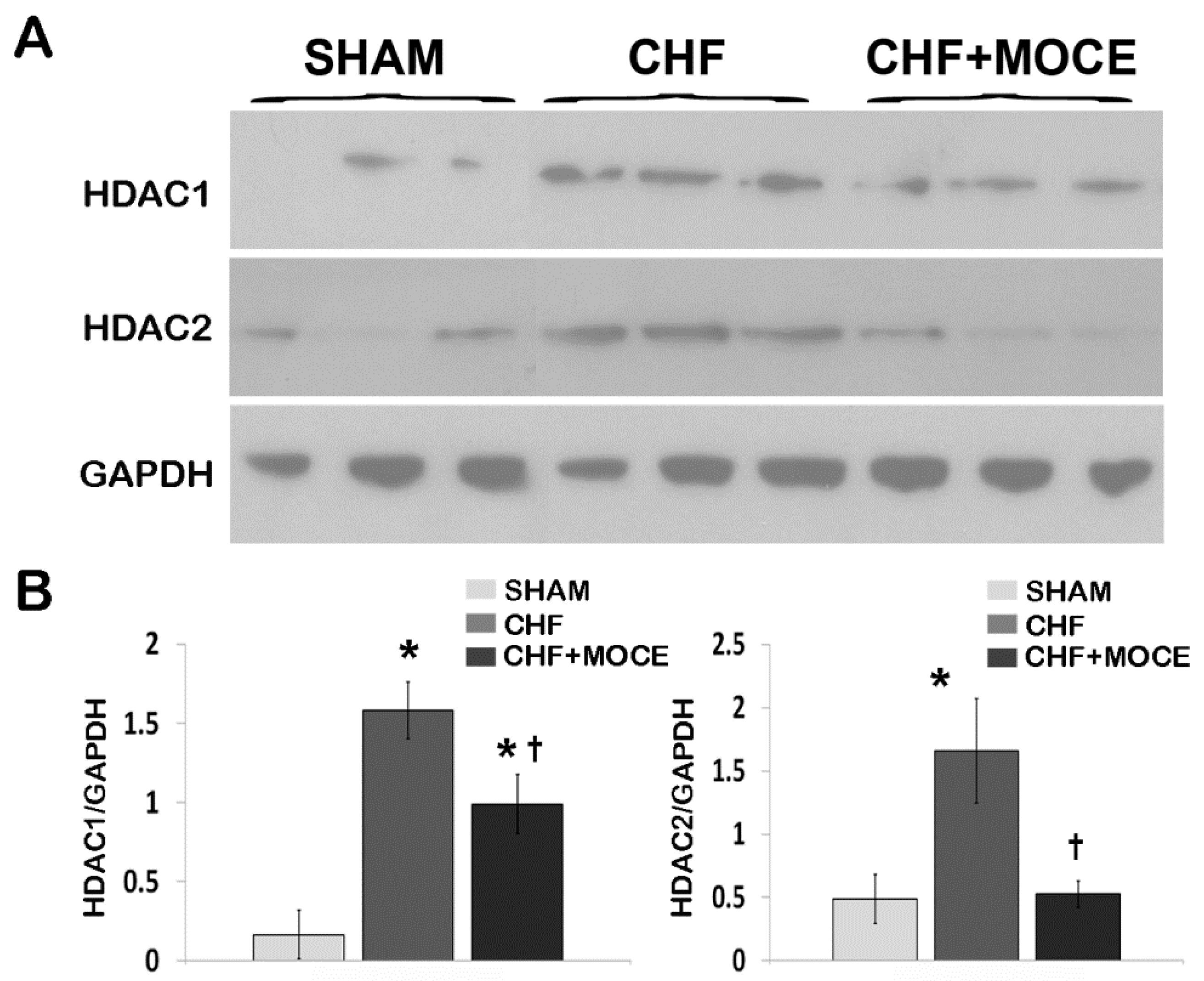
2.1. Mocetinostat Reduced Levels of HDAC1 and HDAC2 in CHF
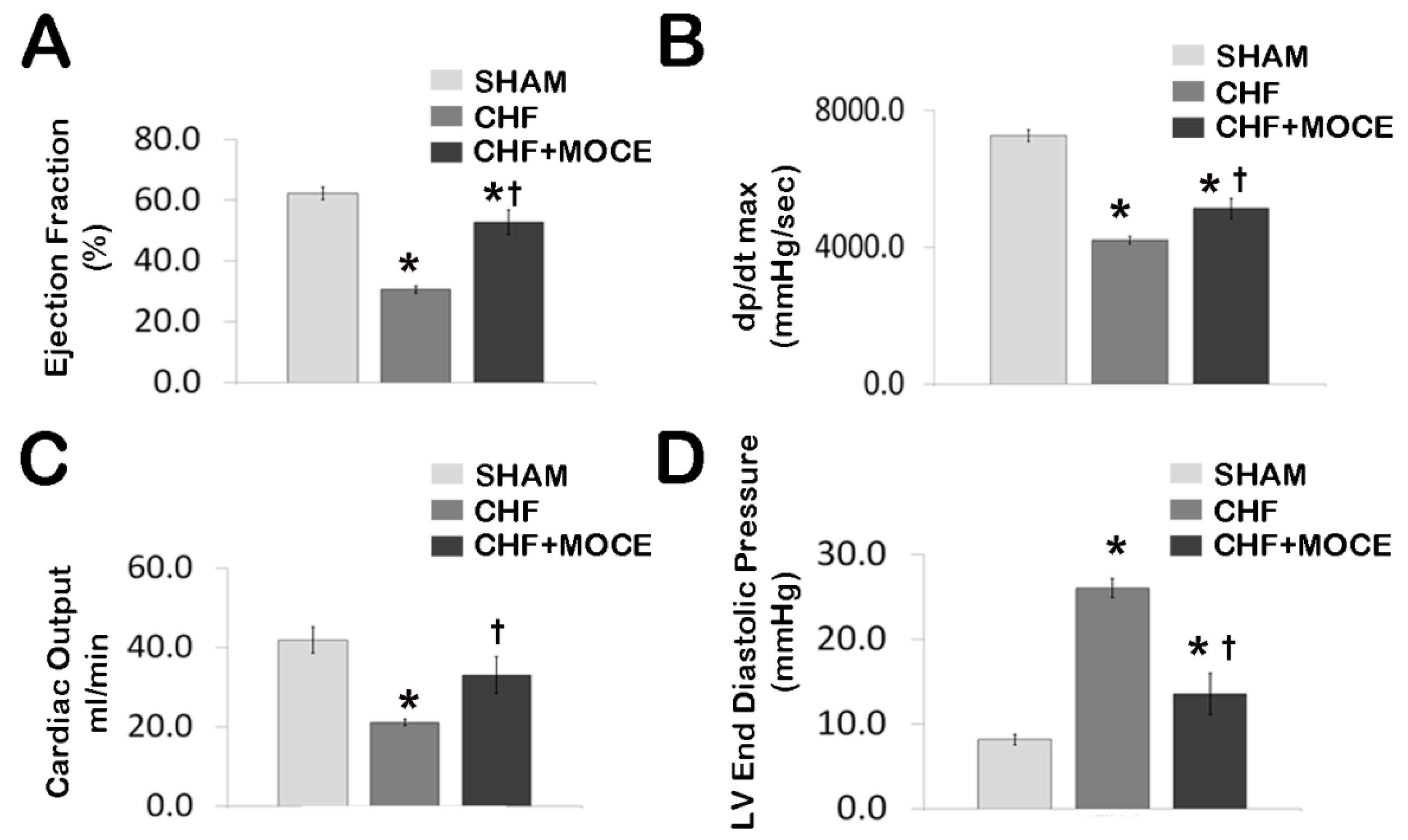
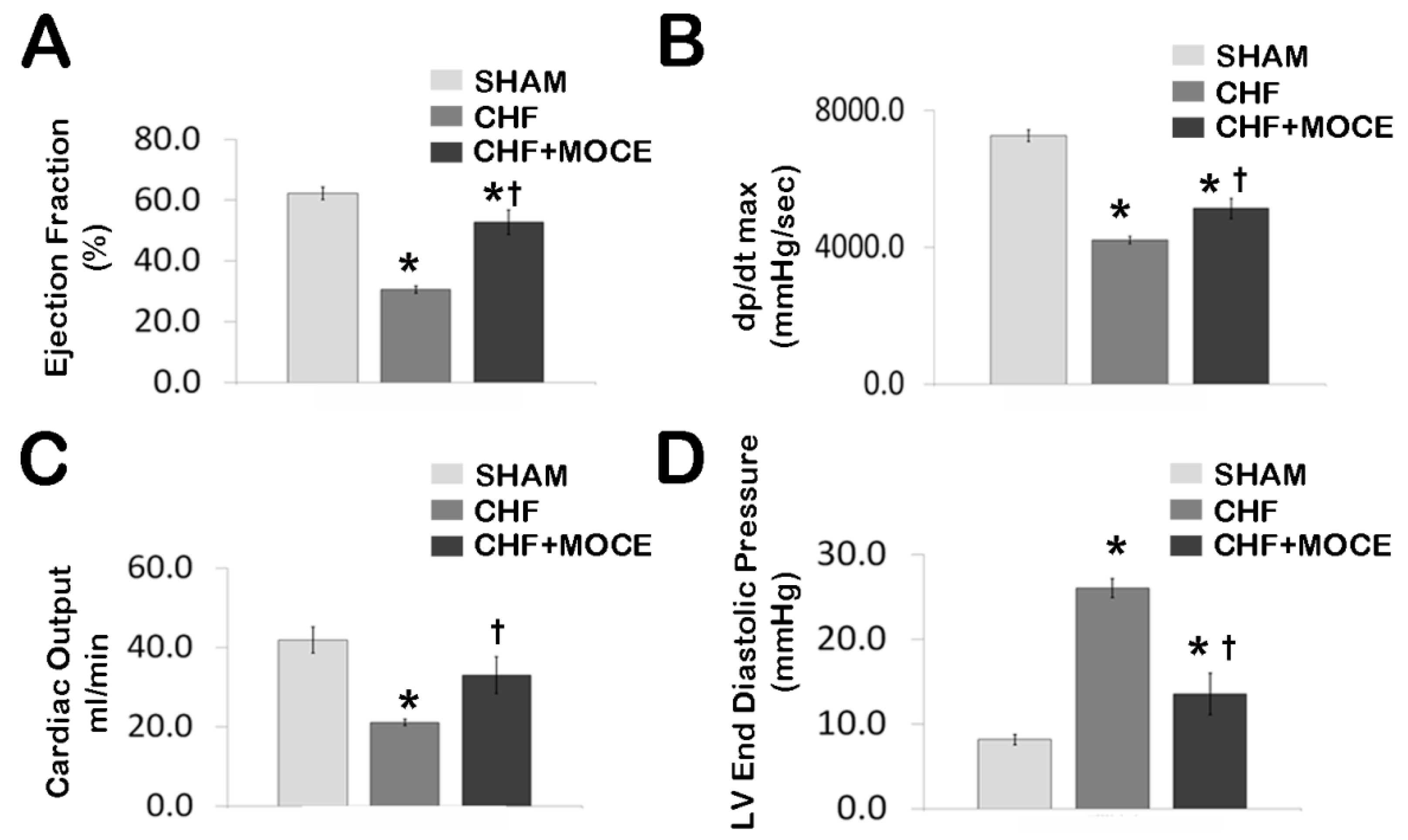
2.2. Mocetinostat at 20 mg/kg Improved Cardiac Function in CHF
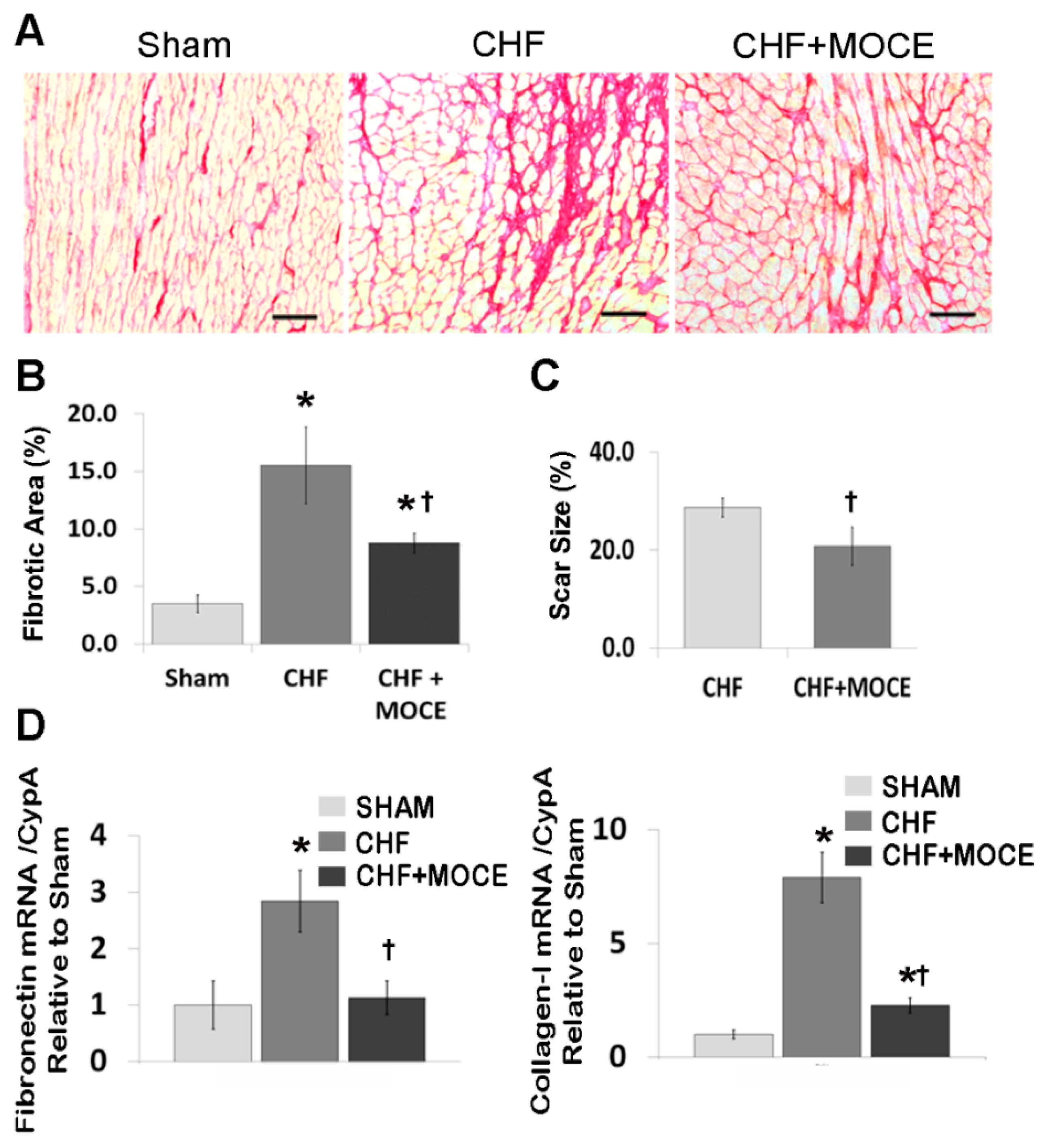
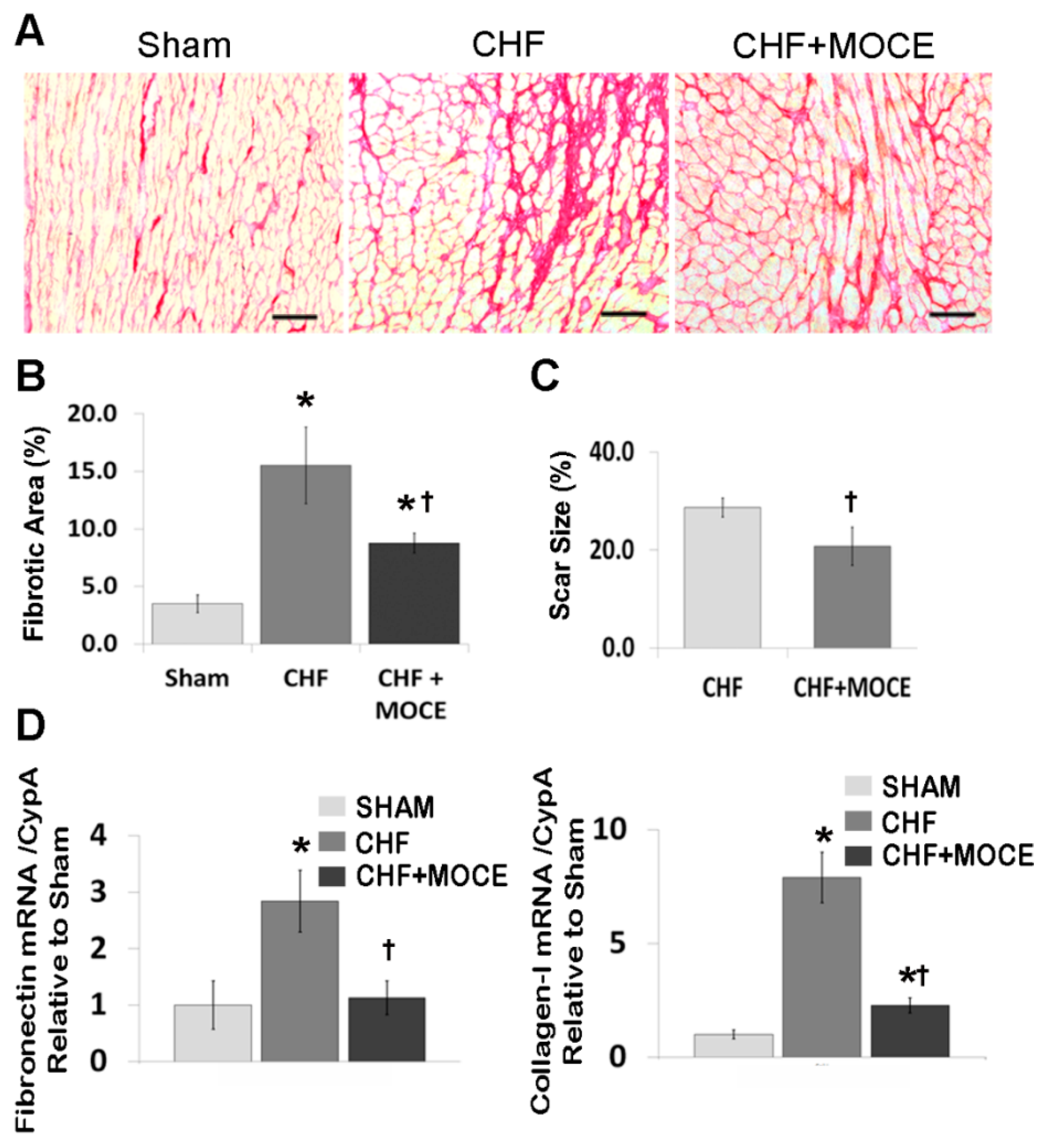
2.3. Mocetinostat at 20 mg/kg/day Reduced Interstitial Fibrosis and Scar Size
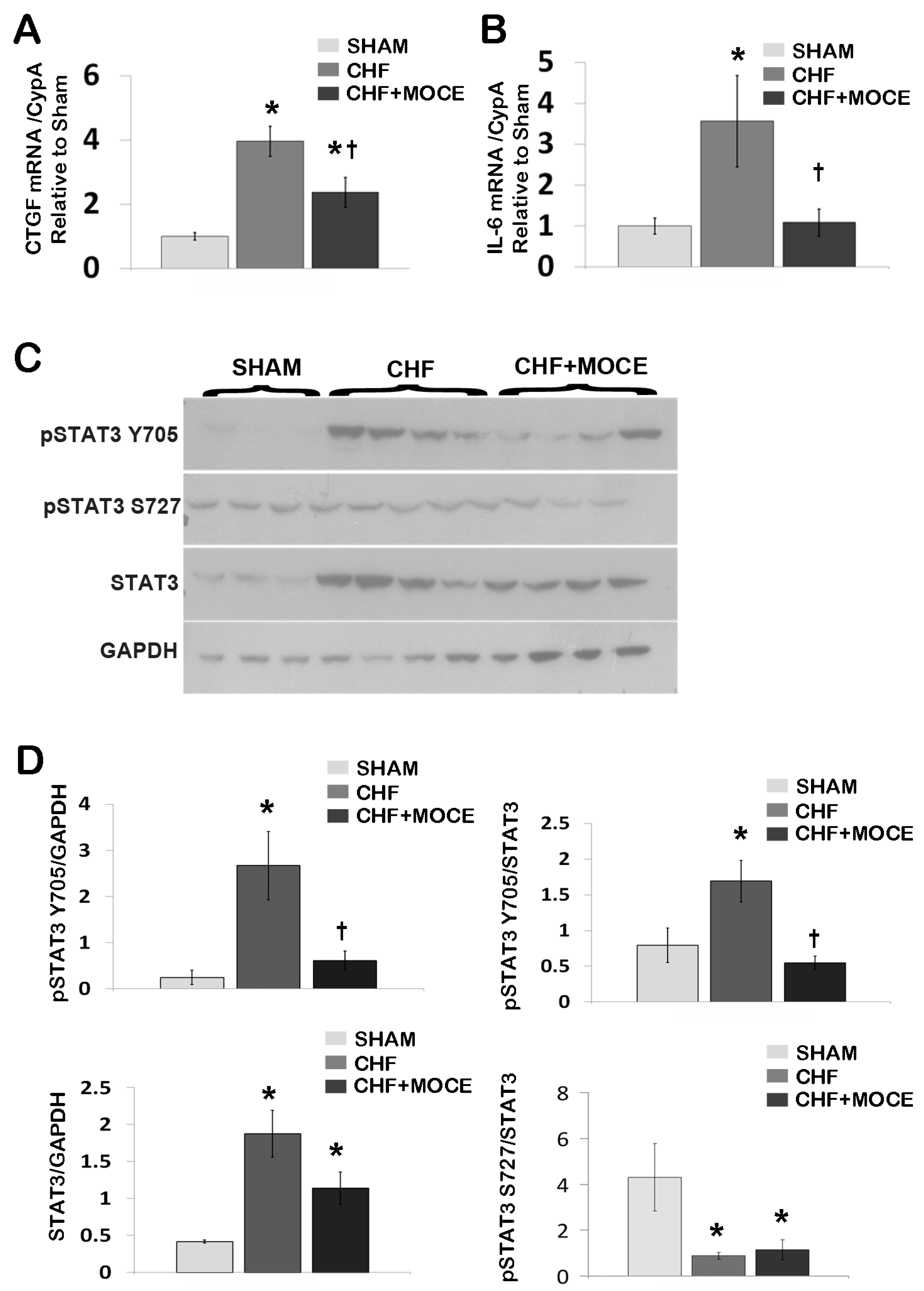
2.4. Mocetinostat Reduced the Expression of Connective Tissue Growth Factor in CHF Myocardium
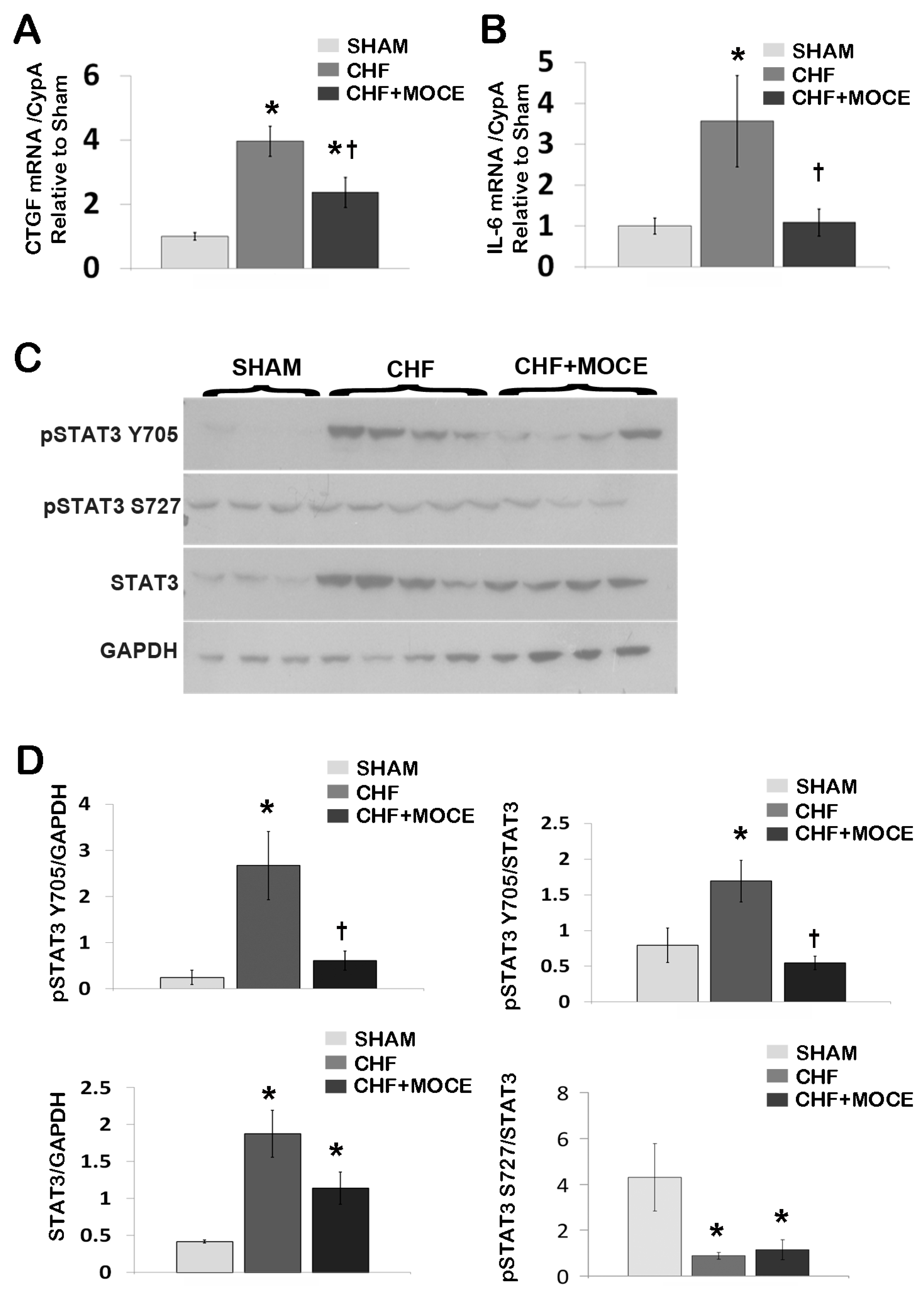
2.5. Mocetinostat Modulates IL-6 and Stat3 Signaling in CHF Myocardium
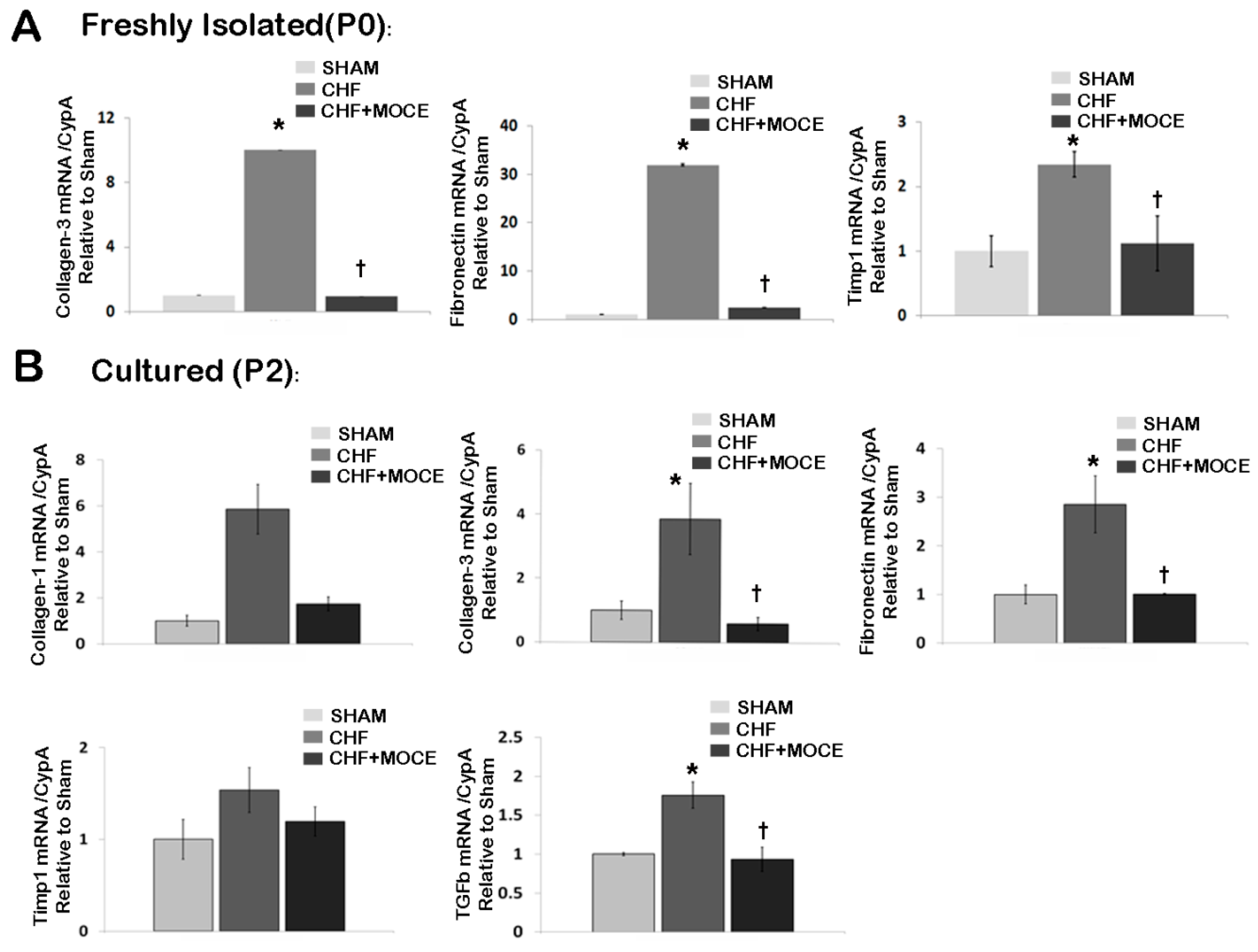
2.6. Mocetinostat Reduced Fibronectin and Collagen Levels in Cardiac Fibroblasts
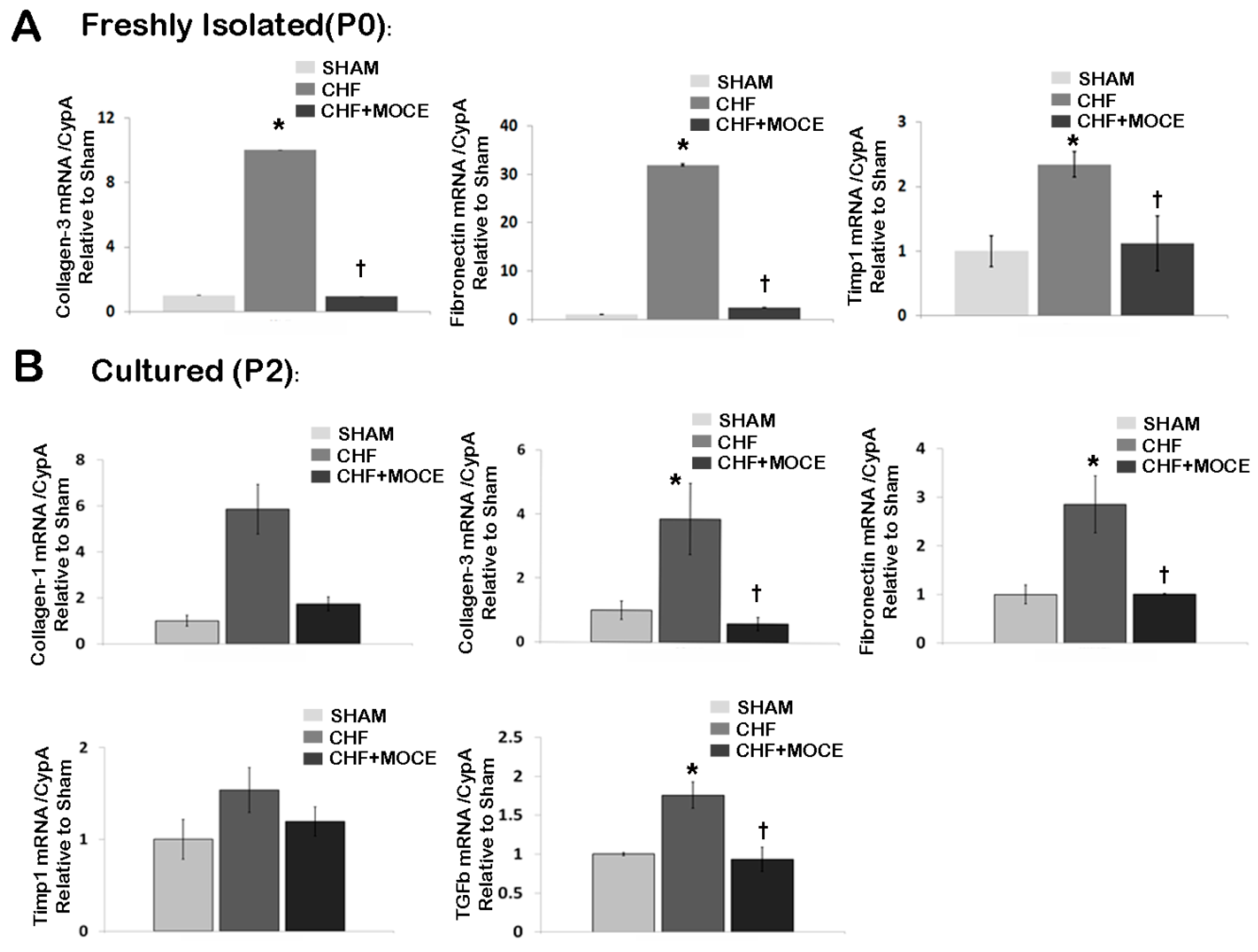
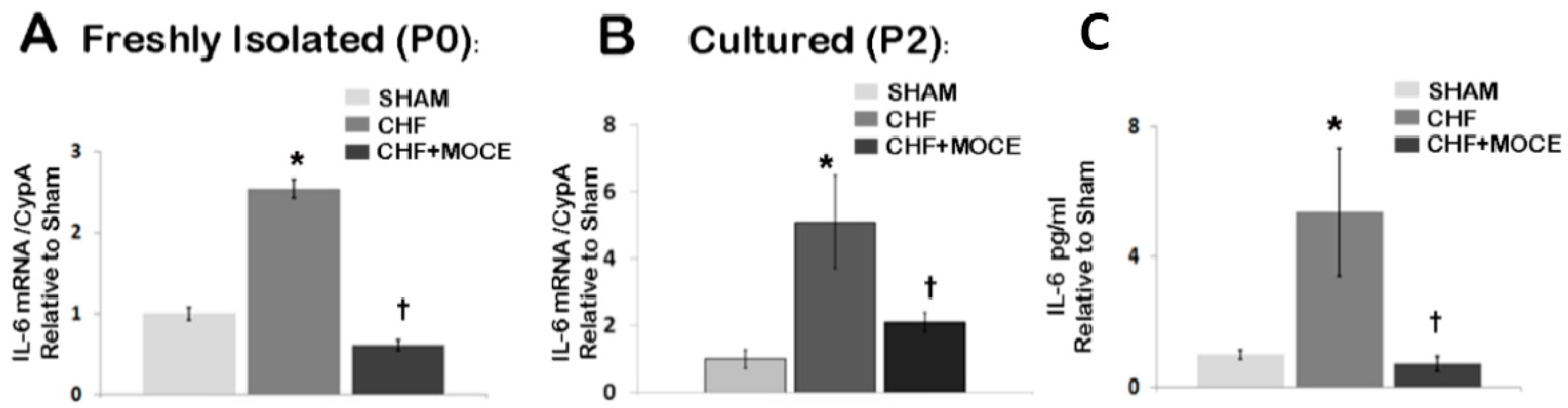
2.7. Mocetinostat Reduced IL-6 Levels in Cardiac Fibroblast
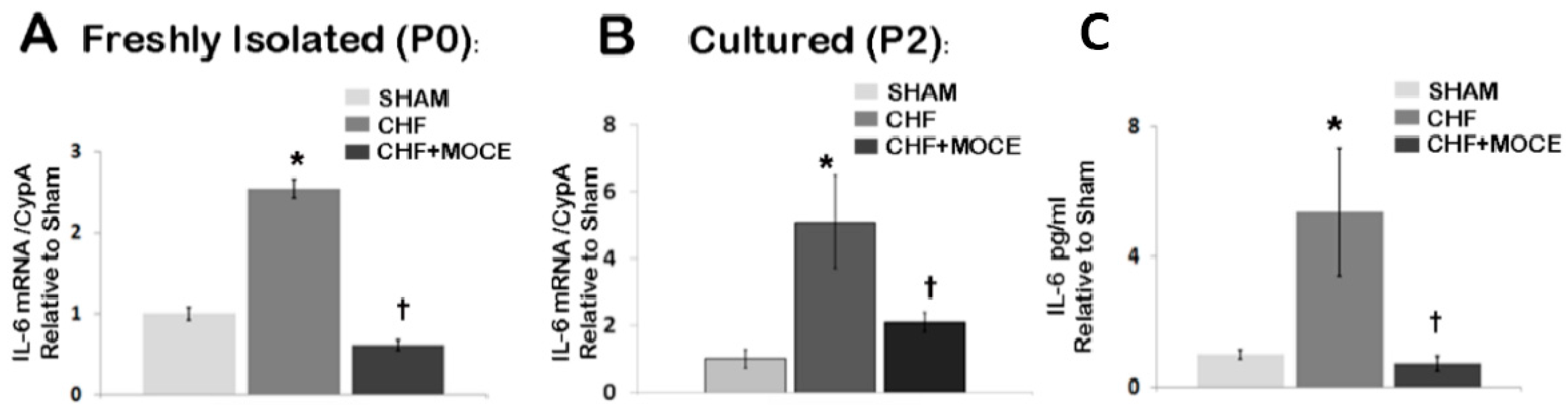
3. Discussion
4. Experimental Section
4.1. Myocardial Infarction and Treatments
4.2. Cell Isolation and Culture
4.3. Immunostaining
4.4. Scar Size Assessment and Collagen Assay
4.5. RNA Isolation and Quantitative Real-Time RT-PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 5'-3' Sequence |
|---|---|
| TGF-β1 F | CGAGGTGACCTGGGCACCATCCATGAC |
| TGF-β1 R | CTGCTCCACCTTGGGCTTGCGACCCAC |
| CTGF F | CAGGCTGGAGAAGCAGAGTCGT |
| CTGF R | CTGGTGCAGCCAGAAAGCTCAA |
| PDGF F | GGACGCGTAGAACAATCGGG |
| PDGF R | TGAACGGGTTGCTCGAGGTC |
| Collagen-1 F | TGCCGTGACCTCAAGATGTG |
| Collagen-1 R | CACAAGCGTGCTGTAGGTGA |
| Collagen-3 F | TCCCAGAACATTACATACCACT |
| Collagen-3 R | GCTATTTCCTTCAGCCTTGA |
| TIMP-1 F | ATAGTGCTGGCTGTGGGGTGTG |
| TIMP-1 R | TGATCGCTCTGGTAGCCCTTCTC |
| Fibronectin F | GATGCCGATCAGAAGTTTGGA |
| Fibronectin R | TCGTTGGTCGTGCAGATCTC |
| IL-6 F | CTCTCCGCAAGAGACTTCCA |
| IL-6 R | GTCTCCTCTCCGGACTTGTG |
| IL-18 F | CGAACAGCCAACGAATCCCA |
| IL-18 R | TAGGGTCACAGCCAGTCCTC |
| Cyp-A F | TATCTGCACTGCCAAGACTGAGTG |
| Cyp-A R | CTTCTTGCTGGTCTTGCCATTCC |
4.6. Western Blotting and ELISA
4.7. Statistical Analysis
Supplementary Materials
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Squires, C.E.; Escobar, G.P.; Payne, J.F.; Leonardi, R.A.; Goshorn, D.K.; Sheats, N.J.; Mains, I.M.; Mingoia, J.T.; Flack, E.C.; Lindsey, M.L. Altered fibroblast function following myocardial infarction. J. Mol. Cell. Cardiol. 2005, 39, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Van den Borne, S.W.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Fenning, A.; Lim, J.; Le, G.T.; Reid, R.C.; Halili, M.A.; Fairlie, D.P.; Brown, L. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats. Br. J. Pharmacol. 2010, 159, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, Y.H.; Liou, J.P.; Chung, C.C.; Lien, G.S.; Kuo, C.C.; Chen, S.A.; Chen, Y.J. Histone deacetylase inhibition improved cardiac functions with direct antifibrotic activity in heart failure. Int. J. Cardiol. 2013, 168, 4178–4183. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Sohn, I.S.; Nam, K.I.; Park, J.E.; Qian, Y.R.; Yin, Z.; Ahn, Y.; Jeong, M.H.; Bang, Y.J.; Kim, N.; et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Nural-Guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D.; Gaballa, M.A. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation Fibrogenesis. Tissue Repair 2014, 7, 10. [Google Scholar] [CrossRef]
- Williams, S.M.; Golden-Mason, L.; Ferguson, B.S.; Douglas, K.B.; Cavasin, M.A.; Demos-Davies, K.; Yeager, M.E.; Stenmark, K.R.; McKinsey, T.A. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J. Mol. Cell. Cardiol. 2013, 67, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Lin, M.S.; Chang, N.C. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats American journal of physiology. Heart Circ. Physiol. 2007, 293, H968–H977. [Google Scholar] [CrossRef]
- Kook, H.; Lepore, J.J.; Gitler, A.D.; Lu, M.M.; Wing-Man Yung, W.; Mackay, J.; Zhou, R.; Ferrari, V.; Gruber, P.; Epstein, J.A. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J. Clin. Investig. 2003, 112, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, B.; Zhao, Y.; Dubielecka, P.M.; Wei, L.; Qin, G.J.; Chin, Y.E.; Wang, Y.; Zhao, T.C. Inhibition of histone deacetylase-induced myocardial repair is mediated by c-kit in infarcted hearts. J. Biol. Chem. 2012, 287, 39338–39348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, X.; Zhao, Y.; Fast, L.; Zhuang, S.; Liu, P.; Cheng, G.; Zhao, T.C. Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. J. Pharmacol. Exp. Ther. 2012, 341, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Kapoun, A.M.; Liang, F.; OʼYoung, G.; Damm, D.L.; Quon, D.; White, R.T.; Munson, K.; Lam, A.; Schreiner, G.F.; Protter, A.A. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: Fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ. Res. 2004, 94, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.; Sarkar, S.; Vellaichamy, E.; Sen, S. Role of myocytes in myocardial collagen production. Hypertension 2001, 37, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Daniels, A.; van Bilsen, M.; Goldschmeding, R.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Connective tissue growth factor and cardiac fibrosis. Acta Physiol. 2009, 195, 321–338. [Google Scholar] [CrossRef]
- Koitabashi, N.; Arai, M.; Kogure, S.; Niwano, K.; Watanabe, A.; Aoki, Y.; Maeno, T.; Nishida, T.; Kubota, S.; Takigawa, M.; et al. Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension 2007, 49, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Haugen, E.; Gan, L.M.; Isic, A.; Skommevik, T.; Fu, M. Increased interleukin-6 but not tumour necrosis factor-alpha predicts mortality in the population of elderly heart failure patients. Exp. Clin. Cardiol. 2008, 13, 19–24. [Google Scholar] [PubMed]
- Hirota, H.; Izumi, M.; Hamaguchi, T.; Sugiyama, S.; Murakami, E.; Kunisada, K.; Fujio, Y.; Oshima, Y.; Nakaoka, Y.; Yamauchi-Takihara, K. Circulating interleukin-6 family cytokines and their receptors in patients with congestive heart failure. Heart Vessel. 2004, 19, 237–241. [Google Scholar] [CrossRef]
- Wollert, K.C.; Drexler, H. The role of interleukin-6 in the failing heart. Heart Fail. Rev. 2001, 6, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Booz, G.W. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J. Cardiovasc. Pharmacol. 2007, 50, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.; Hilfiker-Kleiner, D. Survival pathways in hypertrophy and heart failure: The gp130-STAT3 axis. Basic Res. Cardiol. 2007, 102, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Shukla, P.; Klein, G.; Schaefer, A.; Stapel, B.; Hoch, M.; Muller, W.; Scherr, M.; Theilmeier, G.; Ernst, M.; et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation 2010, 122, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Kothapally, J.; Mao, H.; Tolbert, E.; Ponnusamy, M.; Chin, Y.E.; Zhuang, S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2009, 297, F996–F1005. [Google Scholar] [CrossRef]
- Glauben, R.; Sonnenberg, E.; Wetzel, M.; Mascagni, P.; Siegmund, B. Histone deacetylase inhibitors modulate interleukin 6-dependent CD4+ T cell polarization in vitro and in vivo. J. Biol. Chem. 2014, 289, 6142–6151. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, L.; Mastroeni, D.; Mutlu, N.; Molina, M.; Goldman, S.; Diethrich, E.; Gaballa, M.A. Transplantation of cardiac progenitor cell sheet onto infarcted heart promotes cardiogenesis and improves function. Cardiovasc. Res. 2010, 87, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, F.; Gnemmi, I.; Vallario, A.; Genazzani, A.A.; Canonico, P.L. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br. J. Pharmacol. 2008, 153, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.A.; Chatterjee, A.; Mitra, A.; Pathak, K.; Mahata, S.K.; Sarkar, S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J. Biol. Chem. 2012, 287, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef] [PubMed]
- Fredj, S.; Bescond, J.; Louault, C.; Delwail, A.; Lecron, J.C.; Potreau, D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J. Cell. Physiol. 2005, 204, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Vellaichamy, E.; Young, D.; Sen, S. Influence of cytokines and growth factors in ANG II-mediated collagen upregulation by fibroblasts in rats: Role of myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H107–H117. [Google Scholar] [CrossRef] [PubMed]
- Mandala, T.; Bhowmik, A.; Chatterjee, A.; Chatterjee, U.; Chatterjee, S.; Ghosh, M.K. Reduced phosphorylation of Stat3 at Ser-727 mediated by casein kinase 2—Protein phosphatase 2A enhances Stat3 Tyr-705 induced tumorigenic potential of glioma cells. Cell. Signal. 2014, 26, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E.; et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Fujio, Y.; Nakanishi, T.; Itoh, N.; Yamamoto, Y.; Negoro, S.; Tanaka, K.; Kishimoto, T.; Kawase, I.; Azuma, J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc. Res. 2005, 65, 428–435. [Google Scholar] [CrossRef] [PubMed]
- McGaffin, K.R.; Moravec, C.S.; McTiernan, C.F. Leptin signaling in the failing and mechanically unloaded human heart. Circ. Heart Fail. 2009, 2, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Gaballa, M.A.; Raya, T.E.; Goldman, S. Large artery remodeling after myocardial infarction. Am. J. Physiol. 1995, 268, H2092–H2103. [Google Scholar] [PubMed]
- Gaballa, M.A.; Goldman, S. Gene transfer of endothelial nitric oxide isoform decreases rat hindlimb vascular resistance in vivo. Hum. Gene Ther. 2000, 11, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Hadi, A.M.; Mouchaers, K.T.; Schalij, I.; Grunberg, K.; Meijer, G.A.; Vonk-Noordegraaf, A.; van der Laarse, W.J.; Belien, J.A. Rapid quantification of myocardial fibrosis: A new macro-based automated analysis. Cell Oncol. 2011, 34, 343–354. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nural-Guvener, H.; Zakharova, L.; Feehery, L.; Sljukic, S.; Gaballa, M. Anti-Fibrotic Effects of Class I HDAC Inhibitor, Mocetinostat Is Associated with IL-6/Stat3 Signaling in Ischemic Heart Failure. Int. J. Mol. Sci. 2015, 16, 11482-11499. https://doi.org/10.3390/ijms160511482
Nural-Guvener H, Zakharova L, Feehery L, Sljukic S, Gaballa M. Anti-Fibrotic Effects of Class I HDAC Inhibitor, Mocetinostat Is Associated with IL-6/Stat3 Signaling in Ischemic Heart Failure. International Journal of Molecular Sciences. 2015; 16(5):11482-11499. https://doi.org/10.3390/ijms160511482
Chicago/Turabian StyleNural-Guvener, Hikmet, Liudmila Zakharova, Lorraine Feehery, Snjezana Sljukic, and Mohamed Gaballa. 2015. "Anti-Fibrotic Effects of Class I HDAC Inhibitor, Mocetinostat Is Associated with IL-6/Stat3 Signaling in Ischemic Heart Failure" International Journal of Molecular Sciences 16, no. 5: 11482-11499. https://doi.org/10.3390/ijms160511482